

Abstract
Background
Early Repolarization Syndrome (ERS) is associated with polymorphic ventricular tachycardia (PVT) and fibrillation (VF), leading to sudden cardiac death.
Objective
The present study tests the hypothesis that the Ito-blocking effect of phosphodiesterase-3 (PDE-3) inhibitors plays a role in reversing repolarization heterogeneities responsible for arrhythmogenesis in experimental models of ERS.
Methods
Transmembrane action potentials (AP) were simultaneously recorded from epicardial and endocardial regions of coronary-perfused canine left-ventricular (LV) wedge preparations, together with a transmural pseudo-ECG. The Ito-agonist NS5806 (7–15 μM) and ICa-blocker verapamil (2–3 uM) were used to induce an ER pattern and PVT.
Results
Following stable induction of arrhythmogenesis, the PDE-3 inhibitors cilostazol and milrinone or isoproterenol were added to the coronary perfusate. All were effective in restoring the AP dome in the LV epicardium, thus abolishing the repolarization defects responsible for phase-2-reentry (P2R) and PVT. Arrhythmic activity was suppressed in 7/8 preparations by cilostazol (10 μM), 6/7 by milrinone (2.5 μM) and 7/8 by isoproterenol (0.1–1μM). Using voltage clamp techniques applied to LV epicardial myocytes, both cilostazol (10 μM) and milrinone (2.5 μM) were found to reduce Ito by 44.4% and 40.4%, respectively, in addition to their known effects to augment ICa.
Conclusions
Our findings suggest that PDE-3 inhibitors exert an ameliorative effect in the setting of ERS by producing an inward shift in the balance of current in the early phases of the epicardial AP via inhibition of Ito as well as augmentation of ICa, thus reversing the repolarization defects underlying development of P2R and VT/VF.
Keywords: Phosphodiesterase-3 inhibitors, Transient outward potassium current, Pharmacology, Ventricular fibrillation, Sudden Cardiac Death, Electrophysiology, Early Repolarization
INTRODUCTION
Early Repolarization Syndrome (ERS) has attracted a great deal of attention because of its association with sudden cardiac death (SCD) secondary to development of malignant arrhythmias.1, 2 Case-control and population-based studies point to an association of ER with the development of ventricular tachycardia (VT) and fibrillation (VF) in select individuals. ER pattern (ERP) in the ECG is characterized by a J point elevation ≥ 0.1mV manifesting as a notch or slur on the final 50% of the downslope of the QRS (J wave).1–5. According to the most recent consensus reports by Macfarlane et al., J point is to be measured at the peak of the end-QRS notch or the onset of the end-QRS slur, labelled as Jp. Early case-controlled studies by Haïssaguerre and co-workers, Nam and co-workers and Rosso and co-workers reported an ERP in the ECGs of 31–60 % of their patients presenting with idiopathic ventricular fibrillation6–8. More recent studies report association of ERP with a higher risk for development of arrhythmias during acute myocardial infarction (AMI)9 and therapeutic hypothermia10, 11.
Recent studies from our group have provided evidence in support of the hypothesis that ERS is caused by a preferential accentuation of the AP notch in the LV epicardium secondary to an outward shift in the balance of current contributing to the early phases of the action potential.4, 12 Higher intrinsic levels of Ito were shown to account for the greater sensitivity of the inferior LV wall to development of VT/VF in the setting or ERS.4 The outward shift in the balance of currents was attributable to mutations in genes causing a gain of function in IK-ATP (KCNJ8 and ABCC9) or Ito (KCNE5 mutation and rare polymorphism in DPP10)13–17 or loss of function in ICa (CACNA1C, CACNB2 and CACNA2D1)18, 19 or INa (SCN5A and SCN10A).20, 21 The goal of a pharmacologic approach to therapy should therefore be to produce an inward shift in the balance of current flowing during the early phases of the LV epicardial AP.
In previous studies, we demonstrated that induction of mild ER by an outward shift in the balance of current during the early phases of the AP could potentiate the effect of hypothermia to induce VT and that a pharmacologic-induced inward shift of current, using PDE-3 inhibitors or Ito block with quinidine, exerts an ameliorative effect.12 The present study provides a direct test of the hypothesis that both PDE-3 inhibitors reduce Ito, significantly contributing to their ameliorative effect in J wave syndromes. We examine the effects of two PDE-3 inhibitors in the absence of hypothermia and contrast their actions with isoproterenol which is widely used in the clinic to suppress J wave syndrome-associated arrhythmias. We avoid using ACh in the pharmacologic model so as to exclude the potential anticholinergic effect as a contributor to their antiarrhythmic actions.
METHODS
Our ERS models were created using coronary-perfused wedge preparation isolated from the hearts of adult mongrel dogs of either sex in conformance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication No 85-23, Revised 1996) and approved by the Institutional Animal Care and Use Committee.
Arterially perfused left ventricle wedge preparation
The methods employed were as previously described.22 Wedge preparations were excised from the lateral (25%) and the inferior wall (75%) of the left ventricle and were perfused via distal branches of the left anterior descending artery, left marginal artery or left posterior artery. Floating glass microelectrode techniques were used to simultaneously record action potentials from two epicardial (Epi1, Epi2) and one endocardial (Endo) site. Together with the action potentials, pseudo-ECG was recorded using two AgCl electrodes within the bath positioned along the transmural axis of the preparation. The epicardial microelectrodes were used to map the epicardial surface of the preparation so as to reveal heterogeneities in depolarization and/or repolarization.
Voltage-clamp measurement of Ito
Cardiomyocytes were isolated from the epicardium of the canine left ventricle as described previously23. Ito was measured at 36.5 °C using whole-cell patch clamp techniques.4 Ito was evoked using a series of 370 ms voltage steps from −40 mV and +40 mV ms; holding potential was maintained at −80 mV and 40 ms prepulse to −20 mV was used to discharge the sodium current. Series resistance was compensated ~80% and cells with an Rs values greater than 5 MΩ were discarded from analysis. External solutions contained (in mM): 127 NaCl, 4 KCl, 10 HEPES, 1.8 CaCl2, 1.0 MgCl2, 10 glucose; pH=7.35 with NaOH. The pipette solution contained (in mM): 125 Potassium aspartate, 10 KCl, 10 NaCl, 1 MgCl2, 10 HEPES, 5 EGTA, 5 Mg2ATP; pH = 7.2 with KOH.
AP and ECG parameters were defined and calculated as described in previous studies and briefly outlined in the online supplement12.
Statistical analysis
Results are presented as mean ± S.E.M. Statistical analysis was performed using paired Student’s t-test and one-way ANOVA for repeated measurements followed by pairwise comparisons corrected using the Holm-Sidak method, as appropriate. Statistical significance was considered at P < 0.05.
RESULTS
Induction of ERP and VT/VF
We induced the early repolarization phenotype by adding the transient outward potassium current (Ito) agonist NS5806 (7–15 μM) and the calcium channel blocker verapamil (2–3 μM) to the coronary perfusate. This led to accentuation of the action potential notch in epicardium but not endocardium, thus leading to augmentation of the electrocardiographic J wave secondary to amplification of the transmural voltage gradients (Figures 1–3). Increased concentrations of provocative agents caused a further increase of J wave area and notch-index (Figures 1–4; Table 1), leading to all-or-none repolarization at the end of phase 1 of the Epi AP (Figures 1–4). Loss of the Epi AP dome at some sites but not others resulted in a prominent increase in epicardial dispersion of repolarization (EDR) and transmural dispersion of repolarization (TDR) (Figures 1–4; Table 1). The voltage gradient between the abbreviated Epi AP and the relatively normal Endo AP produced a prominent ST segment elevation (Figures 1–3). A prominent APD gradient developed between sites at which the dome was maintained and where the dome was lost, thus creating a vulnerable window within epicardium as well as between epicardium and endocardium across the left ventricular wall. Propagation of the AP dome from regions at which it was maintained to regions at which it was lost, caused local re-excitation via a P2R mechanism, leading to the development of closely coupled extrasystoles and polymorphic VT/VF (Figures 1–3).
Figure 1.
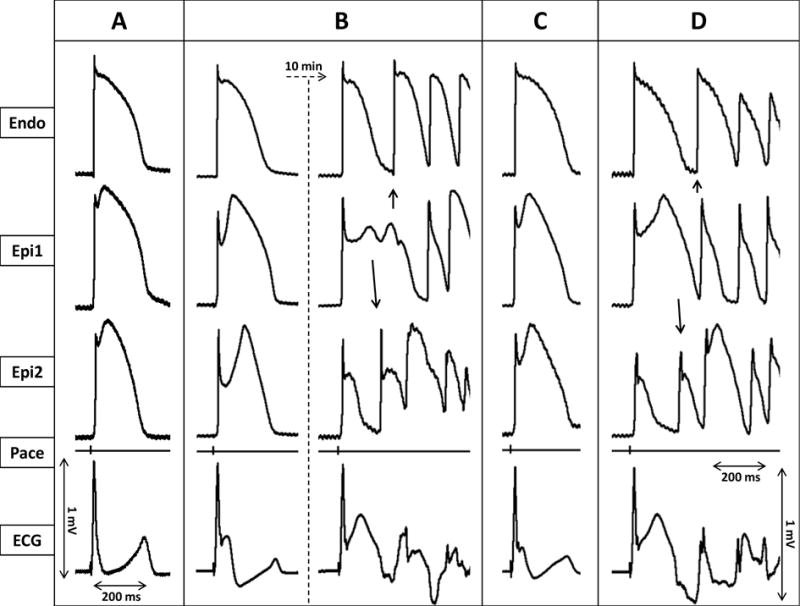
Ameliorative effect of cilostazol (10μM) in an arterially perfused canine left ventricular model of early repolarization syndrome. Each panel shows simultaneously recorded epicardial (Epi1, Epi2) and endocardial (Endo) action potentials, together with a pseudo-ECG. A: Control. B: Recorded 20 min and 30 min after addition of verapamil (2 μM) and NS5806 (7 μM) to the coronary perfusate. C: 15 minutes after addition of 10 μM cilostazol to the coronary perfusate. D: Recorded 15 min after withdrawal of cilostazol.
Figure 3.
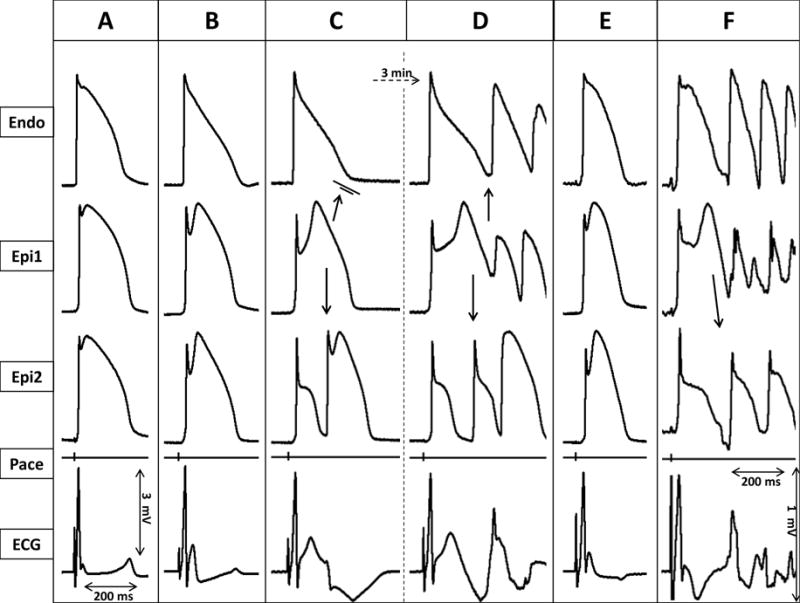
Ameliorative effects of isoproterenol (0.2 μM) in an arterially perfused canine left ventricular model of early repolarization syndrome. Simultaneously recorded epicardial (Epi1, Epi2) and endocardial (Endo) action potentials, together with pseudo-ECG positioned in the transmural axis. A: Control. B: 20 min after addition of the Ito agonist NS5806 (12μM) to the coronary perfusate. C: After 16 min exposure to verapamil (2μM), in addition to NS5806 (12μM). D: 3 minutes later. E: 5 min after the start of isoproterenol (0.2μM) infusion. F: 5 min after the discontinuation of isoproterenol infusion.
Figure 4.
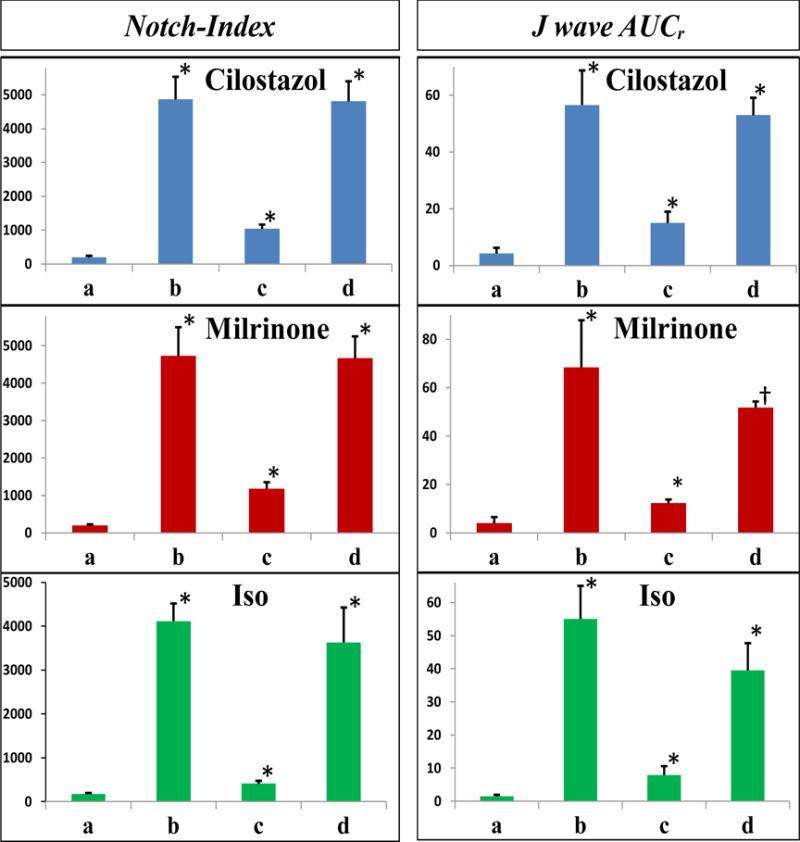
Notch-index (left) and normalized J wave area (right) at each experimental step. In each panel: a. Control; b. Recorded after application of the Ito agonist NS5806 and ICa antagonist verapamil; c. Recorded after addition of cilostazol 10μM (top row), milrinone 2.5μM (middle row), or isoproterenol 1μM (bottom row); d. Recorded after wash-out of drugs. Data are presented as mean ± SEM. Significance is shown relative to previous treatment: * P=<0.001; †P=<0.004. (n = 7 for cilostazol; n = 6 for milrinone; n = 7 for isoproterenol).
Table 1.
Effect of cilostazol, milrinone and isoproterenol on electrophysiological parameters in an experimental model of early repolarization syndrome.
| Notch-Index | EDR (ms) | TDR (ms) | J-w-AUCr (mV × ms) | Epi1 APD90 (ms) | Epi2 APD90 (ms) | Endo APD90 (ms) | |
|---|---|---|---|---|---|---|---|
| Cilostazol | |||||||
| Control | 199.07±44.9 | 16.2±8.2 | 17.8±4.3 | 4.3±2.0 | 203.6±7.8 | 182.8±5.6 | 217.7±5.1 |
| NS (7–15 μM) +Ver. (2–3μM) | 4863.6±669.4* | 137.9±8.1* | 111.8±13.4* | 56.6±12.2* | 252.2±7.8† | 109.4±5.6* | 237.2±14.3¶ |
| + Cilostazol (10μM) | 1041.7±124.2* | 11.9±3.8* | 21.3±3.8* | 15.0±4.0* | 218.4±7.5§ | 205.3±4.4* | 235.6±7.3¶ |
| Washout Cilostazol | 4809.8±590.7* | 123.3±10.6* | 106.9±11.2* | 53.0±6.2* | 252.7±10 | 121.8±9.4* | 237.3±11.5¶ |
| Milrinone | |||||||
| Control | 198.2±287 | 17.3±9.3 | 17.8±3.8 | 4.0±2.5 | 215.7±6.9 | 194.8±5.9 | 227.3±5.92 |
| NS (7–15 μM) +Ver. (2–3μM) | 4732.0±764.8* | 143.0±12.2* | 110±7.9* | 68.4±19.5* | 250.1±14 | 104.9±8* | 230.1±4.7¶ |
| Milrinone (2.5μM) | 1181.8±170.1* | 3.9±0.7* | 11.6±3.7* | 12.268±1.5* | 213.3±6.1¶ | 212.6±6.6* | 232.6±5.9¶ |
| Washout Milrinone | 4668.5±583.6* | 150.4±15.1* | 115.6±12.4* | 51.7±2.6† | 267.8±10.7† | 114.2±9.23* | 243.5±6.3¶ |
| Isoproterenol | |||||||
| Control | 176.8±19.9 | 13.5±5.6 | 14.9±5 | 1.6±0.4 | 206.5±6.9 | 190.7±5.9 | 213.4±4.9 |
| NS (7–15 μM) +Ver. (2–3 μM) | 4114.7±400.8* | 123.9±11.9* | 88.9±5.2 * | 46.0.0±4.9* | 234.5±12¶ | 111.2±9.3* | 198.5±6.3¶ |
| +Iso. (1μM) | 406.9±65.6* | 5.9±1.6* | 13.6±4* | 7.8±2.7* | 164.1±8.3* | 158.6±7.7* | 179.±7.8¶ |
| Washout Iso | 3628.6.±795.9* | 87.7±25.4* | 67.1±15.0* | 39.5±8.2* | 207.9±22.6¶ | 116.0±14.3§ | 195.4±11.5¶ |
NS=NS5806; Ver.=Verapamil; Iso=Isoproterenol; J-w-AUCr=J wave area (normalized) Data are presented as mean ± SEM
P=<0.001
P=<0.004
: P<0.05
P>0.05. n=7 for cilostazol, n=6 for milrinone, n=7 for isoproterenol(Epi1 represents the longer, Epi2 the shorter epicardial APD90)
Induction of ERP was observed in all experiments. Table 1 and Figures 4 and 5 show the effect of the provocative agents to significantly increase notch-index, J wave area, EDR and TDR compared to the controls. PVT and/or VF developed in 26 of 28 LV wedge preparations, compared with 0 of 28 cases under control conditions.
Figure 5.
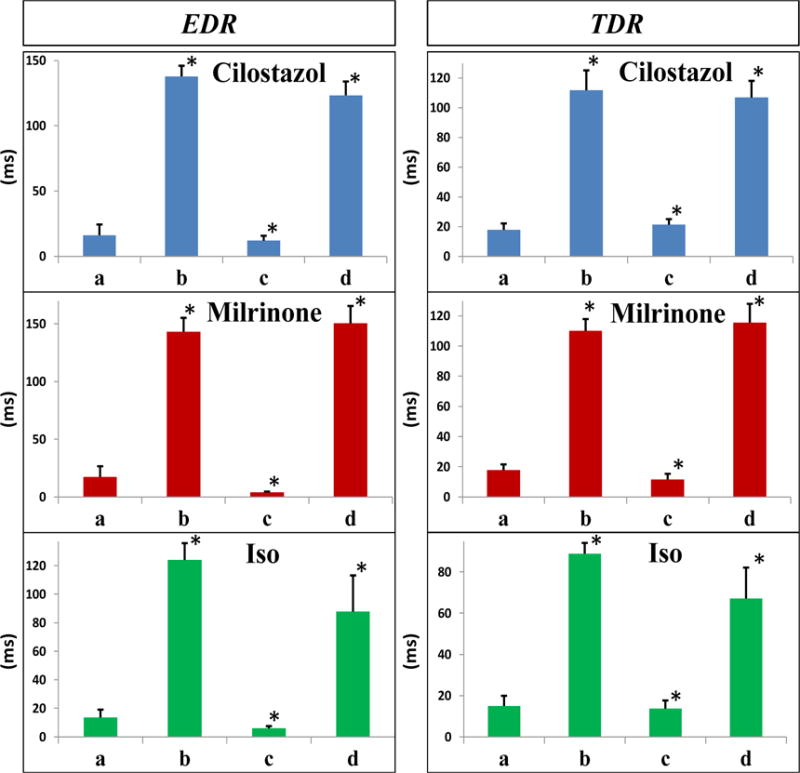
Epicardial (EDR, left) and transmural (TDR, right) dispersion of repolarization at each experimental step. In each panel: a. Control; b. Recorded after application of the Ito agonist NS5806 and ICa antagonist verapamil; c. Recorded after addition of cilostazol 10μM (top row), milrinone 2.5μM (middle row), or isoproterenol 1μM (bottom row); d. Recorded after wash-out of drugs. Data are presented as mean ± SEM. Significance is shown relative to previous treatment: * P=<0.001 (n = 7 for cilostazol; n = 6 for milrinone; n = 7 for isoproterenol).
Effects of cilostazol, milrinone and isoproterenol
Addition of cilostazol (10 μM), milrinone (2.5 μM) or isoproterenol (0.1–1 μM) to the coronary perfusate restored the AP dome at all epicardial sites, reduced epicardial and transmural dispersion of repolarization, decreased J point and ST segment elevation and terminated all arrhythmic activity. Figures 1–3 show representative recordings of APs from LV wedge preparations obtained under baseline conditions, after NS5806 (7–15 μM), + verapamil (2–3 μM), + PDE-3 inhibitor (10 μM cilostazol or 2.5 μM milrinone) or the sympathomimetic agent isoproterenol (0.1–1 μM) and after washout of the therapeutic compounds.
The effects of milrinone, cilostazol and isoproterenol on epicardial AP notch index, J wave area, EDR and TDR as well as APD90 values for Epi1, Epi2 and Endo are summarized in Figures 4–5 and Table 1. All three agents, by virtue of their action to produce an inward shift of balance of currents, reversed the effect of the provocative agents, restoring all electrophysiologic parameters towards normal. Cilostazol (10 μM), milrinone (2.5 μM) and isoproterenol (0.1–1 μM) restored the AP dome at all epicardial sites, thus reducing notch index, J wave area, as well as epicardial and transmural dispersion of repolarization (Figures 1–5 and Table 1).
Figures 1–3 illustrates the development of polymorphic VT following exposure to NS5806 and verapamil and the effect of cilostazol (10 μM), milrinone (2.5 μM) and isoproterenol (0.2 μM) to normalize the ECG and to terminate all arrhythmic activity. Cilostazol (10 μM) abolished VT/VF in 7 of 8 preparations, whereas milrinone (2.5 μM) abolished VT/VF in 6 out of 7 preparations, and isoproterenol terminated VT/VF in 7 of 8. In all cases, washout of the drug resulted in reappearance of arrhythmic activity (Figures 1–3).
Effect of cilostazol and milrinone on Ito in canine ventricular epicardial myocytes
We next evaluated the effect of cilostazol and milrinone on Ito in canine left ventricular epicardial myocytes using whole cell patch clamp techniques. Macroscopic outward potassium current (Ito) was recorded at body temperature from single cardiomyocytes using the whole cell patch-clamp technique. Figure 6A shows representative Ito traces recorded under control conditions (left, panel) and reduction of the current after addition of cilostazol (10 μM)(right panel). Peak Ito was evaluated using a square depolarization pulse to −40 mV to +40 mV applied once every 5 seconds. Figure 6B shows the effect of cilostazol (10 μM) on the I-V relationship. Cilostazol (10 μM) reduced Ito by 44.4 % at +40 mV (n=6, p<0.02; Figure 6C). Figure 7A shows representative Ito traces recorded under control conditions (left, panel) and the reduction of the current after addition of milrinone (2.5 μM)(right panel). Figure 7B shows the effect of milrinone (2.5 μM) on the I-V relationship. Milrinone (2.5 μM) reduced Ito by 40.4 % at +40 mV (n=8, p<0.02; Figure 7C).
Figure 6.
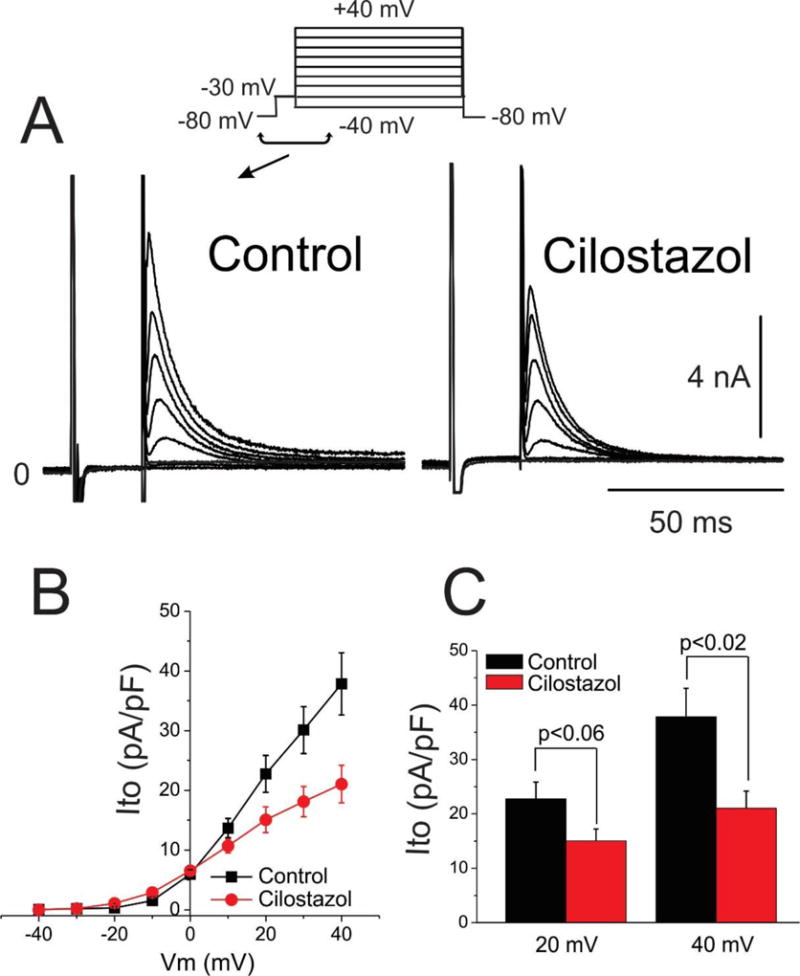
Cilostazol inhibition of transient outward potassium current (Ito). A. Representative macroscopic Ito current traces for control and cilostazol (10 μM). B. Current-Voltage (I–V) relationship for Ito density in control and in response to 10 μM cilostazol (n=6 for each). C. Bar graph showing Ito at following a step to 20 and 40 mV in presence and absence of cilostazol (10 μM). Mean ± SEM.
Figure 7.
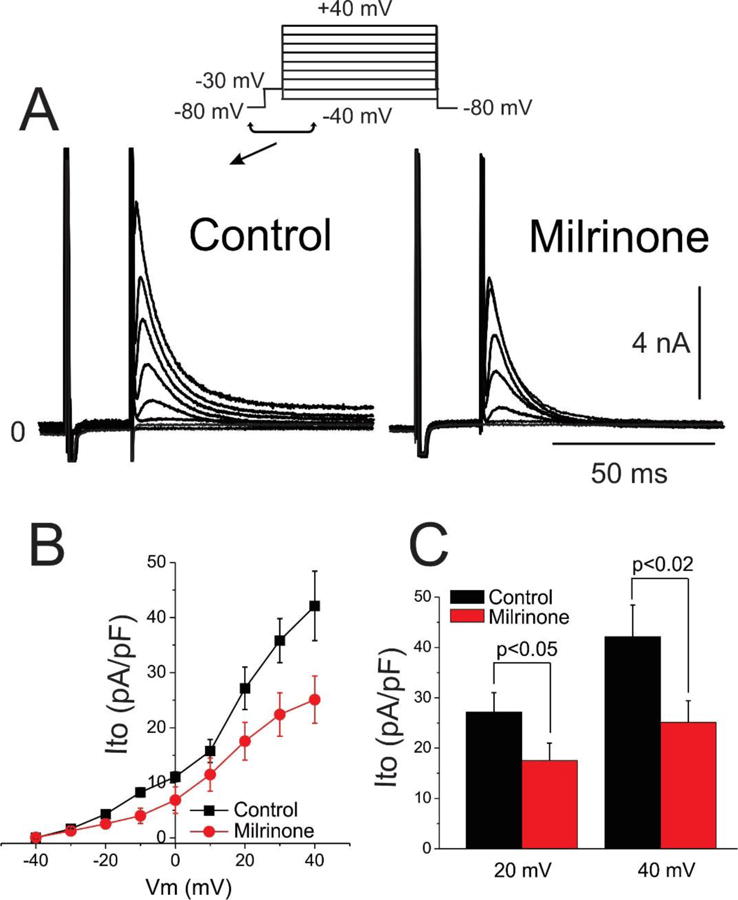
Milrinone inhibition of transient outward potassium current (Ito). A. Representative macroscopic Ito current traces for control and milrinone (2.5 μM). B. Current-Voltage (I–V) relationship for Ito density in control and in response to 2.5 μM milrinone (n=8 for each). C. Bar graph showing Ito at following a step to 20 and 40 mV in presence and absence of milrinone (2.5 μM). Mean ± SEM.
DISCUSSION and CONCLUSION
Our results demonstrate a significant effect of both cilostazol and milrinone to block Ito, pointing to this as an important mechanism for their ameliorative effect in ERS, in addition to their previously demonstrated effects to augment ICa24–26 suggesting that inhibition of Ito may also apply to isoproterenol (and other sympathomimetic) in addition to its boosting effect on ICa via direct stimulation of the β adrenergic receptors. Future experiments should be directed at a test of this hypothesis. It is noteworthy that cilostazol, milrinone and isoproterenol all produce positive inotropic and chronotropic effects. The elevation in heart rate would also be expected to indirectly decrease Ito because the current is relatively slow to recover form inactivation.
The much greater potency of milrinone is consistent with the results of previous studies reporting that the same concentration of milrinone produces a greater increase in cytosolic cAMP than cilostazol.27, 28 possibly due to the fact that milrinone blocks both PDE-3 and PDE4.28
It is noteworthy that all of these agents have the potential to enhance not only automaticity, but triggered activity as well, and thus may promote extrasystolic activity that may have unfavorable outcomes in certain cases.29
Previous reports from our group have provided evidence in support of a preferential accentuation of the AP notch in LV epicardium as the basis for ERS.4, 12 Accentuation of the epicardial AP notch leads to accentuation of transmural gradients across the LV wall and thereby the appearance of prominent J point elevation, distinct J waves, or slurring of the descending limb of the QRS complex. Consistent with an outward shift in the balance of current, ERS has been associated with loss of function mutations in the α1, β2 and α2δ subunits of the cardiac L-type calcium channel (CACNA1C, CACNB2, and CACNA2D1) resulting in a reduction in ICa18, 30, loss of function mutations in sodium channel genes (SCN5A and SCN10A)21, 31 causing a reduction in INa as well as gain of function mutations in IK-ATP channel genes (KCNJ8 and ABCC9) causing an increase in IK-ATP. The J wave syndromes and IVF have also been associated with gain of function mutations in the transient outward potassium current (Ito).17, 32–39
In the present study, we pharmacologically modeled the genetic defects and attendant ionic changes with the use of verapamil to block ICa and NS5806 to augment Ito.
NS5806-induced augmentation of Ito sensitized our preparations to the effects of verapamil consistent with the association of a higher density of this current in the inferior wall with a higher arrhythmic risk.4 Addition of verapamil further accentuates the AP notch, leading to the development of a more prominent J point and ST segment elevation. Increased concentration of these agents can then elicit all-or-none repolarization, leading to loss of the AP dome at some epicardial sites but not others, resulting in an epicardial dispersion of repolarization (EDR) (Figures 1–5; Table 1). Propagation of the AP dome from sites at which it was maintained to sites at which it was lost elicited local re-excitation via a phase 2 reentry mechanism within the left ventricular epicardium. Loss of the dome in the epicardium also creates a transmural dispersion of repolarization (TDR) giving rise to a vulnerable window across the ventricular wall which, when captured by a closely coupled extrasystole generated in the epicardium, induces VT/VF (Figures 1–3).
Early repolarization syndrome is categorized as a J wave syndrome because like Brugada syndrome its electrocardiographic and arrhythmic manifestations are associated with accentuation of J waves.1, 3 It is therefore no surprise that cilostazol, milrinone and isoproterenol have all been found to exert an ameliorative effect in patients with Brugada syndrome and that their mechanisms of action in the setting of Brugada syndrome is similar to their mechanisms of action in ERS.40
STUDY LIMITATIONS
Our pharmacologic models are designed to mimic the genetic defects associated with ERS and to test the effects of ameliorative agents. To test the effects of these compounds on every single known mutation associated with the J wave syndromes is clearly beyond the scope of our (or any) study. Our pharmacological models may not perfectly mimic the effects of every mutation, but they do serve to recapitulate the net result of the ion-current imbalance created and thus provide a reasonable platform to test the effects of PDE inhibitors and β adrenergic agents.
While it would be preferable to study the effect of these agents in transgenic animal models, none at present are capable of recapitulating the arrhythmic and electrcardiographic manifestations of J wave syndromes. In a recent study, Park et al attempted to mimic Brugada syndrome phenotype in Yucatan minipigs by heterozygous expression of a nonsense mutation in SCN5A (E558X) originally identified in a child with Brugada syndrome.41 Myocytes isolated from the SCN5AE558X/+ pigs showed a loss of function of INa. Various conduction abnormalities were observed but not a BrS phenotype, not even after flecainide-test, because pigs lack the transient outward current and action potential notch in ventricular cardiomyocytes. These observations point to the crucial importance of Ito in the development of J wave syndrome phenotype. Transgenic mice are not helpful due to the fundamental differences in repolarization characteristics.
As with any study involving experimental animal models, extrapolation of the data to the clinic must be done with caution.
Supplementary Material
Figure 2.
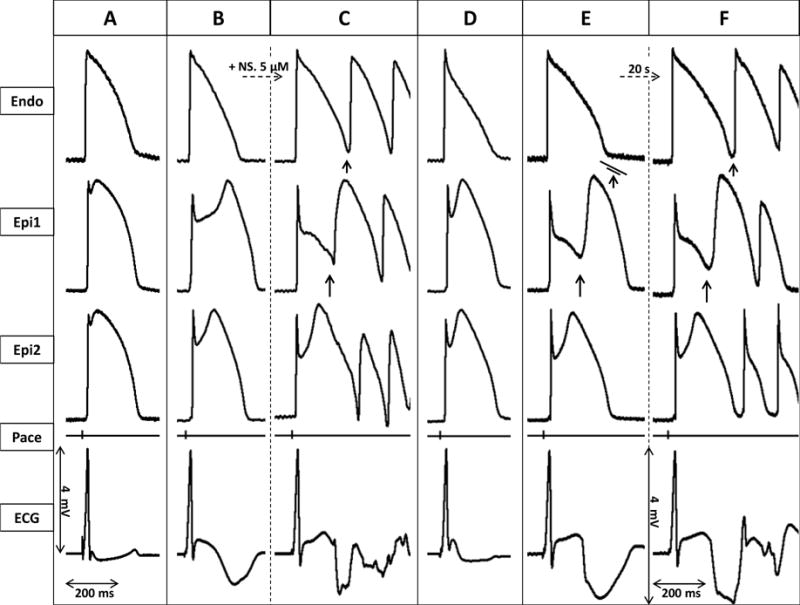
Ameliorative effect of milrinone (2.5μM) in an arterially perfused canine left ventricular model of early repolarization syndrome. Each panel shows simultaneously recorded epicardial (Epi1, Epi2) and endocardial (Endo) action potentials, together with a pseudo-ECG. A: Control. B: Traces recorded 40 min after the addition of Ca2+-channel blocker verapamil (2μM) and Ito-agonist NS5806 (7 μM). C: 15 minutes after raising NS5806 concentration to 12 μM. D: 10 minutes after addition of milrinone 2.5μM to the coronary perfusate. E: 20 minutes after the discontinuation of milrinone infusion. F: 20 seconds later.
Clinical perspectives.
The demonstrated ability of cilostazol and milrinone to inhibit Ito provides support and guidance for the development of new drugs for the management of ERS. Although most agents used in the treatment of J wave syndromes possess Ito-inhibitory effects (e.g., quinidine, cilostazol, bepridil), cardioselective and Ito-specific blockers are not available at present. Further investigation designed to address this significant gap in our antiarrhythmic armamentarium is urgently needed. Despite the similarity between canine and human electrophysiology and our ability to recapitulate the electrocardiographic and arrhythmic manifestations of ERS in this experimental model, translation of our results to humans should be approached with caution.
Acknowledgments
We express our gratitude to José Di Diego MD and Serge Sicouri MD for their generous professional guidance and to Robert Goodrow and Judy Hefferon for their kind technical support and assistance. We also thank Vladislav V. Nesterenko PhD for his substantial contribution to data analysis.
Funding sources: This study was supported by grant HL47678 from NHLBI (CA) and a postdoctoral fellowship grant from the Heart Rhythm Society (IK)
Abbreviations
- AMI
acute myocardial infarction
- AP
transmembrane action potential
- APD
action potential duration
- ATP
adenosine-triphosphate
- cAMP
cyclic adenosine-monophosphate
- ECG
electrocardiogram
- EDR
epicardial dispersion of repolarization
- Endo
endocardium/endocardial
- Epi
epicardium/epicardial
- ER
early repolarization
- ERP
early repolarization pattern
- ERS
early repolarization syndrome
- ICa
L-type calcium current
- IK-ATP
ATP-sensitive potassium current
- INa
cardiac voltage-gated fast sodium current
- Ito
transient outward current
- Jp
peak of the J wave (or the onset of end-QRS slur)
- LV
left ventricle
- P2R
phase 2 reentry
- PDE-3
phosphodiesterase-3
- PKA
protein kinase A (cAMP-dependent protein kinase)
- (P)VT
(polymorphic) ventricular tachycardia
- RV
right ventricle
- SCD
sudden cardiac death
- TDR
transmural dispersion of repolarization
- VF
ventricular fibrillation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None.
References
- 1.Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7:549–558. doi: 10.1016/j.hrthm.2009.12.006. 4/2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gussak I, Antzelevitch C. Early repolarization syndrome: clinical characteristics and possible cellular and ionic mechanisms. J Electrocardiol. 2000;33:299–309. doi: 10.1054/jelc.2000.18106. 10/2000. [DOI] [PubMed] [Google Scholar]
- 3.Antzelevitch C. J wave syndromes: molecular and cellular mechanisms. J Electrocardiol. 2013;46:510–518. doi: 10.1016/j.jelectrocard.2013.08.006. 11/2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koncz I, Gurabi Z, Patocskai B, Panama BK, Szel T, Hu D, Barajas-Martinez H, Antzelevitch C. Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J Mol Cell Cardiol. 2014;68C:20–28. doi: 10.1016/j.yjmcc.2013.12.012. 3/1/2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macfarlane PW, Antzelevitch C, Haissaguerre M, Huikuri HV, Potse M, Rosso R, Sacher F, Tikkanen JT, Wellens H, Yan GX. The Early Repolarization Pattern: A Consensus Paper. J Am Coll Cardiol. 2015 Jul 28;66:470–477. doi: 10.1016/j.jacc.2015.05.033. [DOI] [PubMed] [Google Scholar]
- 6.Haissaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016–2023. doi: 10.1056/NEJMoa071968. 5/8/2008. [DOI] [PubMed] [Google Scholar]
- 7.Nam GB, Kim YH, Antzelevitch C. Augmentation of J waves and electrical storms in patients with early repolarization. N Engl J Med. 2008;358:2078–2079. doi: 10.1056/NEJMc0708182. 5/8/2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosso R, Kogan E, Belhassen B, Rozovski U, Scheinman MM, Zeltser D, Halkin A, Steinvil A, Heller K, Glikson M, Katz A, Viskin S. J-point elevation in survivors of primary ventricular fibrillation and matched control subjects: incidence and clinical significance. J Am Coll Cardiol. 2008;52:1231–1238. doi: 10.1016/j.jacc.2008.07.010. 10/7/2008. [DOI] [PubMed] [Google Scholar]
- 9.Naruse Y, Tada H, Harimura Y, Hayashi M, Noguchi Y, Sato A, Yoshida K, Sekiguchi Y, Aonuma K. Early repolarization is an independent predictor of occurrences of ventricular fibrillation in the very early phase of acute myocardial infarctions. Circ Arrhythm Electrophysiol. 2012;5:506–513. doi: 10.1161/CIRCEP.111.966952. 6/1/2012. [DOI] [PubMed] [Google Scholar]
- 10.Federman NJ, Mechulan A, Klein GJ, Krahn AD. Ventricular fibrillation induced by spontaneous hypothermia in a patient with early repolarization syndrome. J Cardiovasc Electrophysiol. 2013;24:586–588. doi: 10.1111/jce.12030. 5/1/2013. [DOI] [PubMed] [Google Scholar]
- 11.Bastiaenen R, Hedley PL, Christiansen M, Behr ER. Therapeutic hypothermia and ventricular fibrillation storm in early repolarization syndrome. Heart Rhythm. 2010;7:832–834. doi: 10.1016/j.hrthm.2010.02.037. 6/2010. [DOI] [PubMed] [Google Scholar]
- 12.Gurabi Z, Koncz I, Patocskai B, Nesterenko VV, Antzelevitch C. Cellular mechanism underlying hypothermia-induced VT/VF in the setting of early repolarization and the protective effect of quinidine, cilostazol and milrinone. Circ Arrhythm Electrophysiol. 2014;7:134–142. doi: 10.1161/CIRCEP.113.000919. 2/1/2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu D, Barajas-Martinez H, Terzic A, et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol. 2014;171:431–442. doi: 10.1016/j.ijcard.2013.12.084. 2/15/2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barajas-Martinez H, Hu D, Ferrer T, et al. Molecular genetic and functional association of Bugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm. 2012;9:548–555. doi: 10.1016/j.hrthm.2011.10.035. 4/1/2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haissaguerre M, Chatel S, Sacher F, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 2009;20:93–98. doi: 10.1111/j.1540-8167.2008.01326.x. 1/2009. [DOI] [PubMed] [Google Scholar]
- 16.Medeiros-Domingo A, Tan BH, Crotti L, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010;7:1466–1471. doi: 10.1016/j.hrthm.2010.06.016. 10/2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perrin MJ, Adler A, Green S, et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J wave syndrome associated with sudden cardiac death. Circ Cardiovasc Genet. 2014 doi: 10.1161/CIRCGENETICS.114.000623. 9/11/2014. [DOI] [PubMed] [Google Scholar]
- 18.Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al. Mutations in the cardiac L-type calcium channel associated J wave sydnrome and sudden cardiac death. Heart Rhythm. 2010;7:1872–1882. doi: 10.1016/j.hrthm.2010.08.026. 12/2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Napolitano C, Antzelevitch C. Phenotypical manifestations of mutations in the genes encoding subunits of the voltage dependent L-type calcium channel. Circ Res. 2011;108:607–618. doi: 10.1161/CIRCRESAHA.110.224279. 3/4/2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe H, Nogami A, Ohkubo K, et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol. 2011;4:874–881. doi: 10.1161/CIRCEP.111.963983. 12/1/2011. [DOI] [PubMed] [Google Scholar]
- 21.Hu D, Barajas-Martinez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. 2014;64:66–79. doi: 10.1016/j.jacc.2014.04.032. 7/8/2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Diego JM, Sicouri S, Myles RC, Burton FL, Smith GL, Antzelevitch C. Optical and electrical recordings from isolated coronary-perfused ventricular wedge preparations. J Mol Cell Cardiol. 2013;54:53–64. doi: 10.1016/j.yjmcc.2012.10.017. 1/2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zygmunt AC, Goodrow RJ, Antzelevitch C. Sodium effects on 4-aminopyridine-sensitive transient outward current in canine ventricular cells. Am J Physiol. 1997;272:H1–H11. doi: 10.1152/ajpheart.1997.272.1.H1. 1/1997. [DOI] [PubMed] [Google Scholar]
- 24.Atarashi H, Endoh Y, Saitoh H, Kishida H, Hayakawa H. Chronotropic effects of cilostazol, a new antithrombotic agent, in patients with bradyarrhythmias. J Cardiovasc Pharmacol. 1998;31:534–539. doi: 10.1097/00005344-199804000-00010. 4/1998. [DOI] [PubMed] [Google Scholar]
- 25.Endoh M, Yanagisawa T, Taira N, Blinks JR. Effects of new inotropic agents on cyclic nucleotide metabolism and calcium transients in canine ventricular muscle. Circulation. 1986;73:III117–III133. 3/1986. [PubMed] [Google Scholar]
- 26.Matsui K, Kiyosue T, Wang JC, Dohi K, Arita M. Effects of pimobendan on the L-type Ca2+ current and developed tension in guinea-pig ventricular myocytes and papillary muscle: comparison with IBMX, milrinone, and cilostazol. Cardiovasc Drugs Ther. 1999;13:105–113. doi: 10.1023/a:1007779908346. 4/1999. [DOI] [PubMed] [Google Scholar]
- 27.Cone J, Wang S, Tandon N, Fong M, Sun B, Sakurai K, Yoshitake M, Kambayashi J, Liu Y. Comparison of the effects of cilostazol and milrinone on intracellular cAMP levels and cellular function in platelets and cardiac cells. J Cardiovasc Pharmacol. 1999;34:497–504. doi: 10.1097/00005344-199910000-00004. 10/1999. [DOI] [PubMed] [Google Scholar]
- 28.Shakur Y, Fong M, Hensley J, Cone J, Movsesian MA, Kambayashi J, Yoshitake M, Liu Y. Comparison of the effects of cilostazol and milrinone on cAMP-PDE activity, intracellular cAMP and calcium in the heart. Cardiovasc Drugs Ther. 2002;16:417–427. doi: 10.1023/a:1022186402442. 9/2002. [DOI] [PubMed] [Google Scholar]
- 29.Kondo H, Shinohara T, Takahashi N. A case of short-coupled premature ventricular beat-induced ventricular fibrillation with early repolarization in the inferolateral leads. J Arrhythm. 2015 Feb;31:60–63. doi: 10.1016/j.joa.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. 1/15/2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe H, Ohkubo K, Watanabe I, Matsuyama TA, Ishibashi-Ueda H, Yagihara N, Shimizu W, Horie M, Minamino T, Makita N. SCN5A mutation associated with ventricular fibrillation, early repolarization, and concealed myocardial abnormalities. Int J Cardiol. 2013;165:e21–e23. doi: 10.1016/j.ijcard.2012.10.074. 5/10/2013. [DOI] [PubMed] [Google Scholar]
- 32.Barajas-Martinez H, Hu D, Pfeiffer R, Burashnikov E, Powers A, Knilans TK, Antzelevitch C. A genetic variant in DPP10 linked to inherited J-wave syndrome associated with sudden cardiac death by augmentation of Kv4.3 channel current. Heart Rhythm. 2012;9:1919–1920. 11/2012. [Google Scholar]
- 33.Ohno S, Zankov DP, Ding WG, et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhythm Electrophysiol. 2011;4:352–361. doi: 10.1161/CIRCEP.110.959619. 6/1/2011. [DOI] [PubMed] [Google Scholar]
- 34.Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, Ackerman MJ. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011;8:1024–1032. doi: 10.1016/j.hrthm.2011.02.021. 7/2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alders M, Koopmann TT, Christiaans I, et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84:468–476. doi: 10.1016/j.ajhg.2009.02.009. 4/2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delpón E, Cordeiro JM, Núñez L, Thomsen PEB, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Burashnikov A, Christiansen M, Antzelevitch C. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–218. doi: 10.1161/CIRCEP.107.748103. 8/2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giudicessi JR, Ye D, Kritzberger CJ, Nesterenko VV, Tester DJ, Antzelevitch C, Ackerman MJ. Novel mutations in the KCND3-encoded Kv4.3 K+ channel associated with autopsy-negative sudden unexplained death. Hum Mutat. 2012;33:989–997. doi: 10.1002/humu.22058. 6/1/2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu D, Barajas-Martinez H, Medeiros-Domingo A, et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm. 2012;9:760–769. doi: 10.1016/j.hrthm.2011.12.006. 5/1/2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.You T, Mao W, Cai B, Li F, Xu H. Two novel Brugada syndrome-associated mutations increase KV4.3 membrane expression and function. International journal of molecular medicine. 2015 Jul;36:309–315. doi: 10.3892/ijmm.2015.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szel T, Koncz I, Antzelevitch C. Cellular mechanisms underlying the effects of milrinone and cilostazol to supress arrhythmogenesis associated with Brugada syndrome. Heart Rhythm. 2013;10:1720–1727. doi: 10.1016/j.hrthm.2013.07.047. 8/1/2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park DS, Cerrone M, Morley G, et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest. 2015 Jan;125:403–412. doi: 10.1172/JCI76919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.