

Abstract
Human telomerase reverse transcriptase (hTERT) is the catalytic and limiting component of telomerase and also a transcription factor. It is critical to the integrity of the ends of linear chromosomes and to the regulation, extent and rate of cell cycle progression in multicellular eukaryotes. The level of hTERT expression is essential to a wide range of bodily functions and to avoidance of disease conditions, such as cancer, that are mediated in part by aberrant level and regulation of cell cycle proliferation. Value of a gene in regulation depends on its ability to both receive input from multiple sources and transmit signals to multiple effectors. The expression of hTERT and the progression of the cell cycle have been shown to be regulated by an extensive network of gene products and signaling pathways, including the PI3K/Akt and TGF-β pathways. The PI3K inhibitor PX-866 and the competitive estrogen receptor ligand raloxifene have been shown to modify progression of those pathways and, in combination, to decrease proliferation of estrogen receptor positive (ER+) MCF-7 breast cancer cells. We found that combinations of modulators of those pathways decreased not only hTERT transcription but also transcription of additional essential cell cycle regulators such as Cyclin D1. By evaluating known expression profile signatures for TGF-β pathway diversions, we confirmed additional genes such as heparin-binding epidermal growth factor-like growth factor (HB EGF) by which those pathways and their perturbations may also modify cell cycle progression.
Keywords: hTERT, Cyclin D1, Transcription signature, Raloxifene, PX-866
INTRODUCTION
The human telomerase reverse transcriptase (hTERT) gene is believed to have evolved with non-LTR retrotransposons and from reverse transcriptase genes present when DNA was replacing RNA for the maintenance of genomes of eukaryotes and/or their ancestor [1]. Accordingly, a TERT gene is present almost universally in eukaryotes, and few genes in eukaryotes have had as much time, opportunity or necessity to be shaped by evolution in adaptation to changing requirements [1]. Interestingly, few if any human genes have regulation as complex as that of hTERT, and the most extensive regulation of hTERT is at the level of transcription [2-5].
The human TERT protein hTERT is the catalytic and limiting subunit of the telomerase ribonucleoprotein [2] and is required for protection of the ends of linear chromosomes from degradation [3-5]. In the absence of telomerase, the finite length of telomeres leads to limited numbers of cell divisions and senescence [3-4]. In addition, a growing number of genes has been found to be regulated by hTERT as a transcription factor, and as a transcription factor it participates in an hTERT expression positive feedback loop [6-9]. Different patterns of hTERT transcription are required for functions as different as tissue renewal, differentiation, immune cell proliferation and tumor prevention. Accordingly, a complex network of regulatory gene products, signaling pathways and expression overrides is required for hTERT to accommodate its diverse range of responses to a vast range of environmental input [8, 10-12]. Regulation of the cell cycle, cross-linked to regulation of hTERT expression as has been noted [8, 13-14], controls a wide range of bodily functions and development [8-12, 15], and dysregulation accommodates a wide range of human diseases. hTERT is especially valuable as a target for prevention or treatment of the unlimited cell cycle progression, immortality, that sustains cancer [2, 4-5]. Our experiments were designed to test the composite effects of two agents acting on two regulatory pathways as well as the process of selecting relevant pathways based on an understanding of their relationships within the comprehensive regulatory network.
The PI3K/Akt pathway increases hTERT expression through multiple mechanisms [8]. Activated Akt activates the cell cycle by blocking, through phosphorylation, the interaction between MDM2 and p14 (p19) that would prevent ubiquitin-mediated proteolysis of p53 [16-17] and by the mTOR-mediated degradation of cMYC competitor MAD1 [18-19]. Both the canonical and non-canonical NF-κB pathways are activated when Akt phosphorylates and activates IkB kinase (IKK), resulting in the phosphorylation and degradation of IκB [20-22]. The NF-κB pathway is subject to hormone-mediated suppression in estrogen receptor (ER) expressing cells, such as MCF-7, but potentially reversible by antiestrogens, aromatase inhibition, and growth factors or cytokines, including tumor necrosis factor α (TNF α) [23-26]. Additional mechanisms downstream of Akt result in degradation of SMAD4, p53 and p27 [27-30].
In the canonical TGF-β pathway, a complex involving TGF-β, ligand-activated TGF-β receptors, p107, SMAD3, SMAD4 and either E2F-4 or E2F-5 can bind to the cMYC promoter to block transcription [31]. The TGF-β pathway may be inhibited by p107 phosphorylation by complexes of cyclin and cyclin-dependent kinase (cdk) or sustained by cdk inhibitors p27 or p21 [31-33]. Estrogen receptor α (ERα), bound to an estrogen response element (ERE) in the upstream regulatory region of the hTERT promoter and activated by 17 β-estradiol (E2) as a ligand, blocks TGF-β pathway-mediated repression [31, 34-35]. Estrogen has also been reported to block the TGF-β pathway by binding a receptor in the cytoplasm [36].
To inhibit the PI3K/Akt pathway, we used the wortmannin derivative PX-866, specific for PI3K component p110α and currently in Phase II clinical trials [37]. To up-regulate the activity of the TGF-β pathway, we used the selective estrogen receptor modulator (SERM) raloxifene, a competitive ligand for ERα [38-39].
To explore, analyze and screen for potential involvement of treatment-associated and/or cell type-associated mechanisms related to fidelity or diversion from proliferation-limiting canonical TGF-β pathway processing, alternative divergent non-canonical transcription signatures were also examined by real-time PCR. Transcription signatures have proved valuable in associating disease conditions, perturbations and molecular mechanisms [40]. One reported signature of TGF-β pathway misdirection includes Interleukin 11 (IL-11), Cyclin D1 and Axin2 as genes with transcription levels most divergent from normal [41]. Another includes Leukemia inhibitory factor (LIF), HB EGF and ERα with most divergent transcription levels [42].
MATERIALS AND METHODS
Cell cultures, reagents and procedures
Human cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and included ER(+) MCF-7 and ER(−) MDA-MB-231 breast cancer epithelial cells, control non-tumorigenic MCF10A breast epithelial cells and, for contrasting unrelated cells sensitive to proliferation regulated by distinctly different signaling, Jurkat, Clone E6-1, T cell leukemia lymphoblasts. MCF-7 and MDA-MB-231 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA) and 10% APS (amphotericin B, penicillin and streptomycin) (Mediatech) [2]. Before the culture plates were sufficiently confluent (60%) for treatment with harvest assured prior to full confluence, the media for MCF-7 cells was changed to phenol red-free DMEM (Mediatech) supplemented with 0.01 μM β-estradiol (E2) (Sigma-Aldrich, St. Louis, MO) as well as FBS and APS [34]. MCF10A cells were grown in DMEM/F12 medium (Mediatech) supplemented with 5% horse serum, 10 μg/mL insulin, 0.02 μg/mL epidermal growth factor, 100 μg/mL hydrocortisone, 0.001 μg/mL Cholera toxin and 50 μg/mL penicillin-streptomycin. Jurkat cells were grown in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS and 1% penicillin-streptomycin (Mediatech). All cell lines were incubated at 37° C in a humidified environment of 5% CO2 and 95% air.
The MCF-7 cells were treated on three days consecutively, with daily media replenishment, and harvested 18 h following the third treatment. Cells in 10 mL culture plates were individually treated with raloxifene, PX-866, both raloxifene and PX-866 or only 21 μL/10 mL dimethylsulfoxide (DMSO), the statistical median volume in treated plates, as vehicle. Raloxifene (Sigma-Aldrich) stock was kept at room temperature at a concentration of 54.9 mM. PX-866 (BioVision) stock was maintained in the dark at −20° C at a concentration of 190 μM. Treatments included control/vehicle; 1.0 μM raloxifene only; 0.4 μM PX-866 only; or 1.0 μM raloxifene combined with 0.1, 0.4 or 0.8 μM PX-866. The treatment dosages were previously determined to be physiologically relevant and evaluated in previous testing [43-45], and they were chosen based on gel-based PCR in our laboratory as effective in hTERT reduction. Only the higher raloxifene concentration of 1.0 μM was demonstrated in our gel-based PCR to be highly effective (data not shown). The other three cell lines were grown in 10 mL culture plates or T25 flasks (Thermo Fisher Scientific) and harvested untreated for comparison with untreated MCF-7 cells for assessment of treatment-independent, cell type-specific gene expression.
Western blot
Protein was extracted during cell culture harvest using RIPA Lysis Buffer (Upstate Biotechnology, Charlottesville, VA) according to the manufacturer’s protocol. Equal amounts of protein were loaded and electrophoresed using Mini-PROTEAN TGX Precast Gels (Bio-Rad Laboratories, Hercules, CA) and with Precision Plus Protein Kaleidoscope Standards (Bio-Rad Laboratories) for the molecular weight marker. Transfer was performed with the Trans-Blot Turbo Transfer System, as provided by the manufacturer’s protocol (Bio-Rad Laboratories). The SNAP i.d. 2.0 Protein Detection System (EMB Millipore Corporation, Billerica, MA) was used for blocking and incubation with primary and secondary antibodies, according to the manufacturer’s protocol. Primary antibodies included p-MDM2 (Ser 166): sc-293105 (Santa Cruz Biotechnology, Santa Cruz, CA), a rabbit polyclonal IgG against human MDM2 phosphorylated at Serine 166, p21 (C-19): sc-397-G (Santa Cruz Biotechnology), a goat polyclonal IgG against a C-terminal region of human p21, IκBα (C-21): sc-371 (Santa Cruz Biotechnology), a rabbit polyclonal IgG against a C-terminal region of human IκBα and β-actin #4970 (Cell Signaling, Danvers, MA), a rabbit monoclonal IgG against human β-actin. Secondary antibodies included sc-2379 against rabbit IgG and sc-2350 against goat IgG (Santa Cruz Biotechnology) and were conjugated with horseradish peroxidase (HRP). Bio-Rad Clarity Western ECL Substrate (Bio-Rad Laboratories) was used for substrate, and images were developed using ChemiDoc XRS+ System and Image Lab software (Bio-Rad Laboratories). Images from three or more experiments were quantified and normalized using densitometry by ImageJ.
Quantitative real-time PCR
Cultured cells were harvested for total cellular RNA, using RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA) in accordance with the manufacturer’s protocol. Primer pairs used were as previously reported and included the following: hTERT [46]; GAPDH [46]; p21 [47]; cMYC [48]; IL-11 [49]; Cyclin D1 [50]; Axin2 [41]; LIF [51]; HB EGF [52]; ERα [53]; SMAD7 [54]; DEC1 [55]. Real-time PCR was performed for three or more experiments for each combination of treatment and associated gene expression, using the CFX Connect system (Bio-Rad Laboratories).The number of PCR cycles was 40. Annealing temperature was 53° C. Gene expression fold change in treated cells relative to control cells was calculated by the Delta-Delta Ct method [56] using GAPDH as the reference gene.
Statistical analysis
Statistical significance between treatment and control groups and between cell types was evaluated using the relatively conservative ANOVA and Dunnett’s test, which decrease the likelihood of a type 1 error, incorrectly rejecting the null hypothesis of equal means, but increases the likelihood of a type 2 error. Values were provided for means + SE. Significance was determined with p < 0.05 considered significant and p < 0.01 considered highly significant. Reference genes for normalization were GAPDH for mRNA evaluated by real-time PCR and β-actin for protein evaluated by densitometry.
RESULTS
Combined PX-866 and raloxifene treatment decreases phosphorylated MDM2 in MCF-7 cells
We used Western blot to evaluate treatment-associated differences in levels of specific proteins and modified proteins in MCF-7 ER+ breast cancer cells harvested 18 h after the last of three consecutive daily treatments with PX-866 and/or raloxifene or with only DMSO, as vehicle. Protein and modified protein values from Western blot images were quantified by densitometry using ImageJ and normalized to the reference protein β-actin. To assess their effects on MDM2, p53 and p21-mediated regulation of cell cycle proliferation, we examined the results of PX-866 and raloxifene treatment on phosphorylated MDM2 protein and on p21 protein (Figure 1A).
Figure 1.

Immunoblotting analysis revealed p21 and IκBα protein and phosphorylated MDM2 protein following three consecutive days of treatments with raloxifene and/or PX-866. C, control; R1, raloxifene 1.0 μM; PX.1, PX-866 0.1 μM; PX.4, PX-866 0.4 μM; PX.8, PX-866 0.8 μM. (A) Western blot images of phosphorylated MDM2, total p21 protein, IκBα and β-actin. Images displayed are from blots used to derive densitometry averages presented in B-D and are representative of three replicate Western blots. (B) Western blot and densitometry showed a significant level of reduction of phosphorylated MDM2 protein, relative to β-actin, with each level of PX-866 in combination with raloxifene and also with 0.4 μM PX-866 alone. No significant change in phosphorylated MDM2 was seen when raloxifene was used alone. (C) No significant change in p21 protein relative to β-actin was observed. This was in contrast with p21 transcription increases observed, including the significant increase with raloxifene and 0.8 μM PX-866. (D) IκBα protein levels were not significantly increased by raloxifene and/or PX-866. Values represent averages from 3 independent experiments + SEM. * p < 0.05. ** p < 0.01.
As shown in Figure 1B, phosphorylated MDM2 protein was significantly decreased, to less than 40% of control, by treatment with 0.4 μM PX-866 alone or by 1.0 μM raloxifene in combination with any tested PX-866 concentration, either 0.1, 0.4 or 0.8 μM. Phosphorylated MDM2 was not significantly reduced by treatment with raloxifene alone. Expression of p21 protein, expected to be impacted by effects on p21 transcription and the upstream effects on MDM2 phosphorylation, was not significantly increased or decreased by PX-866 and raloxifene, as shown in Figure 1C. In other Western blot findings, IκBα protein was only modestly increased by treatments that included the PI3K inhibitor PX-866 alone or in combination with raloxifene [Figure 1D]. This was likely an indication of relatively high levels of IκBα and relatively low levels of NFκB pathway activity in the control cells.
PX-866 and raloxifene down-regulate hTERT transcription in MCF-7 cells, with support from increased p21 transcription and decreased inhibitory SMAD transcription
As shown in Figure 2A, intervention with PX-866 and raloxifene in two major pathways reported to regulate hTERT transcription resulted in decreases of hTERT transcription, as expected. Significant transcription decrease resulted from 1.0 μM raloxifene combined with either 0.1 or 0.4 μM PX-866 jointly affecting both the TGF-β and PI3K/Akt pathways. No decrease resulted from combined treatment using the highest PX-866 concentration of 0.8 μM, apparently due to feedback from effects on expression of additional genes. Exhibited hTERT down-regulation was 40% and 54%, respectively, of the control cell expression level with 0.1 μM and 0.4 μM PX-866 in combination with raloxifene. While hTERT is regulated by more than the two pathways of present interest, those pathways also each relay input from multiple additional pathways. While the TGF-β and PI3K/Akt pathways do not relay all the environmental signals influencing hTERT expression, additional expansion of interventions may become increasingly less practical.
Figure 2.

Real-time PCR revealed that PX-866 and raloxifene altered some levels of hTERT, p21 and SMAD7 following three consecutive days of treatments. (A) Slight reduction of hTERT mRNA was observed following treatment with either 1.0 μM raloxifene alone or 0.4 μM PX-866 alone. Significant reduction was observed when raloxifene was combined with either 0.1 or 0.4 μM PX-866. No transcription reduction was seen when raloxifene was combined with 0.8 μM PX-866. (B) Significantly increased p21 transcript level was observed with raloxifene in combination with 0.8 μM PX-866. (C) A significant decrease in cMYC transcription, an expected target of canonical TGF-β pathway activity, was not exhibited by real-time PCR. No significant change in cMYC mRNA due to raloxifene and/or PX-866 was exhibited. (D) The contribution of raloxifene to the TGF-β pathway was apparent from a slight reduction of SMAD7 transcription to 58% of control following treatment with raloxifene alone. Each value represents mean + SE after three independent repetitions of the experiment. * p < 0.05. ** p < 0.01.
Increased transcription of the cdk inhibitor p21 is shown in Figure 2B. Significant increase was seen with raloxifene in combination with 0.8 μM PX-866. The effect of p21 transcription increase, anticipated as a consequence of the substantial upstream decrease of phosphorylated MDM2, was modified by the post-transcriptional effects exhibited by Western blot in Figure 1C.
Figure 2C shows no decrease in cMYC transcription to demonstrate PX-866 and/or raloxifene-associated up-regulation of canonical TGF-β pathway processing. A raloxifene-associated enhancement of the TGF-β pathway is exhibited by Figure 2D, showing a significant reduction of transcription of the inhibitory SMAD, SMAD7 to 58% of control, as a result of the use of raloxifene alone in MCF-7 cells.
Transcription signatures reveal additional mechanisms by which raloxifene and PX-866 contribute to down-regulation of hTERT and/or cancer cell proliferation
In testing for PX-866 and raloxifene effects on gene expressions associated with diversion from the proliferation-limiting character of the TGF-β pathway, we observed no increase in treatment-related expression of Axin2. In contrast, we observed (Figure 3A) a slight decrease in Axin2 mRNA using 0.4 μM PX-866 alone and a significant decrease, to 33% of control cell level, using 1.0 μM raloxifene alone. A significant decrease, to 26% of control cell level, was observed with raloxifene in combination with the lowest concentration, 0.1 μM, of PX-866. No treatment-related Cyclin D1 expression increase was observed, as shown in Figure 3B. In contrast to an expression increase, a slight decrease in mRNA expression was seen with 0.4 μM PX-866 alone. Significant decreases were also observed for all three tested PX-866 concentrations in combination with 1.0 μM raloxifene. Expression levels for combinations with 0.1, 0.4 and 0.8 μM PX-866 were 40%, 34% and 36% of control level, respectively.
Figure 3.

Real-time PCR exhibited reductions of Axin2 and Cyclin D1 transcription. (A) Significant reduction in Axin2 transcription was observed with raloxifene alone or in combination with 0.1 μM PX-866. No reduction was observed with highest PX-866 concentration. (B) Significant reduction of Cyclin D1 transcription was observed with each PX-866 concentration in combination with 1.0 μM raloxifene. No decrease in Cyclin D1 was observed with raloxifene alone. Values represent averages from 3 independent experiments + SEM. * p < 0.05. ** p < 0.01.
As shown in Figure 4A and B, expression decreases were seen for LIF and HB EGF and followed the same pattern observed for Axin2, with slight decreases observed with the use of PX-866 alone and significant decreases both with raloxifene alone and with raloxifene in combination with 0.1 μM PX-866. For LIF, expression was reduced to 16% of control level with raloxifene alone and 28% of control level with the combination having 0.1 μM PX-866. For HB EGF, expression was reduced to 34% with raloxifene alone and 37% with the combination having 0.1 μM PX-866. For HB EGF, there was also a slight increase observed with the highest PX-866 concentration, 0.8 μM, reflecting a generally less significant trend seen for several genes at the highest combined treatment level.
Figure 4.

PX-866 and raloxifene altered some transcription levels of LIF, HB EGF and DEC1. (A) A slight decrease in LIF transcription was observed with 0.4 μM PX-866 alone. A significant decrease was seen with 1.0 μM raloxifene either alone or in combination with 0.1 μM PX-866. No significant change in LIF was seen with combinations having higher PX-866 concentrations. (B) Consistent with both Axin2 and LIF expression, the mRNA level of HB EGF was slightly reduced by 0.4 μM PX-866 alone and significantly reduced by 1.0 μM raloxifene either alone or in combination with 0.1 μM PX-866. No decrease of HB EGF was seen with raloxifene in combination with 0.8 μM PX-866. (C) No significant increase or decrease of ERα was seen with raloxifene or PX-866, separately or in combination. (D) A significant increase in DEC1 transcription was seen with raloxifene in combination with the highest PX-866 concentration. Values represent averages from 3 independent experiments + SEM. * p < 0.05. ** p < 0.01.
No significant decrease or increase of ERα expression was observed with raloxifene or PX-866 alone or in combination (Figure 4C). We also tested for PX-866 and raloxifene effects on the transcript level of Differently expressed in chlondrocytes 1 (DEC1) at the downstream end of the TGF-β pathway. As shown in Figure 4D, the level of DEC1 was significantly increased by the highest PX-866 concentration in combination with raloxifene.
Some examined transcription signature genes were also shown to exhibit significantly different cell type-associated expression levels
Significantly altered treatment-associated expression levels are exhibited in Table 1. It displays the pattern among transcription signature genes Axin2, LIF and HB EGF characterized by significant decreases with raloxifene alone and the combination having the lowest PX-866 concentration. It also shows the pattern of higher expression levels associated with higher treatment concentration for some genes. While the reduction of hTERT expression is exhibited with multiple treatments, individual and combined, the most highly significant results are for decreased Cyclin D1 transcription, including a significant decrease at the highest treatment concentration.
Table 1.
Significant relative levels of mRNA, protein and phosphorylated protein following three days of treatment of MCF-7 cells with raloxifene and/or PX-866.
| Micromolar Treatment |
TERT | p21 |
Cyc
D1 |
Axin
2 |
LIF | HB EGF | DEC1 |
|---|---|---|---|---|---|---|---|
| Control | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 1.0 Raloxifene | 0.328 * |
0.158 * |
0.338 * |
||||
| 0.4 PX-866 | |||||||
| Raloxifene + 0.1 PX-866 |
0.396 * |
0.398 * |
0.260 * |
0.283 * |
0.370 * |
||
| Raloxifene + 0.4 PX-866 |
0.539 * |
0.336 * |
|||||
| Raloxifene + 0.8 PX-866 |
2.76 * |
0.356 * |
1.95 * |
| Micromolar Treatment | pMDM2 protein |
|---|---|
| Control | 1.00 |
| 1.0 Raloxifene | |
| 0.4 PX-866 | 0.379 * |
| Raloxifene + 0.1 PX-866 | 0.395 * |
| Raloxifene + 0.4 PX-866 | 0.385 * |
| Raloxifene + 0.8 PX-866 | 0.353 * |
p < 0.5,
p < 0.01
Table 2 exhibits cell type-associated significantly altered expression levels in three cancerous cell lines relative to noncancerous MCF10A cells. Transcription levels were measured by real-time PCR in untreated non-tumorigenic but hTERT-expressing MCF10A breast epithelial cells, in untreated ER+ MCF-7 breast cancer cells, in untreated ER- MDA-MB-231 breast cancer cells and in untreated unrelated Jurkat T cell leukemia lymphoblasts. Worth noting in Table 2 is that the significant decrease of Cyclin D1 in Jurkat cells appears to be balanced by a highly significant decrease in the level of the cdk inhibitor p21. The hormone-mediated regulation of cell cycle proliferation in MCF-7 cells appears to be reflected in a highly significant increase in ERα expression which was in conjunction with a slightly decreased HB EGF (not shown). The gene most significantly expressed differently between cancer cells and noncancerous cells is Cyclin D1. The cell line expression of Cyclin D1 is shown in Figure 5.
Table 2.
Significant relative cell type-specific mRNA expression.
| Cell Type | TERT | p21 |
Cyclin
D1 |
ER
alpha |
|---|---|---|---|---|
| MCF 10A | 1.00 | 1.00 | 1.00 | 1.00 |
| MCF-7 | 6.08 ** |
64.8 ** |
||
| MDA-MB-231 | 3.03 * |
|||
| Jurkat | 3.80 * |
.0075 ** |
.0436 * |
p < 0.5,
p < 0.01
Figuire 5.
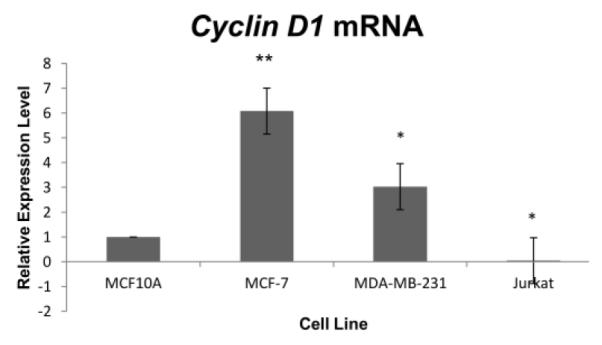
Cell-line transcription levels of Cyclin D1 differed significantly or highly significantly in MCF-7, MDA-MB-231 and Jurkat cells from the level in MCF10A cells. Values represent averages from 3 independent experiments + SEM. * p < 0.05. ** p < 0.01.
Discussion
We previously reported the highly significant decrease of proliferation of MCF-7 cells after three days of treatment with 1.0 μM raloxifene alone or in combination with 0.1, 0.4 or 0.8 μM PX-866 and the absence of any significant decrease of MCF10A cell proliferation following identical treatment [57]. As documented, there are few cellular or intracellular functions even remotely related to cell division, growth or cancer that are hTERT-independent, and this includes gene expression, signaling, survival, mitochondrial function and migration [9]. Here we endeavored to determine (1) whether a significant decrease in hTERT transcription resulted from the same raloxifene and PX-866 treatments as part of the process of proliferation decrease, (2) whether the documented and expected mechanisms of hTERT down-regulation [8] were involved and (3) whether any previously undocumented mechanisms of down-regulation of hTERT expression and/or cell cycle proliferation were involved.
Slight down-regulation of hTERT transcription was achieved with raloxifene alone and with PX-866 alone, and significant decrease was seen with two combinations of raloxifene and PX-866. The approach of hTERT transcription down-regulation by direct intervention in two relevant complementary pathways as vital and comprehensive as the TGF-β and PI3K/Akt pathways is validated. With the continuing discovery of additional hTERT regulators [58], more complete down-regulation might yet require an expanded or next-generation combination.
Decreased hTERT transcription was indirectly [8] due in part to the significant upstream decrease in phosphorylated MDM2, which is phosphorylated by the Akt protein in a process subject to inhibition by the PI3K inhibitor PX-866 [16-17]. Phosphorylation of MDM2 by Akt protects the role of MDM2 in the degradation of p53, a transcription factor that, among other functions, increases the transcription of cdk inhibitor p21 [16-17, 59]. Increases in p21 transcription resulted indirectly from PX-866 interference with MDM2 phosphorylation by Akt.
Excessive accumulation of p21 protein has also been reported to support tumor growth and to result from a post-translational mechanism by which Akt promotes p21 protein stability [60-61]. Accordingly, exhibited p21 protein levels were limited by the PI3K inhibitor PX-866 and Figure 1C showed no increase in p21 protein level despite increased transcription. While exhibited effects on p21 mRNA level and p21 protein level might appear to be in opposition, together they may represent a degree of moderation and control over the excesses of a dysregulated PI3K/Akt pathway.
Canonical TGF-β pathway activity may be confirmed by a decrease in cMYC transcription but is not necessarily otherwise ruled out [43]. The status of the TGF-β pathway is here exhibited, instead, by a modest decrease in SMAD7 mRNA level resulting from raloxifene (Figure 2D), with the decrease limited perhaps due to relatively low levels of NFκB-induced SMAD7 [62] in the ER+ control cells, as suggested by the exhibited relative IκBα levels which generally move in the opposite direction of NF-κB (Figure 1D). SMAD7 is comprehensive in its shutdown of the TGF-β pathway and its downstream effects [62-64]. The TGF-β pathway is blocked by the binding of SMAD7 to activated TGF-β receptor R1 at the beginning of the TGF-β pathway [62]. Transcription of SMAD7 is increased by NF-κB/p65 [62], and raloxifene has been shown to decrease NF-κB transcriptional activity by limiting NF-κB activation, nuclear translocation and DNA binding [65-66].
The down-regulation of genes or mechanisms up-regulated in association with a pathological condition can identify protective mechanisms. At least two transcription signatures have been identified in association with diversion of TGF-β pathway signaling. The TGF-β pathway and Wnt pathway, which normally function in opposition, may instead function in cooperation or synergy in favor of up-regulation of Wnt transcriptional targets, as occurs in the event of mutations in any of a number of genes in early Wnt pathway activity [41]. Regardless of the exact origin of the mutation responsible for β-catenin dysregulation, the associated transcription signature includes an increase in transcription of IL-11, Cyclin D1 and Axin2 [41].
We previously reported that IL-11 transcription was significantly decreased by raloxifene in combination with PX-866 in MCF-7 cells [57]. Recognized contributions of IL-11 to cancer include development, apoptosis evasion, angiogenesis, progression and metastasis in multiple cancer cell types [67].
Both the highest level of statistical significance and the highest level of clinical significance apply to the demonstrated down-regulation of Cyclin D1 transcription. Cyclin D1 binds to cyclin dependent kinases CDK4 and CDK6, and both TGF-β pathway down-regulation and G1 to S phase cell cycle transition result from the phosphorylation of p107 and Rb [32]. In hormone-regulated cells such as MCF-7, transcription of Cyclin E2 requires sufficiently elevated Cyclin D1 expression [68]. SMAD3 activity in cell cycle arrest is blocked by CDK4 and Cyclin D1 in complex [69]. Additional cdk-dependent and cdk-independent effects of Cyclin D1 include suppression of p21 transcription and the accommodation of HER2/Neu and Ras-driven oncogenesis [70-71]. The expression of Cyclin D1, as the critical upstream regulator of pocket protein phosphorylation and E2F transcriptional activity, is a decisive determinant of either proliferation or G1-arrest, in a cell type-dependent manner [32, 68, 71].
Abnormal transcription levels of LIF, HB EGF and ERα are indicated with activation of the actin/megakaryoblastic leulemia 1 (MKL1) pathway, and the resulting phenotype with decreased ERα and increased HB EGF resembles triple-negative breast cancer, with proliferation driven by growth factors rather than hormones [42, 72]. When highly expressed, HB EGF binds to epidermal growth factor receptors to promote growth and more aggressive and invasive forms of cancer [73, 42]. With almost every expression profile gene we examined, the expression level after treatment with PX-866 and/or raloxifene was in direct contrast with that in the pathological expression profile.
With the large number of gene expressions altered by combined treatment through these pathways, the potential for off-target effects is increased [74]. This may be evidenced in the observed dose-dependent changes reflected in the expressions of genes such as LIF, HB EGF and hTERT. At the highest treatment level, we observed a significant increase in expression of DEC1, a transcription product of TGF-β signaling reported to prevent apoptosis and contribute to metastasis [75]. We believe further testing is warranted.
Conclusion
PX-866 and raloxifene down-regulate the PI3K/Akt pathway, up-regulate the TGF-β pathway and, by decreasing transcription of hTERT, Cyclin D1 and other associated genes, decrease proliferation of MCF-7 breast cancer cells. Previously undisclosed genes and mechanisms involved in protective regulation can be discovered from expression signatures associated with pathological conditions. Cell type-associated expression level differences can also forecast the importance of specific genes to conditions specific to those cells.
Highlights.
PX-866 and raloxifene affect the PI3K/Akt and TGF-β pathways.
PX-866 and raloxifene down-regulate genes up-regulated in cancer.
PX-866 and raloxifene decrease transcription of hTERT and Cyclin D1.
Pathological transcription signatures can identify new defense mechanisms.
Acknowledgements
The authors wish to thank Rishabh Kala for valuable technical support. This work was supported in part by grants from the NCI (R01 CA178441) and the American Institute for Cancer Research (316184).
Footnotes
The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Nakamura T, Cech T. Reversing time: origin of telomerase. Cell. 1998;92:587–590. doi: 10.1016/s0092-8674(00)81123-x. [DOI] [PubMed] [Google Scholar]
- [2].Berletch J, Liu C, Love W, Andrews L, Katiyar S, Tollefsbol TO. Epigenetic and genetic mechanisms contribute to telomerase inhibition by EGCG. J. Cell. Biochem. 2008;103:509–519. doi: 10.1002/jcb.21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cong Y, Wen J, Bacchetti S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum. Mol. Genet. 1999;8:137–142. doi: 10.1093/hmg/8.1.137. [DOI] [PubMed] [Google Scholar]
- [4].Poole J, Andrews L, Tollefsbol TO. Activity, function, and gene regulation of the catalytic subunit of telomerase (hTERT) Gene. 2001;269:1–12. doi: 10.1016/s0378-1119(01)00440-1. (2001) [DOI] [PubMed] [Google Scholar]
- [5].Liu L, Lai S, Andrews L, Tollefsbol TO. Genetic and epigenetic modulation of telomerase activity in development and disease. Gene. 2004;340:1–10. doi: 10.1016/j.gene.2004.06.011. [DOI] [PubMed] [Google Scholar]
- [6].Li Y, Tergaonkar V. Noncanonical functions of telomerase: implications in telomerase-targeted cancer therapies. Cancer Res. 2014;74:1639–1644. doi: 10.1158/0008-5472.CAN-13-3568. [DOI] [PubMed] [Google Scholar]
- [7].Wu X-Q, Huang C, He X, Tian Y-Y, Zhou D-X. Feedback regulation of telomerase reverse transcriptase: new insight into the evolving field of telomerase in cancer. Cell. Signal. 2013;25:2462–2468. doi: 10.1016/j.cellsig.2013.08.009. [DOI] [PubMed] [Google Scholar]
- [8].Daniel M, Peek GW, Tollefsbol TO. Regulation of the human catalytic subunit of telomerase (hTERT) Gene. 2012;498:135–146. doi: 10.1016/j.gene.2012.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Saretzki G. Extra-telomeric functions of human telomerase: cancer, mitochondria and oxidative stress. Curr. Pharm. Des. 2014;20:6386–6403. doi: 10.2174/1381612820666140630095606. [DOI] [PubMed] [Google Scholar]
- [10].Bilsland AE, Stevenson K, Liu Y, Hoare S, Cairney CJ, Roffey J, Keith WN. Mathematical model of a telomerase transcriptional regulatory network developed by cell-based screening: analysis of inhibitor effects and telomerase expression mechanisms. PLOS Comput. Biol. 2014;10:e1003448. doi: 10.1371/journal.pcbi.1003448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang F, Chang G-M, Geng X. Bioinformatics analysis of exonic splicing enhancers (ESEs) for predicting potential regulatory elements of hTERT mRNA splicing. Eur. Rev. Med. Pharm. Sci. 2014;18:526–536. [PubMed] [Google Scholar]
- [12].Arancio W, Pizzolanti G, Genovese S, Baiamonte C, Giordano C. CeRNA and interactome bioinformatic analyses on human telomerase. Rejuvenation Res. 2014;17:161–167. doi: 10.1089/rej.2013.1486. [DOI] [PubMed] [Google Scholar]
- [13].Smith LL, Coller HA, Roberts JM. Tolemerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat. Cell Biol. 2003;5:474–479. doi: 10.1038/ncb985. [DOI] [PubMed] [Google Scholar]
- [14].Yang C, Przyborski S, Cooke MJ, Zhang X, Stewart R, Anyfantis G, Atkinson SP, Saretzki G, Armstrong L, Lako M. A key role for telomerase reverse transcriptase unit in modulating human embryonic stem cell proliferation, cell cycle dynamics, and in vitro differentiation. Stem Cells. 2008;26:850–863. doi: 10.1634/stemcells.2007-0677. [DOI] [PubMed] [Google Scholar]
- [15].Testa J, Bellacosa A. AKT plays a central role in tumorigenesis. PNAS. 2001;98:10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou B, Liao Y, Xia W, Zou Y, Spohn B, Hung M. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- [17].Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002;277:21843–21850. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- [18].Chou C-K, Lee D-F, Sun H-L, Li L-Y, Lin C-Y, Huang W-C, Hsu J-M, Kuo H-P, Yamaguchi H, Wang Y-N, Liu M, Wu H-Y, Liao P-C, Yen C-J, Hung M-C. The suppression of MAD1 by AKT-mediated phosphorylation activates MAD1 target genes transcription. Mol. Carcinog. 2009;48:1048–1058. doi: 10.1002/mc.20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. 2008;105:6584–6589. doi: 10.1073/pnas.0802785105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bai D, Ueno L, Vogt P. Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int. J. Cancer. 2009;125:2863–2870. doi: 10.1002/ijc.24748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ozes ON, Mayo LD, Gustin JA, Pfeiffer SR, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;402:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- [22].Wong E, Tergaonkar V. Roles of NF-κB in health and disease: mechanisms and therapeutic potential. Clin. Sci. 2009;116:451–465. doi: 10.1042/CS20080502. [DOI] [PubMed] [Google Scholar]
- [23].Wang W, Nag SA, Zhang R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem. 2015;22:264–289. doi: 10.2174/0929867321666141106124315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Matsumoto G, Namekawa J, Muta M, Nakamura T, Bando H, Tohyama K, Toi M, Umezawa K. Targeting of nuclear factor κB pathways by dehydroxymethylepoxyquinomicin, a novel inhibitor of breast carcinomas: antitumor and antiangiogenic potential in vivo. Clin. Cancer Res. 2005;11:1287–1293. [PubMed] [Google Scholar]
- [25].Zhou Y, Eppenberger-Castori S, Eppenberger U, Benz CC. The NFκB pathway and endocrine-resistant breast cancer. Endocr.-Relat. Cancer. 2005;12:S37–S46. doi: 10.1677/erc.1.00977. [DOI] [PubMed] [Google Scholar]
- [26].Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F, Sonenshein DE. Oestrogen signaling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 2007;9:470–478. doi: 10.1038/ncb1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hsu M-C, Chang H-C, Hung WC. HER-2/neu transcriptionally activates Jab1 expression via the AKT/β-catenin pathway in breast cancer cells. Endocr. Relat. Cancer. 2007;14:655–667. doi: 10.1677/ERC-07-0077. [DOI] [PubMed] [Google Scholar]
- [28].Wan M, Cao X, Wu Y, Bai S, Wu L, Shi X, Wang N, Cao X. Jab1 antagonizes TGF-β signaling by inducing Smad4 degradation. EMBO Rep. 2002;31:171–176. doi: 10.1093/embo-reports/kvf024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Oh W, Lee E-W, Sung Y, Yang MR, Ghim J. Jab1 induces the cytoplasmic localization and degradation of p53 in coordination with Hdm2. J. Biol. Chem. 2006;281:17457–17465. doi: 10.1074/jbc.M601857200. [DOI] [PubMed] [Google Scholar]
- [30].Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato J. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J. Biol. Chem. 2002;277:2302–2310. doi: 10.1074/jbc.M104431200. [DOI] [PubMed] [Google Scholar]
- [31].Chen C-R, Kang Y, Siegel P, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell. 2002;110:19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- [32].Leng X, Noble M, Adams PD, Qin J, Harper JW. Reversal of growth suppression by p107 via direct phosphorylation by cyclin D1/cyclin-dependent kinase 4, Mol. Cell Biol. 2002;22:2242–2254. doi: 10.1128/MCB.22.7.2242-2254.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ray A, James M, Laurochelle S, Fisher R, Blain S. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol. Cell Biol. 2009;29:986–999. doi: 10.1128/MCB.00898-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cherlet T, Murphy L. Estrogen receptors inhibit Smad3 transcriptional activity through Ap-1 transcription factors. Mol. Cell Biochem. 2007;306:33–42. doi: 10.1007/s11010-007-9551-1. [DOI] [PubMed] [Google Scholar]
- [35].Ren Y, Wu L, Frost A, Grizzle X, Cao X, Wan M. Dual effects of TGF-β on ERα-mediated estrogenic transcriptional activity in breast cancer. Mol. Cancer. 2009;8:111. doi: 10.1186/1476-4598-8-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kleuser B, Malek D, Gust R, Pertz H, Potteck H. 17-β-estradiol inhibits transforming growth factor-β signaling and function in breast cancer cells via activation of extracellular signal-regulated kinase through the G protein-coupled receptor 30. Mol. Pharmacol. 2008;74:1533–1543. doi: 10.1124/mol.108.046854. [DOI] [PubMed] [Google Scholar]
- [37].Ihle N, Williams R, Chow S, Chew W, Berggren M, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R, Kirkpatrick L, Powis G. Mol. Pharmacol. and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol. Cancer Ther. 2004;3:763–772. [PubMed] [Google Scholar]
- [38].Wu L, Wu Y, Gathings B, Wan M, Li X, Grizzle W, Liu Z, Lu C, Mao Z, Cao X. Smad4 as a transcription corepressor for estrogen receptor α. J. Biol. Chem. 2003;278:15192–15200. doi: 10.1074/jbc.M212332200. [DOI] [PubMed] [Google Scholar]
- [39].Kawagoe J, Ohmichi M, Takahashi T, Ohshima C, Mabuchi S, Takahashi K, Igarashi H, Mori-Abe A, Saitoh M, Du B, Ohta T, Kimura A, Kyo S, Inoue M, Kurachi H. Raloxifene inhibits estrogen-induced up-regulation of telomerase activity in a human breast cancer cell line. J. Biol. Chem. 2003;278:43363–43372. doi: 10.1074/jbc.M304363200. [DOI] [PubMed] [Google Scholar]
- [40].Lamb J, Crawford E, Peck D, Middel J, Blat I, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggerty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- [41].Labbe E, Lock L, Letamendia A, Gorska A, Gryfe R, Gallinger S, Moses HL, Attisano L. Transcriptional cooperation between the transforming growth factor-β and Wnt pathways in mammary and intestinal tumorigenesis. Cancer Res. 2007;67:75–84. doi: 10.1158/0008-5472.CAN-06-2559. [DOI] [PubMed] [Google Scholar]
- [42].Kerdivel G, Boudot A, Habauzit D, Percevault F, Demay F, Pakdel F, Flouriot G. Activation of the MKL1/actin pathway induces hormonal escape in estrogen-responsive breast cancer cell lines Mol. Cell Endocrinol. 2014;390:34–44. doi: 10.1016/j.mce.2014.03.009. [DOI] [PubMed] [Google Scholar]
- [43].Sasaki H, Hayakawa J, Terai Y, Kanemura M, Tanabe-Kimura A, Kamegai H, Seino-Noda H, Ezoe S, Matsumura I, Kanakura Y, Sakata M, Tasaka K, Ohmichi M. Difference between genomic actions of estrogen versus raloxifene in human ovarian cancer cell lines. Oncogene. 2008;27:2737–2745. doi: 10.1038/sj.onc.1210926. [DOI] [PubMed] [Google Scholar]
- [44].Ihle N, Paine-Murrieta G, Berggren M, Baker A, Tate W, Wipf P, Abraham RT, Kirkpatrick DL, Powis G. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor inhibitor getitinib in A-549 human non-small cell lung cancer xenographs. Mol. Cancer Ther. 2005;4:1349–1357. doi: 10.1158/1535-7163.MCT-05-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Koul D, Shen R, Kim Y-W, Kondo K, Lu Y, Bankson J, Ronen SM, Kirkpatrick DL, Powis G, Yung WKA. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastom. Neuro-Oncol. 2010;12:559–569. doi: 10.1093/neuonc/nop058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Meeran S, Patel S, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLOS ONE. 2010;5:e11457. doi: 10.1371/journal.pone.0011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Saramaki A, Banwell C, Campbell M, Carlberg C. Regulation of the human p21 gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006;34:543–554. doi: 10.1093/nar/gkj460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kumar N, Basundra R, Maiti S. Elevated polyamines induce c-MYC overexpression by perturbing quadruplex-WC duplex equilibrium. Nucleic Acids Res. 2009;37:3321–331. doi: 10.1093/nar/gkp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Onnis B, Fer N, Rapisarda A, Perez V, Melillo G. Autocrine production of IL-11 mediates tumorigenicity in hypoxic cancer cells. J. Clin. Investig. 2013;123:1615–1629. doi: 10.1172/JCI59623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Masuda M, Suzui M, Yasumatu R, Nakashima T, Kuratomi Y, Azuma K, Tomita K, Komiyama S, WeinStein IB. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res. 2002;62:3351–3355. [PubMed] [Google Scholar]
- [51].Sabry D, Nouh O, Marzouk S, Hassouna A. Pilot study on molecular quantitation and sequencing of endometrial cytokines gene expression and their effect on the outcome of in vitro fertilization (IVF) cycle. J. Adv. Res. 2013;5:595–600. doi: 10.1016/j.jare.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Luo S, Peng Z, Zheng Y, Zhang L, Feng Y, Wang G. Synergistic effects of acitretin and narrow-band UVB on inducing the expression of heparin-binding epidermal-growth-factor-like growth factor in normal human keratinocytes. Arch. Dermatol. Res. 2007;299:409–413. doi: 10.1007/s00403-007-0768-3. [DOI] [PubMed] [Google Scholar]
- [53].Poola I, Yue Q. Estrogen receptor alpha (ERα) mRNA copy numbers Inimmunohistochemically ERα-positive-, and negative breast cancer tissues. BMC Cancer. 2007;7:56. doi: 10.1186/1471-2407-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jungert K, Buck A, Buchholz M, Wagner M, Adler G, Gress TM, Ellenrieder V. Smad-Sp1 complexes mediate TGFβ-induced early transcription of oncogenic Smad7 in pancreatic cancer cells. Carcinog. 2006;27:2392–2401. doi: 10.1093/carcin/bgl078. [DOI] [PubMed] [Google Scholar]
- [55].Kawamoto T, Noshiro M, Sato F, Maemura K, Takeda N, Nagai R, Iwata T, Fujomoto K, Furukawa M, Miyazaki K, Honma S, Honma K, Kato Y. A novel autofeedback loop of Dec1 transcription involved in circadian rhythm regulation. Biochem. Biophys. Res. Commun. 2004;313:117–1124. doi: 10.1016/j.bbrc.2003.11.099. [DOI] [PubMed] [Google Scholar]
- [56].Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- [57].Peek GW, Tollefsbol TO. Combinatorial PX-866 and raloxifene decrease Rb phosphorylation, Cyclin E2 transcription and proliferation of MCF-7 breast cancer cells. J. Cell. Biochem. 2015 doi: 10.1002/jcb.25462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. Went/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336:1549–1554. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- [59].Lai S, Cunningham A, Huynh V, Andrews L, Tollefsbol TO. Evidence of extra-telomeric effects of hTERT and its regulation involving a feedback loop. Exp. Cell Res. 2007;313:322–330. doi: 10.1016/j.yexcr.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Erber R, Klein W, Andl T, Enders C, Born AI, Conradt C, Bartek J, Bosch FX. Aberrant p21CIP1/WAF1 protein accumulation in head-and-neck cancer. Int. J. Cancer. 1997;74:383–389. doi: 10.1002/(sici)1097-0215(19970822)74:4<383::aid-ijc4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- [61].Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhance protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 2002;277:11352–11361. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- [62].Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg A. A mechanism of suppression of TGF-β/SMAD signaling by NF-κB/RelA. Genes Dev. 2000;14:187–197. [PMC free article] [PubMed] [Google Scholar]
- [63].Guo X, Wang X-F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Seoane J. Escaping from the TGFβ anti-proliferative control. Carcinog. 2012;27:2148–2156. doi: 10.1093/carcin/bgl068. [DOI] [PubMed] [Google Scholar]
- [65].Oliver S, Close P, Castermans E, de Leval L, Tabruyn S, Chariot A, Malaise M, Merville M-P, Bours V, Franchimont N. Raloxifene-induced myeloma cell apoptosis: a study of nuclear factor-κB inhibition and gene expression signature. Mol. Pharmacol. 2006;69:1615–1623. doi: 10.1124/mol.105.020479. [DOI] [PubMed] [Google Scholar]
- [66].Li R, Xu W, Chen Y, Qin J, Harper J. Raloxifene suppresses experimental autoimmune encephalomyelitis and NF-κB-dependent CCL20 expression in reactive astrocytes. PLOS One. 2014;9:e94320. doi: 10.1371/journal.pone.0094320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Johnstone CN, Chand A, Putoczki TL, Ernst M. Emerging roles for IL-11 signaling in cancer development and progression: focus on breast cancer. Cytokine Growth Factor Rev. 2015;889:489–498. doi: 10.1016/j.cytogfr.2015.07.015. [DOI] [PubMed] [Google Scholar]
- [68].Caldon CE, Sergio CM, Schutte J, Boersma MN, Sutherland RL, Carroll JS, Musgrove EA. Estrogen regulation of Cyclin E2 requires Cyclin D1 but not c-Myc. Mol. Cell. Biol. 2009;29:4623–4639. doi: 10.1128/MCB.00269-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zelivianski S, Cooley A, Kall R, Jeruss J. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol. Cancer Res. 2010;8:1375–1387. doi: 10.1158/1541-7786.MCR-09-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bienvenu F, Barre B, Giraud S, Coqueret O. Transcriptional regulation by a DNA -associated form of cyclin D1. Mol. Biol. Cell. 2005;16:1850–1858. doi: 10.1091/mbc.E04-08-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- [72].Selvaraj A, Prywes R. Expression profiling of serum inducible genes identifies a subset of SRF target genes that are MKL dependent. BMC Mol. Biol. 2004;5:13. doi: 10.1186/1471-2199-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Miyamoto S, Yagi H, Yotsumoto F, Kawarabayashi T, Mekada E. Heparin-binding epidermal growth factor-like growth factor as a novel targeting molecule for cancer therapy. Cancer Sci. 2006;97:341–347. doi: 10.1111/j.1349-7006.2006.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Connolly EC, Freimuth J, Akhurst RJ. Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 2012;8:964–978. doi: 10.7150/ijbs.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ehata S, Hanyu A, Hayashi M, Aburatani H, Kato Y, Fujime M, Saitoh M, Miyazawa K, Imamura T, Miyazono K. Transforming growth factor-β promotes survival of mammary carcinoma cells through introduction of antiapoptotic transcription factor DEC1. Cancer Res. 2007;67:9694–9703. doi: 10.1158/0008-5472.CAN-07-1522. [DOI] [PubMed] [Google Scholar]