

Abstract
The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited and incurable neurodegenerative disorders primarily afflicting the pediatric population. Current treatment regimens offer only symptomatic relief and do not target the underlying cause of the disease. Although the underlying pathophysiology that drives disease progression is unknown, several small molecules have been identified with diverse mechanisms of action that provide promise for the treatment of this devastating disease. This review aims to summarize the current cellular and animal models available for the identification of potential therapeutics and presents the current state of knowledge on small molecule compounds that demonstrate in vitro and/or in vivo efficacy across the NCLs with an emphasis on targets of action.
Graphical abstract

INTRODUCTION
The neuronal ceroid lipofuscinoses (NCLs), commonly grouped together as Batten disease, are the most common neurodegenerative diseases of the pediatric population.1,2 The incidence of NCL is estimated at 1 in 12 500;3 however, this representation may not be accurate due to the rarity of the disease. Although rare, the disease often strikes multiple offspring in the same family that carry the defective NCL gene. Enzyme replacement therapy, stem cell therapy, gene therapy, dietary supplements, and small molecules have been tested in various cell and animal models and in humans. No cure for NCL has yet been realized; however, there is pharmacological intervention in clinical trials4–6 that can slow disease progression and/or improve quality of life.7–10 The need for the identification of new drug candidates that are disease-modifying in the NCLs is unmet and urgent, yet is hampered by the largely unknown pathophysiology of NCL progression.
The disease presents with early vision problems and/or seizures progressing to mental impairment, seizures of increased severity, progressive loss of sight and motor skills, and increasing spasticity. Children reaching the end-stages of the disease become blind, bedridden, spastic, and demented with poorly controlled seizures before expiring in their teens or early twenties.2,11,12 The NCLs progress through an accumulation of lipofuscin in body tissues. Fat, but primarily protein deposits (termed storage material), accumulates in the brain, retina, and other tissues, which specifically locate to the lysosomes.13,14 Accelerated apoptosis,15,16 impaired autophagy, and secondary destructive inflammation have been documented. Accumulation of subunit c of mitochondrial ATP synthase in lysosome-derived organelles is observed in NCLs; however, the exact biochemical mechanism of accumulation is unknown.14,17
Most forms of NCL are autosomal recessive disorders, with the exception of some families with adult Kufs disease manifesting autosomal dominant inheritance.11 The NCLs are linked to 13 genes; mutations in any of the genes named as CLN1–14 can lead to NCLs. Proteins encoded by the majority of the NCL genes (CLN1, CLN2, CLN3, CLN5, CLN7, CLN10, CLN12, and CLN13) are primarily localized to the lysosome. Some of these proteins are also known to have extralysosomal functions.18,19 Mutations in the CLN3 gene cause juvenile NCL (JNCL) and encode for the CLN3 protein, which has been localized to Golgi, lipid rafts and plasma membrane, and lysosomes. However, the overall function of the CLN3 protein is unknown; mutations lead to altered lysosomal pH, defective arginine transport, and malfunction in transport across lysosomal and vacuolar membranes.20–22 In CLN3 knockdown SH-SY5Y cells, calcium induced cell death was mediated by calsenilin, a calcium sensor known to be proapoptotic in neurons. Calsenilin binds to the C-terminal of CLN3, which is known to be cytoprotective, leading to a reduction in calsenilin induced cell death.23 It has been proposed for JNCL that different receptors undergo dysregulation in an age dependent fashion as observed in Cln3Δex1–6 mice. This same process has been observed in a mouse model of Huntington’s disease, the cellular and molecular mechanism changing during disease progression.24,25
Another common NCL variant is CLN2 disease or late infantile NCL (LINCL), attributed to a defective tripeptidyl peptidase I (TPP1) enzyme, the protein product of the healthy CLN2 gene. Infantile NCL (INCL) is the most severe form of NCL. Patients have a defective palmitoyl protein thioesterase 1 (PPT1) enzyme, caused by a mutated CLN1 gene.26 PPT1 is responsible for the cleavage of palmitate from S-acylated proteins,27 whereas TPP1 removes tripeptides from the N-terminal of small polypeptides.28 However, the native substrates of these enzymes remain unidentified.1 CLN4 disease is caused by mutations in the DNAJC5 gene which encodes for the protein cysteine-string protein α (CSPα).29 CSPα has been shown to play a role as a chaperone in folding of proteins and in synaptic vesicle exocytosis and endocytosis.30,31 The cathepsin D (CTSD, CLN10) gene in the physiological state codes for the protein cathepsin D, an aspartyl endopeptidase, which plays several roles in apoptosis. CLN10 disease is caused by mutation in the cathepsin D gene.1,18 Mutation in the cathepsin F (CTSF, CLN13) gene leads to adult-onset NCL, also known as type B Kufs disease.32 Cathepsin F is a cysteine protease highly expressed in neurons and is shown to be involved in autophagy and proteasomal degradation.1,18 The functions of proteins encoded by genes CLN5–9, CLN11, CLN12, and CLN14 are unknown.1,18 Some of the CLN proteins demonstrate interactions with each other, but their physiological relevance is unknown.1
Apoptosis, among other cellular death mechanisms, has been proposed as one of the mechanisms of neuron death in the NCLs based on in vitro experiments.33 One mechanism of survival for neurons in LINCL and JNCL is upregulation of the apoptotic regulator protein Bcl-2. Likewise the CLN3 gene, mutated in the disease state, is known to exhibit antiapoptotic properties, excerpting this effect by upregulation of the apoptosis suppressor Bcl-2.33–35 The gene that mutates in LINCL, CLN2, was also shown to be antiapoptotic; hNT neurons transduced with sense-CLN2-AAV2 virus showed protection against etoposide induced apoptosis, whereas NT2 cells transduced with antisense-CLN2-AAV2 showed an increased rate of apoptosis.34 Mutations in CLN1,36 CLN5, CLN6, and CLN8 genes also result in accelerated apoptosis contributing to the manifestation of NCL at different ages.37 Wild-type CLN3 has also been implicated in down-regulating ceramide generation, a proapoptotic lipid second messenger.16 It was believed that the deposition of storage material in specific parts of the brain led to death of neurons. However, experiments conducted in a CLN6 sheep model show no correlation between the deposition of storage material and death of neurons, suggesting that other mechanisms of cell death may also play a role.38,39 This also suggests that reduction in storage material may not be a therapeutic approach to treat Batten disease nor a reliable biomarker to measure treatment effect.
While the exact pathophysiology of the NCLs is unknown, the basic understanding of disease processes elucidated to date has allowed the creation of cellular and animal models that mimic the phenotype of deficient CLN genes. This in turn has led to the evaluation of several small molecule candidate therapeutics for use in the treatment of this devastating disease (Table 1).
Table 1.
Summary of Mechanism of Action and Preclinical Testing of Candidate Therapeutics for NCLs
| Structure | Mechanism of Action |
Comments | Current Status | References |
|---|---|---|---|---|
 |
NMDA receptor antagonist, upregulation of Bcl-2. Other mechanisms possible | Active in in vitro models including CLN1, CLN2, CLN3 and CLN6 patient cells | Survey based study performed in patients was inconclusive. No in vivo experiments performed | 34, 59, 60 |
 |
L-type Calcium channel blocker | Active in in vitro models including patient cells | No follow up studies of amlodipine in vivo. No follow up studies of other calcium channel blockers | 47, 62 |
 |
AMPA receptor antagonist | Active in vivo in Cln3Δex1–6 mouse model | No follow up study in clinical trials of any AMPA receptor antagonist | 63, 64 |
 |
NMDA receptor antagonist | Active in vivo in Cln3Δex1–6 and Ppt1−/− mice models but further experiments are required. May be useful in INCL patients who are symptomatic | No follow up studies in other in vivo models or in humans | 24, 70 |
 |
Immunosuppressant acting by inhibiting inosine monophosphate dehydrogenase | Active in vivo in Cln3−/− mouse model | Currently in Phase II clinical trials | 71, 72 |
 |
Antioxidant | Active in in vitro models including patient cells | Pilot study in patients showed a beneficial effect. More studies required | 5, 45 |
 |
Mimics PPT1 enzyme | Active in in vitro models including INCL patient cells as well as in vivo Ppt1−/− mouse model | No follow-up studies | 44 |
 |
Mimics PPT1 enzyme | Active in in vitro models including INCL patient cells | Inconclusive results when tested in 4 INCL patients | 4, 43, 77 |
 |
Mimics PPT1 enzyme | Active in in vitro models including INCL patient cells | No follow-up studies | 77, 78 |
 |
Antioxidant | Active in in vitro models including patient cells as well as in vivo mouse model | No clinical trials performed | 79, 81 |
 |
Promotes read through of premature stop codon | Active in vitro in CLN2 stem cell model and INCL patient cells | No follow up studies | 84 |
 |
Unknown | Beneficial effect in a single LINCL patient | No other studies performed | 90 |
 |
Promotes read through of premature stop codon | Active in vitro in LINCL patient cells | No in vivo experiments performed | 92 |
 |
Upregulates TPP1 mRNA via PPARα/RXRα | Active in vitro; however no experiments conducted in phenotypic model. No effect in stem cell derived model | No follow-up studies | 48, 94 |
 |
Upregulates TPP1 mRNA via PPARα/RXRα | Active in vitro; however no experiments conducted in phenotypic model. No effect in stem cell derived model | No follow-up studies | 48, 94 |
 |
Unknown | No effect in vivo in CLN6 sheep model | No follow-up studies | 96 |
 |
AMPA receptor antagonist | Active in vivo in CLN8 mouse model | No follow-up studies | 97, 99 |
 |
AMPA receptor antagonist | Active in vivo in mnd mouse model | No follow-up studies | 100 |
 |
Known β2 agonist but probably not acting by this mechanism in NCL | Active in vivo in mnd mouse model | No follow-up studies | 103 |
| Lithium | IMPase inhibition | Active in vitro in phenotypic CLN3 mutant knock-in cerebellar cells | No follow-up studies | 74 |
| Pharmacological Chaperones | Stabilize three dimensional conformation of misfolded protein | Active in vitro in INCL patient cells | No follow-up studies | 88 |
CELLULAR MODELS
Several phenotypic models mimicking the NCL disease state in humans have been developed to aid drug discovery efforts and to help determine the underlying pathophysiology. Several researchers have reported the use of NCL patient lymphoblasts and fibroblasts, along with neurons derived from animal models of NCL disease.40–45 A phenotypic model of the human neuroblastoma cell line SH-SY5Y was developed using siRNA knockdown of the CLN3 gene, the gene known to mutate in JNCL.46 A similar model was developed in primary rat cortical neurons by CLN3 knockdown using a siRNA based method.47 Two cell models of LINCL and JNCL were created, where hNT neurons were transduced with AS-CLN2-AAV2 and Ad-AS-CLN3, resulting in loss of CLN2 and CLN3 expression, respectively.34
Human induced pluripotent stem cell (iPSC) models have gained increasing attention, having the advantage that the majority of the genotypic and phenotypic changes observed in human NCL patients are reciprocated by this model. Fibroblasts obtained from LINCL and JNCL patients were reprogrammed to iPSCs; however, no changes were observed in the Golgi or mitochondria in both CLN2 and CLN3 patient iPSCs, but lysosomal changes were observed without deposition of storage material and mitochondrial ATP synthase subunit c. The iPSCs derived from LINCL patients did, however, exhibit reduced TPP1 activity.48
Neural progenitor cells (NPCs) generated in turn from iPSCs did exhibit differences in structure of the Golgi body and lysosomal membranes along with deposition of storage-like material different from the storage material observed in NCL patients; morphological anomalies in mitochondria were also observed. The NPCs obtained from CLN2 patients showed diminished TPP1 activity, while CLN3 patient NPCs showed slightly elevated TPP1 activity. Deposition of subunit c of mitochondrial ATP synthase was observed similar to NCL patients in both sets of NPCs.48
Mature CLN2 and CLN3 patient neurons were also generated from NPCs. Mature neurons showed aberrations in the Golgi body, mitochondria, endoplasmic reticulum (ER), and lysosomal membrane. Storage material similar to NCL patients was also observed. An increase in disease progression was observed in the iPSC model when moving from iPSC to mature neurons.48 However, iPSCs suffer from high degrees of variation between attempts to reprogram a single fibroblast line or between cells derived from different patients, complicating their use in early drug discovery screens. Finally, a yeast model for the study of NCL has been developed, as the organism contains a gene, BTN1, which is homologous to the human CLN3 gene.49
ANIMAL MODELS
Currently there are several animal models available for different types of NCLs including Drosophila,50 mouse,51–54 sheep, cow, ferret, cat, horse, goat, pig, parrot, monkey, duck, and canine.51,54 Of particular significance are the PPT1 knockout mice models mimicking the CLN1 disease state, and the Cln3Δex1–6 mimicking the CLN3 disease state, as the majority of small molecule therapeutic invetsigation for NCLs are performed using these models. Other mice models for CLN3 disease such as Cln3Δex7/8 knockout, Cln3Δex7/8 knock-in, and Cln3LacZ β-galactosidase have also been developed.55 Several other mice models are also available: three for CLN1, two for CLN2, and one each for CLN5–8 and CLN10.52
PPT1 Knockout Mice as a Model for CLN1 Disease
PPT1 gene disruption was verified using Southern blot and immunoblotting and was confirmed by background levels of PPT1 enzymatic activity. A 50% mortality rate was observed in PPT1 knockout mice by 7 months of life, and seizures were observed from 3–4 months of age. Significant increase in autofluorescence was observed at 4 weeks of age, and granular osmiophilic deposits (GRODs) were readily observed along with thinning of the hippocampus, ballooning defragmentation of neurons, and apoptotic nuclear fragments.56
Cln3Δex1–6 Mouse Model
A Cln3 gene disrupted mouse model in which several neuropathological changes are observed is now extensively utilized in the field of NCL disease.24,57 Disruption of the CLN3 gene was confirmed using reverse transcription polymerase chain reaction (RT-PCR) and Southern blotting. Significant increase in accumulation of storage material in the neurons of Cln−/− mice was observed. Autofluorescent lipopigments were detected at 3 months of age, and a progressive increase in quantity was observed. Autofluorescent lipopigment was distributed in several parts of the brain including the cortex, hippocampus, basal ganglia, and brainstem. A reduction in the brain mass was noted that was not statistically significant; however, a decrease in the cortex volume was also observed that was statistically significant. CLN2 protease activity was elevated in the brain similar to that activity observed in NCL patients.57 α-Amino-3-hydroxy-5-methyl-4-isooxazolepropionic acid (AMPA) and N-methyl-d-aspartate (NMDA)-type glutamate receptor dysfunction has been observed in this model as well as in JNCL patients.24,58
SMALL MOLECULE THERAPEUTICS EVALUATED IN CLN3 DISEASE (JNCL)
Flupirtine (1), a non-opioid analgesic approved for the treatment of seizures in Europe, is known to be neuroprotective via the upregulation of Bcl-2 and antagonism at the NMDA receptor.34,59 Flupirtine reduced the rate of apoptosis in CLN1-, CLN2-, CLN3-, and CLN6-deficient NCL patient lymphoblasts. The compound also showed a neuroprotective effect in hNT neurons transduced with AS-CLN2-AAV2 and Ad-AS-CLN3 and from etoposide induced apoptosis in hNT neurons and hNT neurons transduced with sense-CLN2-AAV2. In addition, flupirtine rescued hNT neurons from NMDA activated neuronal apoptosis.34 In a follow-up study to assess the effect of flupirtine in JNCL patients, a survey was conducted among the parents of patients who were undergoing treatment with flupirtine. The unified Batten disease rating scale (UBDRS) was used to assess five aspects of JNCL: physical ability, seizure activity, behavioral problems, functional capacity, and global clinical impression of disease. Parents reported a beneficial effect from flupirtine; however, statistical analysis showed no difference between the treatment and control groups. This study was not a clinical trial and was solely based on the survey filled out by the parents of JNCL patients.60 The initial study reported that flupirtine was neuroprotective in vitro at >20 µM, a concentration difficult to achieve and maintain within the human body.34,60

It is well established that elevated intracellular calcium levels induce apoptosis.16 Elevated levels of CLN3 protein, as opposed to deficient levels as seen in JNCL patients, have been shown to reverse apoptosis induced by abnormal intracellular calcium accumulation.61 In JNCL, CLN3 gene mutation causes calcium induced cell death.23 In a primary rat cortical neuron model with CLN3 knockdown using siRNA, amlodipine (2) reversed the elevated calcium levels and thus prevented apoptosis.47,62 Amlodipine functions as a calcium channel blocker; however, follow-up studies on other calcium channel blockers have not been reported.
Hyperactivity of the AMPA receptor in cerebellar neurons in Cln3Δex1–6 mice has been suggested as one of the biochemical dysfunctions of the NCLs.58 The selective, noncompetitive AMPA receptor antagonist 3 (EGIS-8332) at 1 mg/kg, ip, improved motor coordination in 1 month-old Cln3Δex1–6 mice.

Higher doses of 3 mg/kg and 10 mg/kg, however, did not show any enhancement in motor coordination.63 Compound 3 (1 mg/kg, ip) did not show any improvement in motor skills immediately after treatment but did show a delayed response after 4 days in 6- to 7-month-old Cln3Δex1–6 mice. The compound did not improve the total neuronal count and was ineffective on astrocytic and microglial activation in cortex and the cerebellum.64

Anomalous glutaminergic activity has been observed in various mouse models and human patients of neurodegenerative disorders including infantile, late infantile, and juvenile NCL.65–69 Acute treatment with the NMDA receptor antagonist memantine (4) (1 mg/kg and 5 mg/kg, ip) showed no improvement in motor coordination in 1-month-old Cln3Δex1–6 mice; however, 1 mg/kg memantine showed significant improvement in motor coordination after 1 day and 5 mg/kg showed a delayed response with an improvement in motor coordination after 4 days of treatment in 6- to 7-month-old Cln3Δex1–6 mice. To assess the age-dependent effect, repeated doses of memantine (1 mg/kg) and 3 (1 mg/kg) were administered once a week for 4 weeks. Seven-month-old Cln3Δex1–6 mice treated with compound 3, in comparison to memantine treated mice, showed an enhanced performance on the rotarod test. Neither of the compounds showed an increase in neuron numbers in the thalamus or cerebellum. The authors suggested that acute inhibition of the NMDA receptor (a known mechanism of action of flupirtine (1) reviewed above) could serve as a therapeutic target for NCL.24
Neurons isolated from Ppt1−/− mice have shown NMDA receptor hyperfunction.67 The effect of memantine was examined in 3-month-, 5-month-, and 7-month-old Ppt1−/− mice using an accelerating rotarod. A dose of 10 mg/kg did not show any significant effect. A dose of 20 mg/kg did not show a significant effect in 3- and 5-month-old Ppt1−/− mice; however, it did show a significant effect in motor learning in 7-month-old Ppt1−/− mice. The authors conclude that further tests are required but memantine may be useful in INCL patients who have already developed disease symptoms.70
Mice with the Cln3−/−/µMT mutation were developed that were incapable of producing immunoglobulin G. The mice demonstrated ameliorated levels of neuroinflammation. At postnatal day 60 (P60), Cln3−/−/µMT mice showed significantly improved motor coordination as compared to Cln3−/− mice; however, this difference was not observed at P100 and P180. This Cln3−/−/µMT mutation was used as a model to assess the effect of immunosuppressant activity on reduction of neuroinflammation prior to the evaluation of small molecules.

Mycophenolate mofetil (5), an immunosuppressant acting by inhibiting inosine monophosphate dehydrogenase and thus B and T cell proliferation, was evaluated as a potential therapeutic in JNCL. Treatment with compound 5 resulted in a significant increase in motor coordination in Cln3−/− mice and reduced neuroinflammation at 60 mg/kg dose compared to untreated Cln3−/− mice.71 A phase II clinical trial has recently been initiated to determine the safety and tolerability of short-term mycophenolate mofetil administration in patients with JNCL.72
Lithium, which has been shown to be neuroprotective,73 albeit with an unknown mechanism of action, reduces autophagosomes in CLN3 mutant knock-in cerebellar cells (CbCln3Δex7/8/Δex7/8). Lithium also reduced the accumulation of subunit c of mitochondrial ATP synthase and autofluorescence and prevented cell death induced by amino acid deprivation. The mechanism of action leading to the neuroprotective effect of lithium in CbCln3Δex7/8/Δex7/8 cells was shown to be via IMPase inhibition.74
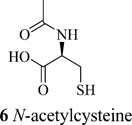
CLN3 has been shown to have a protective effect against oxidative stress induced neuronal cell death in a Drosophila model.75 Mutated CLN3 is responsible for reduced lysosomal arginine levels which would stimulate the urea cycle, increasing nitric oxide (NO) byproducts resulting in generation of reactive oxygen species and DNA damage. Increased levels of biomarkers including carbamoyl phosphate synthetase 1 (CPS1), 8-oxoguanine DNA glycosylase 1 (OGG1), and DNA polymerase β were observed in NCL patient lymphoblasts. Treatment with N-acetylcysteine (6), a known antioxidant, precursor, and analogue of glutathione, reduced expression levels of CPS1, OGG1, and DNA polymerase β in cells derived from Batten disease patients.45 A pilot study was conducted in nine INCL patients to determine the beneficial effects of combination of cysteamine bitartrate and N-acetylcysteine. Decrease in the number and size of GRODs was observed after the first posttreatment follow-up and in the subsequent follow-up GROD levels were undetectable indicating that the treatment had beneficial effects in patients.5
SMALL MOLECULE THERAPEUTICS EVALUATED IN CLN1 DISEASE (INCL)
N-(tert-Butyl)hydroxylamine (NtBuHA) (7) was found to be nontoxic to INCL patient lymphoblasts and fibroblasts, mimics PPT1 by cleaving thioester linkage in palmitoyl-CoA and palmitoylated proteins, reduces GRODs which correlate to ceroid depositions in the cells of INCL patients, ameliorates ER and oxidative stress, and suppresses oxidative stress mediated apoptosis.44

Similarly, NtBuHA in Ppt1−/− mice does not stimulate the production of methemoglobin. Unlike the structurally related compound hydroxylamine, the compound crosses the blood–brain barrier (BBB), depletes GRODs and ceroid deposition, and therefore reduces autofluorescence and apoptosis, preserving motor function and prolonging lifespan. While this compound represents one of the most beneficial small molecules ever evaluated in NCL patient cells and murine models, NtBuHA is only effective in depalmitoylating S-acetylated proteins and is unlikely to help in the depalmitoylation of N-palmitoylated proteins, as the palmitate is linked to the cysteine residues by an amide bond.44

Cysteamine (8), the simplest stable aminothiol, was shown to cleave palmitoyl-CoA in vitro.76 A structure–activity relationship (SAR) of various aminothiols against PPT substrate accumulation was derived. The aminothiols were active at slightly alkaline pH and inactive at acidic pH. The sulfhydryl group was shown to be essential, as it functions to cleave the thioester linkage of the substrate. Cysteamine reduces the quantity of storage material present in INCL lymphoblasts; however, the closely related analogue phosphocysteamine (9) does not. Neither did cysteamine lower the levels of saposin D, a biomarker found in INCL lymphoblasts.43,77 Two analogues of cysteamine, 2-(aminopropyl)aminoethanethiol and dimethylaminoethanethiol, which were reactive in cleaving palmitoyl-CoA, showed elevated levels of saposin D. The authors concluded that cysteamine and its analogues were insufficiently active to produce a therapeutic effect.43 Cysteamine was also tested in four patients with CLN1 mutation in a 7-year open label, nonrandomized trial. Cysteamine showed a decrease in storage material in the patients. However, the reduction in storage material was reversible with decrease or discontinuation of cysteamine. Slower disease progression was observed in two individuals, but the authors could not determine if the slower disease progression was due to the treatment, as slower progression was seen in the same two patients even before treatment began.4

Phosphocysteamine (9) and N-acetylcysteine (6) cleave the thioester linkages in [14C]palmitoyl-CoA in a concentration- and time-dependent fashion in vitro. Phosphocysteamine at 0.5 mM concentration disrupted the thioester linkages in S-acetylated proteins in immortalized lymphoblasts of INCL, LINCL, and JNCL patients with PPT1 deficiency. Phosphocysteamine reduced GROD formation after 3 weeks of treatment in INCL patient fibroblasts and lymphoblasts; however, withdrawal of phosphocysteamine led to reaccumulation of ceroid deposits. Phosphocysteamine also reduced levels of saposin A and D in INCL patient lymphoblasts;77 elevated levels of saposin A and D are observed in PPT1-deficient INCL cells.78 INCL patient lymphoblasts were rescued from apoptosis by treatment with phospocysteamine.77 Phosphocysteamine, an analogue of cysteamine, was used in this study because phosphocysteamine retains the pharmacological properties of cysteamine and does not have a disagreeable odor, unlike cysteamine.

Oxidative stress in neurological disorders can lead to downregulation of antioxidant enzymes such as superoxide dismutase 1 (SOD-1).79 Experiments performed in post-mortem brain tissues from INCL patients showed augmented levels of glucose regulated protein 78 (GRP78/BiP) due to elevated ER stress and cleaved poly ADP-ribose polymerase (PARP), indicating apoptosis.79,80 Resveratrol (10), a potent antioxidant found in grapes, amplified SOD-1 expression in NCL patient lymphoblasts along with reduction in the ER stress marker protein GRP78/BiP and apoptosis marker proteins apoptosis-inducing factor (AIF), cytochrome c, and cleaved PARP.79 In 6-month-old Ppt1-knockout mice reduced levels of claudin 5, occludin, junctional adhesion molecule 1 (JAM1), and claudin-1 indicated that BBB integrity was breached. Elevated levels of IL-17A-positive TH17 lymphocytes, observed in the spleen and brain of Ppt1-knockout mice, activate metalloproteinases (MMPs) which are responsible for disruption of the BBB. Elevated levels of MMP-2, MMP-3, and MMP-9 proteins were observed in both Ppt1 knockout (KO) mice brain and in post-mortem brain tissues of INCL patients. Ppt1-KO mice on a resveratrol diet showed reduced levels of IL-17A, MMP-2, MMP-3, and MMP-9 and elevated levels of occludin and claudin 5. Resveratrol suppressed differentiation of TH17 lymphocytes from naive CD4+ T cells isolated from the spleen of Ppt1-KO mice in a dose- and time-dependent manner.81

PTC124 (Ataluren) (11) is currently under clinical trials for diseases such as cystic fibrosis and Duchenne muscular dystrophy, all caused by nonsense mutations.82,83 Compound 11 causes ribosomal read-through of premature stop codons and is ineffective against normal stop codons. In patient derived INCL fibroblasts, 11 induced PPT1 enzyme activity in a dose- and time-dependent fashion. PPT1 activity was restored to that of 1.3% of normal fibroblasts. Compound 11 was not toxic in INCL patient fibroblasts unlike gentamicin (13). Low levels of PPT1 induced by 11 decreased ceroid deposition and GRODs and protected INCL patient fibroblasts from apoptosis.84 Compound 11 significantly elevated TPP1 protein and enzyme levels in CLN2 patient NPCs and showed a reduction in deposition of subunit c of mitochondrial ATP synthase.48
Pharmacological chaperones have shown a beneficial effect in lysosomal storage disorders resulting from mis-sense mutation and misfolded proteins.85–87 The use of several pharmacological chaperones has been reported in NCL, resulting in the restoration of 2-fold PPT1 activity in INCL patient lymphoblasts with residual enzyme activity but with no observable effect in lymphoblasts with inactive enzyme.88 However, only patients with specific types of mutations can be treated with chaperones, limiting this therapy to a subset of patients.89
SMALL MOLECULE THERAPEUTICS EVALUATED IN CLN2 DISEASE (LINCL)
A single case has been reported wherein a 5-year-old boy was diagnosed with Jansky–Bielschowsky disease (LINCL), caused by TPP1 deficiency. Δ9-Tetrahydrocannabinol (12) showed a “considerable improvement” in the child’s condition.90 No further details have been reported to date.

Aminoglycoside antibiotics like gentamicin (13) can promote read-through of premature stop codons and can be beneficial in disorders caused by nonsense mutations such as cystic fibrosis.91 Premature stop codon mutations are observed in approximately half of the children diagnosed with LINCL92 and in 31% of children diagnosed with INCL.84 Treatment with gentamicin showed approximately 5% and 7% increase in TPP1 levels in cell lines with heterozygous Arg208Stop and heterozygous Arg127Stop mutations, respectively, while it had no effect on a stop codon at Gln66.92

Mutation of the CLN2 gene leads to defective TPP1 causing LINCL. However, a few copies of the normal gene are expressed in patients, supported by the hypothesis that residual TPP1 activity is present.93 The FDA approved lipid lowering drugs gemfibrozil (14) and fenofibrate (15) upregulate TPP1 mRNA and protein in mouse brain cells, such as primary astrocytes, cortex, hippocampus, and striatum neurons, and in human astrocytes and the SH-SY5Y cell line. The fibrate drugs mediated upregulation of TPP1 via the activation of peroxisome proliferator-activated receptor α/retinoid X receptor α (PPARα/RXRα) heterodimer as observed in mouse astrocytes and in vivo in mice. PPARβ and PPARγ were not involved in TPP1 upregulation.94 It should be noted, however, that these experiments were performed in mice brain cells without the phenotype for LINCL, translating these observations into patients remains to be studied. Gemfibrozil and fenofibrate failed to increase TPP1 levels in CLN2 patient NPCs, the stem cell model discussed earlier.48
SMALL MOLECULE THERAPEUTICS EVALUATED IN CLN6 DISEASE (KUFS DISEASE TYPE A)
Minocycline (16), an antibiotic belonging to the tetracycline family, has been shown to have a beneficial effect on neuroinflammation.95 However, in South Hampshire sheep with ovine CLN6 disorder, treatment with minocycline had no preventive effect on the development of blindness, the ratio of gross brain atrophy was unaffected, and microglial and astrocytic activation were not inhibited. The authors concluded that treatment with minocycline does not target chronic inflammation.96

SMALL MOLECULE THERAPEUTICS EVALUATED IN CLN8 DISEASE
A motor neuron disease (mnd) mouse model, similar to a CLN8 gene mutation in humans, shows AMPA receptor mediated excitotoxicity and accumulation of lysosomal storage material.97,98 Chronic treatment with a selective AMPA receptor antagonist 17 6-nitro-7-sulfamoylbenzo(F)quinoxaline-2,3-dione (NBQX) showed an improvement in a range of different tests of motor performance.97,99

Dosing of 70 mg/kg, but not 140 mg/kg, of the noncompetitive AMPA receptor antagonist 18 (ZK 187638) in mnd mice showed significant enhancement in motor neuron function when assessed by different semiquantitative and quantitative neurological evaluation tests as compared to mnd mice with no treatment. Compound 18 showed 3-fold elevated brain concentration as compared to plasma concentration.100
The β2 agonist clenbuterol (19) has previously been shown to be neuroprotective.101 Treatment with clenbuterol significantly delayed motor neuron dysfunction in mnd mice as compared to control when assessed by behavioral tests.102 Clenbuterol treated mnd mice showed significantly reduced eccentrically nucleated motor neurons as compared to untreated mnd mice.103

CONCLUSIONS
Small molecule therapeutics, both experimental and clinically approved, demonstrate potential for the eventual development of a disease-modifying therapy for the treatment of the NCLs (Table 1). Several therapies such as enzyme replacement therapy, gene therapy, and stem cell based therapy have been developed for CLN1 and CLN2 diseases and have reached clinical trial. However, the advantage of small molecules is that they can penetrate the BBB while macromolecules cannot. While the BBB is disrupted in several neurological disorders including Batten disease, normal function would be expected to be restored due to the effect of the therapeutic agent, which may make macromolecules less effective over time.104 Although the exact pathophysiology that drives the disease is still largely unknown, a variety of targets have emerged that provide hope for drug discovery and development. Active compounds exhibit mechanisms of action including enzyme mimics and upregulators, Bcl-2 upregulators, NMDA and AMPA receptor antagonists, calcium channel blockers, immunosuppressants, antioxidants, pharmacological chaperones, and compounds that promote the read-through of premature stop codons. Many of these modes of action are common across all neurodegenerative diseases, and lessons learned from the study of adult diseases will transfer to the development of compounds to treat the NCLs with the hope of speeding the process to fruition by learning from history.
Many of the compounds under evaluation for efficacy in the NCLs are clinically approved drugs for other indications such as the analgesic flupirtine, the Alzheimer’s disease drug memantine, the immunosuppressant mycophenolate mofetil, and the cholesterol lowering drugs gemfibrozil and fenofibrate. A robust drug repurposing program may allow the rapid advancement of these compounds to clinical trial and allow patients access to the latest research in a much decreased time span.105
Of equal importance are the hit structures that these compounds represent for further drug discovery efforts. While the initial compounds may prove ineffective due to a high dosage requirement or manifestation of undesirable side effects, medicinal chemistry lead optimization programs are underway in our lab and others to enhance the activity of these compounds while simultaneously removing the undesirable side effects.
While the community awaits the results of the phase II clinical trial of mycophenolate mofetil, research continues on the polypharmacology of the use of two or more agents in synergy, a tactic developed for the treatment of adult neurodegenerative diseases.106 This approach may well result in measurable improvements in the condition of NCL patients; the combined effects of two or more drugs delivered in synergy are often more efficacious than the individual drugs.
While the NCLs are an orphan disease and lack the investment of more prolific diseases such as cancer, the academic community has embraced the challenge to understand the pathophysiology of the disease, develop robust animal and cellular models and has begun the process of designing and evaluating potential small molecule therapeutics for the treatment of this devastating disease.
Acknowledgments
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award UL1TR001105. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
ABBREVIATIONS USED
- AIF
apoptosis-inducing factor
- AMPA
α-amino-3-hydroxy-5-methyl-4-isooxazolepropionic acid
- BBB
blood–brain barrier
- CSPα
cysteine-string protein α
- CPS1
carbamoyl phosphate synthetase 1
- ER
endoplasmic reticulum
- GROD
granular osmiophilic deposit
- INCL
infantile neuronal ceroid lipofuscinosis
- iPSC
induced pluripotent stem cell
- JAM1
junctional adhesion molecule 1
- JNCL
juvenile neuronal ceroid lipofuscinosis
- KO
knockout
- LINCL
late infantile neuronal ceroid lipofuscinosis
- MMP
matrix metalloproteinase
- mnd
motor neuron disease
- NBQX
6-nitro-7-sulfamoylbenzo-(F)quinoxaline-2,3-dione
- NCL
neuronal ceroid lipofuscinosis
- NMDA
N-methyl-d-aspartate
- NO
nitric oxide
- NPC
neural progenitor cell
- NtBuHA
N-(tert-butyl)hydroxylamine
- OGG1
8-oxoguanine DNA glycosylase 1
- PARP
poly ADP-ribose polymerase
- PPAR
peroxisome proliferator-activated receptor
- PPT1
palmitoyl protein thioesterase 1
- RT-PCR
reverse transcription polymerase chain reaction
- RXR
retinoid X receptor
- SAR
structure–activity relationship
- SOD-1
superoxide dismutase 1
- TPP1
tripeptidyl peptidase I
- UBDRS
unified Batten disease rating scale
Biographies
Nihar Kinarivala obtained his Bachelors in Pharmacy from Nirma University, India, in 2013. He is currently a Ph.D. student in the laboratory of Paul C. Trippier in the Pharmaceutical Sciences Department at Texas Tech University Health Science Center (TTUHSC), Amarillo, TX, where his dissertation research is focused on the synthesis of novel neuroprotective molecules which could be useful for the treatment of Batten disease.
Paul C. Trippier received his D.Phil in Organic Chemistry from Merton College, University of Oxford, U.K., under the supervision of Mark G. Moloney. His research concerned the total synthesis of the natural product oxazolomycin. He carried out postdoctoral research at Cardiff University, U.K., with Chris McGuigan and at Northwestern University, IL, with Richard B. Silverman. At Northwestern his research involved the design, synthesis, and mode of action determination of small molecules active in murine models of amyotrophic lateral sclerosis. Since 2012 Dr. Trippier has been an Assistant Professor at the School of Pharmacy of TTUHSC and a member of the Center for Chemical Biology where his research focuses on small molecule drug discovery for cancer and neurodegenerative diseases.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Kollmann K, Uusi-Rauva K, Scifo E, Tyynela J, Jalanko A, Braulke T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim. Biophys. Acta Mol. Basis Dis. 2013;1832:1866–1881. doi: 10.1016/j.bbadis.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 2.Mink JW, Augustine EF, Adams HR, Marshall FJ, Kwon JM. Classification and natural history of the neuronal ceroid lipofuscinoses. J. Child Neurol. 2013;28:1101–1105. doi: 10.1177/0883073813494268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santavuori P. Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 1988;10:80–83. doi: 10.1016/s0387-7604(88)80075-5. [DOI] [PubMed] [Google Scholar]
- 4.Gavin M, Wen GY, Messing J, Adelman S, Logush A, Jenkins EC, Brown WT, Velinov M. Substrate reduction therapy in four patients with milder CLN1 mutations and juvenile-onset Batten disease using cysteamine bitartrate. JIMD Rep. 2013;11:87–92. doi: 10.1007/8904_2013_226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levin SW, Baker EH, Zein WM, Zhang Z, Quezado ZM, Miao N, Gropman A, Griffin KJ, Bianconi S, Chandra G, Khan OI, Caruso RC, Liu A, Mukherjee AB. Oral cysteamine bitartrate and N-acetylcysteine for patients with infantile neuronal ceroid lipofuscinosis: A pilot study. Lancet Neurol. 2014;13:777–787. doi: 10.1016/S1474-4422(14)70142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selden NR, Al-Uzri A, Huhn SL, Koch TK, Sikora DM, Nguyen-Driver MD, Guillaume DJ, Koh JL, Gultekin SH, Anderson JC, Vogel H, Sutcliffe TL, Jacobs Y, Steiner RD. Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J. Neurosurg. Pediatr. 2013;11:643–652. doi: 10.3171/2013.3.PEDS12397. [DOI] [PubMed] [Google Scholar]
- 7.Hobert JA, Dawson G. Neuronal ceroid lipofuscinoses therapeutic strategies: Past, present and future. Biochim. Biophys. Acta Mol. Basis Dis. 2006;1762:945–953. doi: 10.1016/j.bbadis.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Sands MS. Considerations for the treatment of infantile neuronal ceroid lipofuscinosis (infantile Batten disease) J. Child Neurol. 2013;28:1151–1158. doi: 10.1177/0883073813495960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawkins-Salsbury JA, Cooper JD, Sands MS. Pathogenesis and therapies for infantile neuronal ceroid lipofuscinosis (infantile CLN1 disease) Biochim. Biophys. Acta Mol. Basis Dis. 2013;1832:1906–1909. doi: 10.1016/j.bbadis.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong AM, Rahim AA, Waddington SN, Cooper JD. Current therapies for the soluble lysosomal forms of neuronal ceroid lipofuscinosis. Biochem. Soc. Trans. 2010;38:1484–1488. doi: 10.1042/BST0381484. [DOI] [PubMed] [Google Scholar]
- 11.Haltia M, Goebel HH. The neuronal ceroid-lipofuscinoses: A historical introduction. Biochim. Biophys. Acta Mol. Basis Dis. 2013;1832:1795–1800. doi: 10.1016/j.bbadis.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Schulz A, Kohlschutter A, Mink J, Simonati A, Williams R. NCL diseases - clinical perspectives. Biochim. Biophys. Acta Mol. Basis Dis. 2013;1832:1801–1806. doi: 10.1016/j.bbadis.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim. Biophys. Acta Mol. Basis Dis. 2013;1832:1807–1826. doi: 10.1016/j.bbadis.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Palmer DN, Barry LA, Tyynela J, Cooper JD. NCL disease mechanisms. Biochim. Biophys. Acta Mol. Basis Dis. 2013;1832:1882–1893. doi: 10.1016/j.bbadis.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 15.Lockhart EM, Warner DS, Pearlstein RD, Penning DH, Mehrabani S, Boustany RM. Allopregnanolone attenuates N-methyl-D-aspartate-induced excitotoxicity and apoptosis in the human NT2 cell line in culture. Neurosci. Lett. 2002;328:33–36. doi: 10.1016/s0304-3940(02)00448-2. [DOI] [PubMed] [Google Scholar]
- 16.Persaud-Sawin DA, Boustany RM. Cell death pathways in juvenile Batten disease. Apoptosis. 2005;10:973–985. doi: 10.1007/s10495-005-0733-6. [DOI] [PubMed] [Google Scholar]
- 17.Boustany RM. Lysosomal storage diseases- the horizon expands. Nat. Rev. Neurol. 2013;9:583–598. doi: 10.1038/nrneurol.2013.163. [DOI] [PubMed] [Google Scholar]
- 18.Carcel-Trullols J, Kovacs AD, Pearce DA. Cell biology of the NCL proteins: What they do and don’t do. Biochim. Biophys. Acta Mol. Basis Dis. 2015;1852:2242–2255. doi: 10.1016/j.bbadis.2015.04.027. [DOI] [PubMed] [Google Scholar]
- 19.Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease) Biochim. Biophys. Acta Mol. Basis Dis. 2015;1852:2237–2241. doi: 10.1016/j.bbadis.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rakheja D, Narayan SB, Bennett MJ. The function of CLN3P, the Batten disease protein. Mol. Genet. Metab. 2008;93:269–274. doi: 10.1016/j.ymgme.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Rakheja D, Narayan SB, Bennett MJ. Juvenile neuronal ceroid-lipofuscinosis (Batten disease): A brief review and update. Curr. Mol. Med. 2007;7:603–608. doi: 10.2174/156652407781695729. [DOI] [PubMed] [Google Scholar]
- 22.Phillips SN, Benedict JW, Weimer JM, Pearce DA. CLN3, the protein associated with Batten disease: Structure, function and localization. J. Neurosci. Res. 2005;79:573–583. doi: 10.1002/jnr.20367. [DOI] [PubMed] [Google Scholar]
- 23.Chang JW, Choi H, Kim HJ, Jo DG, Jeon YJ, Noh JY, Park WJ, Jung YK. Neuronal vulnerability of CLN3 deletion to calcium-induced cytotoxicity is mediated by calsenilin. Hum. Mol. Genet. 2007;16:317–326. doi: 10.1093/hmg/ddl466. [DOI] [PubMed] [Google Scholar]
- 24.Kovacs AD, Saje A, Wong A, Ramji S, Cooper JD, Pearce DA. Age-dependent therapeutic effect of memantine in a mouse model of juvenile Batten disease. Neuropharmacology. 2012;63:769–775. doi: 10.1016/j.neuropharm.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham RK, Pouladi MA, Joshi P, Lu G, Deng Y, Wu NP, Figueroa BE, Metzler M, Andre VM, Slow EJ, Raymond L, Friedlander R, Levine MS, Leavitt BR, Hayden MR. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. J. Neurosci. 2009;29:2193–2204. doi: 10.1523/JNEUROSCI.5473-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, Hofmann SL, Peltonen L. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–587. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- 27.Camp LA, Hofmann SL. Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J. Biol. Chem. 1993;268:22566–22574. [PubMed] [Google Scholar]
- 28.Lin L, Sohar I, Lackland H, Lobel P. The human CLN2 protein/tripeptidyl-peptidase I is a serine protease that autoactivates at acidic pH. J. Biol. Chem. 2001;276:2249–2255. doi: 10.1074/jbc.M008562200. [DOI] [PubMed] [Google Scholar]
- 29.Noskova L, Stranecky V, Hartmannova H, Pristoupilova A, Baresova V, Ivanek R, Hulkova H, Jahnova H, van der Zee J, Staropoli JF, Sims KB, Tyynela J, Van Broeckhoven C, Nijssen PC, Mole SE, Elleder M, Kmoch S. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011;89:241–252. doi: 10.1016/j.ajhg.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mastrogiacomo A, Parsons SM, Zampighi GA, Jenden DJ, Umbach JA, Gundersen CB. Cysteine string proteins: A potential link between synaptic vesicles and presynaptic Ca2+ channels. Science. 1994;263:981–982. doi: 10.1126/science.7906056. [DOI] [PubMed] [Google Scholar]
- 31.Sharma M, Burre J, Sudhof TC. CSP[alpha] promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011;13:30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- 32.Smith KR, Dahl H-HM, Canafoglia L, Andermann E, Damiano J, Morbin M, Bruni AC, Giaccone G, Cossette P, Saftig P, Grötzinger J, Schwake M, Andermann F, Staropoli JF, Sims KB, Mole SE, Franceschetti S, Alexander NA, Cooper JD, Chapman HA, Carpenter S, Berkovic SF, Bahlo M. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2013;22:1417–1423. doi: 10.1093/hmg/dds558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lane SC, Jolly RD, Schmechel DE, Alroy J, Boustany RM. Apoptosis as the mechanism of neurodegeneration in Batten’s disease. J. Neurochem. 1996;67:677–683. doi: 10.1046/j.1471-4159.1996.67020677.x. [DOI] [PubMed] [Google Scholar]
- 34.Dhar S, Bitting RL, Rylova SN, Jansen PJ, Lockhart E, Koeberl DD, Amalfitano A, Boustany RM. Flupirtine blocks apoptosis in Batten patient lymphoblasts and in human postmitotic CLN3-and CLN2-deficient neurons. Ann. Neurol. 2002;51:448–466. doi: 10.1002/ana.10143. [DOI] [PubMed] [Google Scholar]
- 35.Puranam K, Qian WH, Nikbakht K, Venable M, Obeid L, Hannun Y, Boustany RM. Upregulation of Bcl-2 and elevation of ceramide in Batten disease. Neuropediatrics. 1997;28:37–41. doi: 10.1055/s-2007-973664. [DOI] [PubMed] [Google Scholar]
- 36.Cho S, Dawson PE, Dawson G. Role of palmitoyl-protein thioesterase in cell death: Implications for infantile neuronal ceroid lipofuscinosis. Eur. J. Paediatr. Neurol. 2001;5(Suppl. A):53–55. doi: 10.1053/ejpn.2000.0435. [DOI] [PubMed] [Google Scholar]
- 37.Haddad SE, Khoury M, Daoud M, Kantar R, Harati H, Mousallem T, Alzate O, Meyer B, Boustany RM. CLN5 and CLN8 protein association with ceramide synthase: Biochemical and proteomic approaches. Electrophoresis. 2012;33:3798–3809. doi: 10.1002/elps.201200472. [DOI] [PubMed] [Google Scholar]
- 38.Oswald MJ, Palmer DN, Kay GW, Shemilt SJA, Rezaie P, Cooper JD. Glial activation spreads from specific cerebral foci and precedes neurodegeneration in presymptomatic ovine neuronal ceroid lipofuscinosis (CLN6) Neurobiol. Dis. 2005;20:49–63. doi: 10.1016/j.nbd.2005.01.025. [DOI] [PubMed] [Google Scholar]
- 39.Kay GW, Jay NP, Palmer DN. The specific loss of GnRH-positive neurons from the hypothalamus of sheep with CLN6 neuronal ceroid lipofuscinosis occurs without glial activation and has only minor effects on reproduction. Neurobiol. Dis. 2011;41:614–623. doi: 10.1016/j.nbd.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Schulz A, Dhar S, Rylova S, Dbaibo G, Alroy J, Hagel C, Artacho I, Kohlschutter A, Lin S, Boustany RM. Impaired cell adhesion and apoptosis in a novel CLN9 Batten disease variant. Ann. Neurol. 2004;56:342–350. doi: 10.1002/ana.20187. [DOI] [PubMed] [Google Scholar]
- 41.Vidal-Donet JM, Carcel-Trullols J, Casanova B, Aguado C, Knecht E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One. 2013;8:e55526. doi: 10.1371/journal.pone.0055526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sleat DE, Gin RM, Sohar I, Wisniewski K, Sklower- Brooks S, Pullarkat RK, Palmer DN, Lerner TJ, Boustany RM, Uldall P, Siakotos AN, Donnelly RJ, Lobel P. Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am. J. Hum. Genet. 1999;64:1511–1523. doi: 10.1086/302427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu JY, Hofmann SL. Inefficient cleavage of palmitoylprotein thioesterase (PPT) substrates by aminothiols: Implications for treatment of infantile neuronal ceroid lipofuscinosis. J. Inherited Metab. Dis. 2006;29:119–126. doi: 10.1007/s10545-006-0225-z. [DOI] [PubMed] [Google Scholar]
- 44.Sarkar C, Chandra G, Peng S, Zhang Z, Liu A, Mukherjee AB. Neuroprotection and lifespan extension in Ppt1(−/−) mice by NtBuHA: Therapeutic implications for INCL. Nat. Neurosci. 2013;16:1608–1617. doi: 10.1038/nn.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JB, Lim N, Kim SJ, Heo TH. N-acetylcysteine normalizes the urea cycle and DNA repair in cells from patients with Batten disease. Cell Biochem. Funct. 2012;30:677–682. doi: 10.1002/cbf.2849. [DOI] [PubMed] [Google Scholar]
- 46.Narayan SB, Rakheja D, Tan L, Pastor JV, Bennett MJ. CLN3P, the Batten’s disease protein, is a novel palmitoyl-protein Delta-9 desaturase. Ann. Neurol. 2006;60:570–577. doi: 10.1002/ana.20975. [DOI] [PubMed] [Google Scholar]
- 47.Warnock A, Tan L, Li C, An Haack K, Narayan SB, Bennett MJ. Amlodipine prevents apoptotic cell death by correction of elevated intracellular calcium in a primary neuronal model of Batten disease (CLN3 disease) Biochem. Biophys. Res. Commun. 2013;436:645–649. doi: 10.1016/j.bbrc.2013.04.113. [DOI] [PubMed] [Google Scholar]
- 48.Lojewski X, Staropoli JF, Biswas-Legrand S, Simas AM, Haliw L, Selig MK, Coppel SH, Goss KA, Petcherski A, Chandrachud U, Sheridan SD, Lucente D, Sims KB, Gusella JF, Sondhi D, Crystal RG, Reinhardt P, Sterneckert J, Scholer H, Haggarty SJ, Storch A, Hermann A, Cotman SL. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum. Mol. Genet. 2014;23:2005–2022. doi: 10.1093/hmg/ddt596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pearce DA, Sherman F. A yeast model for the study of Batten disease. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6915–6918. doi: 10.1073/pnas.95.12.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hickey AJ, Chotkowski HL, Singh N, Ault JG, Korey CA, MacDonald ME, Glaser RL. Palmitoyl-protein thioesterase 1 deficiency in Drosophila melanogaster causes accumulation of abnormal storage material and reduced life span. Genetics. 2006;172:2379–2390. doi: 10.1534/genetics.105.053306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bond M, Holthaus SM, Tammen I, Tear G, Russell C. Use of model organisms for the study of neuronal ceroid lipofuscinosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013;1832:1842–1865. doi: 10.1016/j.bbadis.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 52.Shacka JJ. Mouse models of neuronal ceroid lipofuscinoses: Useful pre-clinical tools to delineate disease pathophysiology and validate therapeutics. Brain Res. Bull. 2012;88:43–57. doi: 10.1016/j.brainresbull.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Shacka JJ, Roth KA. Cathepsin deficiency as a model for neuronal ceroid lipofuscinoses. Am. J. Pathol. 2005;167:1473–1476. doi: 10.1016/S0002-9440(10)61233-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Faller KME, Gutierrez-Quintana R, Mohammed A, Rahim AA, Tuxworth RI, Wager K, Bond M. The neuronal ceroid lipofuscinoses: Opportunities from model systems. Biochim. Biophys. Acta Mol. Basis Dis. 2015;1852:2267–2278. doi: 10.1016/j.bbadis.2015.04.022. [DOI] [PubMed] [Google Scholar]
- 55.Osorio NS, Sampaio-Marques B, Chan CH, Oliveira P, Pearce DA, Sousa N, Rodrigues F. Neurodevelopmental delay in the Cln3Deltaex7/8 mouse model for Batten disease. Genes, Brain Behav. 2009;8:337–345. doi: 10.1111/j.1601-183X.2009.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitchison HM, Bernard DJ, Greene ND, Cooper JD, Junaid MA, Pullarkat RK, de Vos N, Breuning MH, Owens JW, Mobley WC, Gardiner RM, Lake BD, Taschner PE, Nussbaum RL. Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. The Batten Mouse Model Consortium [corrected] Neurobiol. Dis. 1999;6:321–334. doi: 10.1006/nbdi.1999.0267. [DOI] [PubMed] [Google Scholar]
- 58.Kovacs AD, Weimer JM, Pearce DA. Selectively increased sensitivity of cerebellar granule cells to AMPA receptor-mediated excitotoxicity in a mouse model of Batten disease. Neurobiol. Dis. 2006;22:575–585. doi: 10.1016/j.nbd.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 59.Kornhuber J, Bleich S, Wiltfang J, Maler M, Parsons CG. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. J. Neural Transm. 1999;106:857–867. doi: 10.1007/s007020050206. [DOI] [PubMed] [Google Scholar]
- 60.Cialone J, Augustine EF, Newhouse N, Adams H, Vierhile A, Marshall FJ, de Blieck EA, Kwon J, Rothberg PG, Mink JW. Parent-reported benefits of flupirtine in juvenile neuronal ceroid lipofuscinosis (Batten disease; CLN3) are not supported by quantitative data. J. Inherited Metab. Dis. 2011;34:1075–1081. doi: 10.1007/s10545-011-9346-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Narayan SB, Rakheja D, Pastor JV, Rosenblatt K, Greene SR, Yang J, Wolf BA, Bennett MJ. Over-expression of CLN3P, the Batten disease protein, inhibits PANDER-induced apoptosis in neuroblastoma cells: Further evidence that CLN3P has anti-apoptotic properties. Mol. Genet. Metab. 2006;88:178–183. doi: 10.1016/j.ymgme.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 62.An Haack K, Narayan SB, Li H, Warnock A, Tan L, Bennett MJ. Screening for calcium channel modulators in CLN3 siRNA knock down SH-SY5Y neuroblastoma cells reveals a significant decrease of intracellular calcium levels by selected L-type calcium channel blockers. Biochim. Biophys. Acta Gen. Subj. 2011;1810:186–191. doi: 10.1016/j.bbagen.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kovacs AD, Pearce DA. Attenuation of AMPA receptor activity improves motor skills in a mouse model of juvenile Batten disease. Exp. Neurol. 2008;209:288–291. doi: 10.1016/j.expneurol.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovacs AD, Saje A, Wong A, Szenasi G, Kiricsi P, Szabo E, Cooper JD, Pearce DA. Temporary inhibition of AMPA receptors induces a prolonged improvement of motor performance in a mouse model of juvenile Batten disease. Neuropharmacology. 2011;60:405–409. doi: 10.1016/j.neuropharm.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gardoni F, Di Luca M. New targets for pharmacological intervention in the glutamatergic synapse. Eur. J. Pharmacol. 2006;545:2–10. doi: 10.1016/j.ejphar.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 66.Macauley SL, Wozniak DF, Kielar C, Tan Y, Cooper JD, Sands MS. Cerebellar pathology and motor deficits in the palmitoyl protein thioesterase 1-deficient mouse. Exp. Neurol. 2009;217:124–135. doi: 10.1016/j.expneurol.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Finn R, Kovacs AD, Pearce DA. Altered glutamate receptor function in the cerebellum of the Ppt1−/− mouse, a murine model of infantile neuronal ceroid lipofuscinosis. J. Neurosci. Res. 2012;90:367–375. doi: 10.1002/jnr.22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pears MR, Salek RM, Palmer DN, Kay GW, Mortishire-Smith RJ, Griffin JL. Metabolomic investigation of CLN6 neuronal ceroid lipofuscinosis in affected South Hampshire sheep. J. Neurosci. Res. 2007;85:3494–3504. doi: 10.1002/jnr.21343. [DOI] [PubMed] [Google Scholar]
- 69.Seitz D, Grodd W, Schwab A, Seeger U, Klose U, Nägele T. MR imaging and localized proton MR spectroscopy in late infantile neuronal ceroid lipofuscinosis. Am. J. Neuroradiol. 1998;19:1373–1377. [PMC free article] [PubMed] [Google Scholar]
- 70.Finn R, Kovacs AD, Pearce DA. Treatment of the Ppt1(−/−) mouse model of infantile neuronal ceroid lipofuscinosis with the N-methyl-D-aspartate (NMDA) receptor antagonist memantine. J. Child Neurol. 2013;28:1159–1168. doi: 10.1177/0883073813494480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seehafer SS, Ramirez-Montealegre D, Wong AM, Chan CH, Castaneda J, Horak M, Ahmadi SM, Lim MJ, Cooper JD, Pearce DA. Immunosuppression alters disease severity in juvenile Batten disease mice. J. Neuroimmunol. 2011;230:169–172. doi: 10.1016/j.jneuroim.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cellcept for treatment of juvenile neuronal ceroid lipofuscinosis (JUMP) [accessed June 25, 2015]; https://www.clinicaltrials.gov/ct2/show/NCT01399047.
- 73.Chuang DM. The antiapoptotic actions of mood stabilizers: Molecular mechanisms and therapeutic potentials. Ann. N. Y. Acad. Sci. 2005;1053:195–204. doi: 10.1196/annals.1344.018. [DOI] [PubMed] [Google Scholar]
- 74.Chang JW, Choi H, Cotman SL, Jung YK. Lithium rescues the impaired autophagy process in CbCln3(Deltaex7/8/Deltaex7/8) cerebellar cells and reduces neuronal vulnerability to cell death via IMPase inhibition. J. Neurochem. 2011;116:659–668. doi: 10.1111/j.1471-4159.2010.07158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tuxworth RI, Chen H, Vivancos V, Carvajal N, Huang X, Tear G. The Batten disease gene CLN3 is required for the response to oxidative stress. Hum. Mol. Genet. 2011;20:2037–2047. doi: 10.1093/hmg/ddr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu JY, Verkruyse LA, Hofmann SL. The effects of lysosomotropic agents on normal and INCL cells provide further evidence for the lysosomal nature of palmitoyl-protein thioesterase function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2002;1583:35–44. doi: 10.1016/s1388-1981(02)00158-0. [DOI] [PubMed] [Google Scholar]
- 77.Zhang Z, Butler JD, Levin SW, Wisniewski KE, Brooks SS, Mukherjee AB. Lysosomal ceroid depletion by drugs: Therapeutic implications for a hereditary neurodegenerative disease of childhood. Nat. Med. 2001;7:478–484. doi: 10.1038/86554. [DOI] [PubMed] [Google Scholar]
- 78.Tyynela J, Palmer DN, Baumann M, Haltia M. Storage of saposins A and D in infantile neuronal ceroid-lipofuscinosis. FEBS Lett. 1993;330:8–12. doi: 10.1016/0014-5793(93)80908-d. [DOI] [PubMed] [Google Scholar]
- 79.Yoon DH, Kwon OY, Mang JY, Jung MJ, Kim do Y, Park YK, Heo TH, Kim SJ. Protective potential of resveratrol against oxidative stress and apoptosis in Batten disease lymphoblast cells. Biochem. Biophys. Res. Commun. 2011;414:49–52. doi: 10.1016/j.bbrc.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 80.Kim SJ, Zhang Z, Hitomi E, Lee YC, Mukherjee AB. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 2006;15:1826–1834. doi: 10.1093/hmg/ddl105. [DOI] [PubMed] [Google Scholar]
- 81.Saha A, Sarkar C, Singh SP, Zhang Z, Munasinghe J, Peng S, Chandra G, Kong E, Mukherjee AB. The blood-brain barrier is disrupted in a mouse model of infantile neuronal ceroid lipofuscinosis: Amelioration by resveratrol. Hum. Mol. Genet. 2012;21:2233–2244. doi: 10.1093/hmg/dds038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Study of ataluren in nonsense mutation cystic fibrosis (ACT CF) [accessed June 25, 2015]; https://clinicaltrials.gov/ct2/show/NCT02139306.
- 83.Phase 3 study of ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD) [accessed June 25, 2015]; https://clinicaltrials.gov/ct2/show/NCT01826487.
- 84.Sarkar C, Zhang Z, Mukherjee AB. Stop codon read-through with PTC124 induces palmitoyl-protein thioesterase-1 activity, reduces thioester load and suppresses apoptosis in cultured cells from INCL patients. Mol. Genet. Metab. 2011;104:338–345. doi: 10.1016/j.ymgme.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 86.Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C, Kinnunen PK, Nylandsted J, Jaattela M. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–553. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 87.Boyd RE, Lee G, Rybczynski P, Benjamin ER, Khanna R, Wustman BA, Valenzano KJ. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 2013;56:2705–2725. doi: 10.1021/jm301557k. [DOI] [PubMed] [Google Scholar]
- 88.Dawson G, Schroeder C, Dawson PE. Palmitoyl:protein thioesterase (PPT1) inhibitors can act as pharmacological chaperones in infantile Batten disease. Biochem. Biophys. Res. Commun. 2010;395:66–69. doi: 10.1016/j.bbrc.2010.03.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Mol. Med. 2009;1:268–279. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lorenz R. A casuistic rationale for the treatment of spastic and myocloni in a childhood neurodegenerative disease: Neuronal ceroid lipofuscinosis of the type Jansky-Bielschowsky. Neuro. Endocrinol. Lett. 2002;23:387–390. [PubMed] [Google Scholar]
- 91.Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat. Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- 92.Sleat DE, Sohar I, Gin RM, Lobel P. Aminoglycoside-mediated suppression of nonsense mutations in late infantile neuronal ceroid lipofuscinosis. Eur. J. Paediatr. Neurol. 2001;5(Suppl. A):57–62. doi: 10.1053/ejpn.2000.0436. [DOI] [PubMed] [Google Scholar]
- 93.Walus M, Kida E, Golabek AA. Functional consequences and rescue potential of pathogenic missense mutations in tripeptidyl peptidase I. Hum. Mutat. 2010;31:710–721. doi: 10.1002/humu.21251. [DOI] [PubMed] [Google Scholar]
- 94.Ghosh A, Corbett GT, Gonzalez FJ, Pahan K. Gemfibrozil and fenofibrate, Food and Drug Administration-approved lipid-lowering drugs, up-regulate tripeptidyl-peptidase 1 in brain cells via peroxisome proliferator-activated receptor alpha: Implications for late infantile Batten disease therapy. J. Biol. Chem. 2012;287:38922–38935. doi: 10.1074/jbc.M112.365148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav. Brain Res. 2009;196:168–179. doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 96.Kay GW, Palmer DN. Chronic oral administration of minocycline to sheep with ovine CLN6 neuronal ceroid lipofuscinosis maintains pharmacological concentrations in the brain but does not suppress neuroinflammation or disease progression. J. Neuroinflammation. 2013;10:97. doi: 10.1186/1742-2094-10-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mennini T, Cagnotto A, Carvelli L, Comoletti D, Manzoni C, Muzio V, Rizzi M, Vezzani A. Biochemical and pharmacological evidence of a functional role of AMPA receptors in motor neuron dysfunction in mnd mice. Eur. J. Neurosci. 1999;11:1705–1710. doi: 10.1046/j.1460-9568.1999.00588.x. [DOI] [PubMed] [Google Scholar]
- 98.Bronson RT, Lake BD, Cook S, Taylor S, Davisson MT. Motor neuron degeneration of mice is a model of neuronal ceroid lipofuscinosis (Batten’s disease) Ann. Neurol. 1993;33:381–385. doi: 10.1002/ana.410330408. [DOI] [PubMed] [Google Scholar]
- 99.Callahan LM, Wylen EL, Messer A, Mazurkiewicz JE. Neurofilament distribution is altered in the mnd (Motor Neuron Degeneration) mouse. J. Neuropathol. Exp. Neurol. 1991;50:491–504. doi: 10.1097/00005072-199107000-00009. [DOI] [PubMed] [Google Scholar]
- 100.Elger B, Schneider M, Winter E, Carvelli L, Bonomi M, Fracasso C, Guiso G, Colovic M, Caccia S, Mennini T. Optimized synthesis of AMPA receptor antagonist ZK 187638 and neurobehavioral activity in a mouse model of neuronal ceroid lipofuscinosis. Chem Med Chem. 2006;1:1142–1148. doi: 10.1002/cmdc.200600144. [DOI] [PubMed] [Google Scholar]
- 101.Semkova I, Schilling M, Henrich-Noack P, Rami A, Krieglstein J. Clenbuterol protects mouse cerebral cortex and rat hippocampus from ischemic damage and attenuates glutamate neurotoxicity in cultured hippocampal neurons by induction of NGF. Brain Res. 1996;717:44–54. doi: 10.1016/0006-8993(95)01567-1. [DOI] [PubMed] [Google Scholar]
- 102.Messer A, Flaherty L. Autosomal dominance in a late-onset motor neuron disease in the mouse. J. Neurogenet. 1986;3:345–355. doi: 10.3109/01677068609106858. [DOI] [PubMed] [Google Scholar]
- 103.Zeman RJ, Peng H, Etlinger JD. Clenbuterol retards loss of motor function in motor neuron degeneration mice. Exp. Neurol. 2004;187:460–467. doi: 10.1016/j.expneurol.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 104.Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: Lessons from 20 years of experience and remaining challenges. Annu. Rev. Genomics Hum. Genet. 2012;13:307–335. doi: 10.1146/annurev-genom-090711-163739. [DOI] [PubMed] [Google Scholar]
- 105.Chong CR, Sullivan DJ., Jr New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 106.Trippier PC, Jansen Labby K, Hawker DD, Mataka JJ, Silverman RB. Target- and mechanism-based therapeutics for neurodegenerative diseases: Strength in numbers. J. Med. Chem. 2013;56:3121–3147. doi: 10.1021/jm3015926. [DOI] [PMC free article] [PubMed] [Google Scholar]