

Abstract
Development of colorectal cancer occurs via a number of key pathways, with the clinicopathological features of specific subgroups being driven by underlying molecular changes. Mutations in key genes within the network of signalling pathways have been identified; however, therapeutic strategies to target these aberrations remain limited. As understanding of the biology of colorectal cancer has improved, this has led to a move toward broader genomic testing, collaborative research and innovative, adaptive clinical trial design. Recent developments in therapy include the routine adoption of wider mutational spectrum testing prior to use of targeted therapies and the first promise of effective immunotherapy for colorectal cancer patients. This review details current biomarkers in colorectal cancer for molecular stratification and for treatment allocation purposes, including open and planned precision medicine trials. Advances in our understanding, therapeutic strategy and technology will also be outlined.
Keywords: Colorectal cancer, Biomarker stratification, Targeted therapy, Pathways, Molecular subtypes, Personalized medicine trials, Microsatellite instability, RAS, BRAF, EGFR, c-MET, HER2
Background
Treatment of metastatic colorectal cancer (CRC) with traditional 5-fluorouracil (5FU)-based chemotherapy has been modified, with addition of oxaliplatin, irinotecan and monoclonal antibodies targeting vascular endothelial growth factor (VEGF) and the epidermal growth factor receptor (EGFR). These modifications have led to improved overall survival for patients to over 40 months from diagnosis [1–3]. However, considerable heterogeneity exists within CRC due to the varied genetic and epigenetic mechanisms involved in differing pathways of colorectal carcinogenesis [4]. Improved understanding of common tumours, for example breast cancer, has resulted in stratification for prognostication and treatment purposes being used for over a decade [5]. In non-small cell lung cancer (NSCLC), single gene “driver” mutations predict for high response rates using molecularly targeted agents [6, 7]. However, attempts to stratify CRC using clinicopathological and molecular features for prognostic and predictive purposes have had limited success, and correlation between subtyping strategies has been poor [8–10]. This review will discuss molecularly defined subtypes of CRC and implications for current and future patient management.
Colorectal Carcinogenesis
Fearon and Vogelstein proposed a stepwise model for colorectal carcinogenesis highlighting the role of critical tumour suppressor genes and oncogenes in adenoma and subsequent carcinoma development with inactivation of the adenomatous polyposis coli (APC) tumour suppressor gene as the initiating event [11]. This is understood to be the predominant mechanism for development of CRC. Subsequently, less frequently occurring tumourigenic pathways have been identified. The processes have been classified as either chromosomal instability (CIN) or mutator phenotype that includes DNA repair defects and aberrant DNA methylation [4]. Characteristic genomic changes are associated with each of these (Fig. 1a).
Fig. 1.
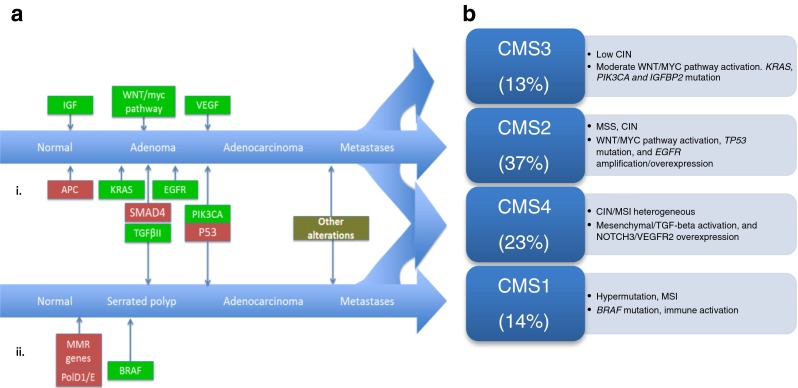
a Genetic models of CRC: i. Chromosomal Instability; ii. Mutator phenotype. Stepwise carcinogenesis occurs due to differing molecular changes in each model. b Resulting core genomic subtypes by molecular subtyping. MMR mismatch repair, CIN chromosomal instability, MSS microsatellite stable, MSI microsatellite instable
Microsatellite Instability
Protective mechanisms exist to facilitate replication of normal cellular DNA. Genes encoding for DNA damage repair (DDR) proteins are commonly mutated in cancer. Dysregulation of this process permits an accumulation of somatic mutations and contributes to genomic instability, now recognised as critical in cancer development and metastasis [12]. As a pathway involved in both hereditary and sporadic forms of CRC, the mutator phenotype with mismatch repair deficiency (dMMR) is of particular interest.
Postreplicative mismatch repair (MMR) maintains genomic stability by eliminating single-base mismatches and insertion-deletion loops of short repeated nucleotide sequences (microsatellites) occurring during DNA replication [13]. Normal interaction of critical MMR proteins comprising MLH1, MSH2, MSH3, MSH6 and PMS2 is required for proofreading and correction of these insertion-deletion loops. Mutation of genes encoding for these MMR proteins or hypermethylation of the MLH1 promoter causes gene silencing, with dMMR and subsequent microsatellite instability (MSI). Due to lack of recognition and clearance of damaged cells during replication, dMMR status results in an accumulation of mutations within tumour cells compared to those with proficient MMR (pMMR) or microsatellite stability (MSS) that facilitates tumour growth [14].
The MSI subgroup constitutes approximately 15 % of CRC overall. Germline mutation of MMR genes may occur as an autosomal dominant syndrome termed hereditary non-polyposis colorectal cancer (HNPCC), or Lynch syndrome, and contributes to 2–5 % of CRC. MLH1 is most frequently involved either by mutation in 60 % of inherited cases or epigenetic inactivation in association with the CpG island methylator phenotype (CIMP) [15]. MSI CRCs are more frequently located in the proximal bowel, with poor differentiation, mucinous subtype and dense lymphocytic infiltration, suggestive of a strong antitumoural immune response [16]. Patients with sporadic MSI CRC are older and the tumours are characterised by frequent BRAF (V600E) mutation and absence of MLH1 and PMS2 proteins [17]. Loss of MLH1 expression increases with age, resulting in CRC development in the elderly, in contrast to earlier disease presentation in patients with Lynch syndrome [18, 19].
DNA polymerases δ and ε are involved in DNA reconstruction following damage. Germline variants in the exonuclease domain of the DNA polymerases POLE and POLD1 predispose to cancer including CRC, by impairing polymerase proofreading and greatly increasing the rate of base substitution mutations. The resulting tumour phenotype has similarities to that caused by dMMR, including high immunogenicity and favourable prognosis but with mutation burden significantly higher than that found in MSI CRC [20, 21]. These are a rare subtype (0.25–1.5 % of CRC) and no therapeutic strategies currently exist to target these specific genomic aberrations, although such investigation is planned (Table 1 and Table 2).
Table 1.
Selected molecular subtypes of colorectal cancer and associated clinical trials utilising targeted agents
| Tumour subtype | Investigative strategies | Agents | Phase of trial | Clinicaltrials.gov identifier |
|---|---|---|---|---|
| Mismatch repair deficient/microsatellite instable | PARP inhibition | Olaparib | II | NCT00912743 |
| Immune checkpoint inhibition (PD-L1) | Durvalumab (MEDI4736) | II | NCT02227667 | |
| Immune checkpoint inhibition (PD-L1) | Atezolizumab (MPDL3280A) | II | NCT02291289 | |
| Immune checkpoint inhibition (PD-1) | Nivolumab ± Ipilimumab | I/II | NCT02060188 | |
| Immune checkpoint inhibition (PD-1) | Pembrolizumab (MK-3475) | II | NCT02460198 | |
| ± standard chemotherapy | III | NCT02563002 | ||
| RAS mutation | Pan-RAF inhibition | BMS-908662 ± Cetuximab | I/II | NCT01086267 |
| AKT and MEK inhibition | MK-2206 and AZD6244 (Selumetinib) | II | NCT01333475 | |
| HDAC inhibition | 4SC-201 (Resminostat) + FOLFIRI | II | NCT01277406 | |
| ERK inhibition | CC-90003 | I | NCT02313012 | |
| NOTCH inhibition | RO4929097 + Cetuximab | I | NCT01198535 | |
| MEK and MET inhibition | PD-0325901 + Crizotinib | I | NCT02510001 | |
| Multikinase inhibition | Regorafenib | II | NCT02175654 | |
| MEK and BCL2 inhibition | Trametinib and Navitoclax (ABT-263) | I/II | NCT02079740 | |
| PI3K and MEK inhibition | BKM120 + MEK162 | I | NCT01363232 | |
| Resistant to EGFR inhibition | RAF inhibition | BMS-908662 ± Cetuximab | I/II | NCT01086267 |
| NOTCH inhibition | RO4929097 + Cetuximab | I | NCT01198535 | |
| BRAF mutation | PI3K and MEK inhibition | BKM120 + MEK162 | I | NCT01363232 |
| BEZ235 + MEK162 | I | NCT01337765 | ||
| BRAF, MEK and EGFR inhibition | Trametanib, Dabrafenib + Panitumumab | II | NCT01750918 | |
| RAF inhibition | BMS-908662 ± cetuximab | I/II | NCT01086267 | |
| BRAF and EGFR inhibition | Irinotecan, Cetuximab + Vemurafenib | II | NCT02164916 | |
| BRAF, PI3K and EGFR inhibition | LGX818, BYL719, and Cetuximab | I/II | NCT01719380 | |
| BRAF, WNT and EGFR inhibition | LGX818, WNT974 and Cetuximab | I/II | NCT02278133 | |
| ERK inhibition | BVD-523 | I | NCT02313012 | |
| ERK inhibition | CC-90003 | I | NCT02313012 | |
| PI3K mutation/PTEN depletion | MEK and PI3K/mTOR inhibition | Pimasertib + SAR245409 | I | NCT01390818 |
| AKT and MEK inhibition | MK-2206 and AZD6244 (selumetinib) | II | NCT01333475 | |
| HER2 amplification | HER2 dual inhibition | Trastuzumab + Lapatinib | II | EudraCT Number: 2012-002128-33 |
| Trastuzumab + Pertuzumab | II | |||
| MET amplification | MEK and MET inhibition | PD-0325901 + Crizotinib | I | NCT02510001 |
Table 2.
FOCUS4 trial cohorts: an example of a trial designed with treatment stratification based on molecular subtyping within colorectal cancer
| Study arm | Molecular aberration | Treatment |
|---|---|---|
| FOCUS-A | BRAF mutation | EGFR/BRAF/MEK inhibitors vs observation |
| FOCUS-B | PIK3CA mutation | Aspirin vs placebo |
| FOCUS-C | P53 and RAS dual mutation or H3K36me3 loss | WEE1 inhibitor vs placebo |
| FOCUS-D | BRAF/RAS/PI3KCA WT and no PTEN loss | HER-1, 2, 3 inhibitor vs placebo |
| FOCUS-E | Mismatch repair deficiency or POLD1/POLE mutations | PD-L1 inhibitor vs placebo |
| FOCUS-F | ATM loss | ATR inhibitor vs placebo |
| FOCUS-N | No other cohort available | Capecitabine vs observation |
Prognostic and Predictive Implications for MSI
MSI status has been evaluated as a prognostic biomarker in CRC, with conferral of prognostic benefit for patients with early MSI CRC [22]. Molecular subtyping strategies have highlighted a lower disease-specific mortality in patients with MSI CRC regardless of the initiating defect [23]. This benefit appears to hold true in the adjuvant setting. However, a subsequent study of patients with stage II and III disease showed no significant difference in overall survival (OS) or progression-free survival (PFS) due to dMMR, but demonstrated statistically poorer OS following administration of 5FU-based chemotherapy (82.4 % compared with 89.5 % for pMMR) [24]. It has been proposed that adjuvant 5FU-based chemotherapy should be avoided in stage II MSI CRC where toxicity may be prevented. Whether the addition of oxaliplatin may abrogate the detrimental effect of 5FU for patients with MSI CRC remains unclear [25–29]. The prognostic benefit of MSI in early stage disease is lost at time of relapse with rapid disease progression and reduced OS [30].Given their possibly poorer response to 5FU-based chemotherapy, use of targeted agents is of great interest in this subgroup.
Poly [ADP-ribose] polymerase-1 (PARP-1) localizes to sites of DNA damage and is involved in repair of these defects. In MLH1- and MSH3-deficient CRC cell lines, the combination of PARP inhibition and irinotecan therapy was more effective than in those with functional MLH1 [31, 32]. Consequently, there is interest in use of PARP inhibition for treatment of MSI CRC (Table 1).
Lymphocytic infiltration as a characteristic feature of MSI CRC has prompted further investigation due to the increasing availability of effective immunotherapies. The reasons for this immunogenic phenotype are unclear. However, it is postulated that this may occur due to the creation of tumour-specific neoantigens during accumulation of mutations [33]. Analysis of primary tumour tissue from this patient subset identified high levels of infiltration of activated CD8+ cytotoxic T-cells and activated Th1 cells with interferon-γ (IFNγ) and T-box expressed in T cells (TBET), a Th1 transcription factor. In addition, upregulated expression of the immune checkpoints cytotoxic T lymphocyte-associated antigen 4 (CTLA4), programmed cell-death-1 (PD-1), programmed cell-death ligand-1 (PD-L1), lymphocyte activation gene 3 (LAG3) and indoleamine 2,3-dioxygenase (IDO) was noted in MSI tumours [34•]. The critical role of the immune system in tumour regulation has been widely highlighted [12] and lymphocytic infiltration in CRC is associated with lower rate of relapse and improved prognosis [35]. Current immunotherapeutic strategies incorporate antibodies to directly inhibit the CTLA4 and PD-1 pathways. Use of the PD-1 inhibitor pembrolizumab has been associated with responses in patients with MSI CRC [36••]. Further studies of immune checkpoint inhibitors are ongoing in this patient population (Table 1).
Individual Somatic Mutations
RAS Mutation
The mitogen-activated protein kinase (MAPK) pathway comprised of the RAS-RAF-MEK-ERK signalling cascade is deregulated due to somatic gene mutation in >50 % of CRC, with 40 % of these due to activating mutations in KRAS [37]. This pathway lies downstream of the epidermal growth factor receptor (EGFR). Activating mutations in this pathway will result in transcription of the gene for transforming growth factor-α (TGFα), a ligand of the EGFR. This creates an autocrine signalling loop that contributes to tumoural resistance to the anti-EGFR monoclonal antibodies (mAbs), cetuximab and panitumumab [38]. In combination with chemotherapy, these therapies are the only approved biomarker-driven therapies currently licensed for treatment of CRC and have extended survival up to 41.3 months in patients with tumours wild type (WT) for KRAS and NRAS [3, 39]. However, despite improved survival for a subset of patients, no benefit is seen for those with KRAS/NRAS mutated tumours. Absence of this mutation is not an accurate predictor of response, as only 40–50 % of patients with KRAS/NRAS WT disease respond to the anti-EGFR monoclonal mAbs [40]. Binding of amphiregulin (AREG) and epiregulin (EREG) ligands to the EGFR stimulate mitogenesis [41]. In KRAS WT CRC, high expression of AREG and EREG messenger RNA (mRNA) is predictive of improved response rate, progression-free survival and overall survival in patients treated with cetuximab [42–44]. High EREG mRNA expression levels may predict for response in patients irrespective of EGFR mutation status [44]. However, these biomarkers are not validated for use in clinical practice.
Initially, mutations conferring resistance to anti-EGFR mAbs were identified in codons 12 and 13 of exon 2 of the KRAS gene. However, retrospective analysis of multiple trials demonstrated the presence of further resistance mutations in exons 2, 3 and 4 of KRAS/NRAS, resulting in a recommendation to utilise wider RAS mutational testing prior to treatment selection and inclusion in the license for panitumumab and cetuximab [45]. The KRAS G13D mutation conveys sensitivity to EGFR inhibitors in preclinical models, and retrospective clinical reports suggested possible treatment benefit with cetuximab similar to KRAS wild-type CRC. However, the prospective international ICECREAM trial recently confirmed lack of efficacy of cetuximab in this molecular subgroup [46].
Acquired mechanisms of resistance may limit treatment efficacy of EGFR inhibitors. Use of circulating tumour DNA (ctDNA) has identified emergence of RAS mutations following cetuximab administration, conferring resistance to EGFR-targeted therapy. Interestingly, introduction of an MEK inhibitor in combination with cetuximab resensitised the tumour to anti-EGFR therapy [47].
A number of other primary resistance mechanisms against anti-EGFR mAbs exist, including downstream activation of BRAF and MEK. In addition, EGFR activation may occur via an alternate signalling pathway within the interlinked RAS-MAPK-PI3K or may be mediated by type 1 insulin-like growth factor receptor (IGF-1R). Reduced affinity of the EGFR ligand or mutations within the gene, resulting in a conformational change to the binding site, may also physically impair the ability of the monoclonal antibody to effectively bind to and inhibit activation of the EGFR [48]. Based upon preclinical research demonstrating HER2 as a resistance pathway to EGFR inhibitors, the HERACLES trial investigates dual inhibition of the HER2 pathway and the efficacy of dual HER2 targeting in patient-derived xenografts [49]. The rate of HER2 positivity was 5.4 % in the first cohort, with a 34 % response rate and 44 % stable disease from trastuzumab and lapatinib in 23 patients. Trials investigating additional targeted agents aimed at reducing resistance to anti-EGFR inhibitors are currently underway (Table 1).
Due to the relative frequency of RAS mutation in CRC and its use as a negative predictor for current molecularly stratified treatment, identification of an effective therapy to target this subgroup has potential clinical benefit. Treatment directed at RAS mutated CRC has been targeted against downstream MEK signalling, but has been less successful than treatment for KRAS WT disease. Inhibition of MEK signalling may cause upregulation of signalling via the PI3K-AKT-mTOR pathway [50]. RAS is also an oncogenic driver of the PI3K-AKT-mTOR pathway, providing rationale for the combination of RAS and MEK pathway inhibition as a potential therapeutic strategy (Table 1).
BRAF Mutation
An additional activating mutation implicated in tumourigenesis within the MAPK signalling cascade is BRAF V600E mutation. This occurs in 8–10 % of CRC, very rarely occurs in conjunction with RAS mutations, and confers resistance to anti-EGFR therapy [51]. Patients with these tumours have a poor prognosis in the metastatic setting with aggressive tumour biology. This mutation is strongly associated with sporadic MSI CRC [30]. Targeted BRAF blockade using single-agent tyrosine kinase inhibition was attempted for this disease subtype. However, this approach was unsuccessful, with preclinical data suggesting therapeutic failure was due to aberrant upstream signalling via MEK and activation of the PTEN-PI3K-AKT pathway [52]. It was further demonstrated that BRAF-mutated cells acquired resistance to vemurafenib by stimulation of EGFR, thereby perpetuating cell proliferation [53•]. Based upon these findings, a triple combination of targeted agents concurrently inhibiting BRAF, MEK and EGFR is currently under investigation for treatment of BRAF-mutant CRC (Table 1.). Other approaches undergoing trials include use of ERK inhibitors and combinations of irinotecan, BRAF and EGFR inhibitors; BRAF, EGFR and PI3K inhibitors; BRAF, EGFR and WNT pathway inhibitors and FOLFOXIRI with bevacizumab.
PIK3CA and PTEN
Resistance to EGFR inhibition may also be driven by the PI3K-PTEN-AKT pathway. PI3K signalling is negatively regulated by PTEN, and may be activated by PIK3CA mutation. This gene encodes for the catalytic subunit of PI3K and is present in 10–18 % CRC. However, activation of this pathway may also occur due to loss of PTEN expression in 30 % CRC. KRAS and PIK3CA mutation frequently coexist, with activation of parallel pathways in a single tumour. Similar to MEK inhibition, single-agent targeting of this pathway has demonstrated lack of activity due to development of resistance feedback loops [50]. Early phase clinical trials are investigating the combination of MEK inhibition with AKT, PI3K and mTOR inhibition, but results to date have been disappointing (Table 1).
Interestingly, retrospective analysis of aspirin use in patients with established CRC identified the subgroup of patients with PIK3CA exon 9 and 20 mutations as deriving a survival benefit [54]. Although the reason for this interaction remains poorly understood, this may provide a clinical utility of this genomic biomarker. Aspirin is being prospectively investigated in this subgroup in the UK in the FOCUS4 trial (first-line setting for metastatic disease) and in the adjuvant Add-Aspirin trial (stage II/III CRC).
MET Gene
The mesenchymal-epithelial transition (MET) proto-oncogene encodes for c-MET, a receptor with tyrosine-kinase activity targeting hepatocyte growth factor (HGF). Activation of this pathway has been implicated in metastatic progression of CRC. EGFR and MET are coexpressed in CRC and MET activation has been implicated in resistance to the anti-EGFR mAb cetuximab [55]. c-MET is overexpressed in 50–60 %, amplified in 10 % and mutated in 5 % CRC [56, 57]. The MET inhibitor crizotinib was trialled as a single agent for treatment of MET-amplified CRC as part of The French National AcSé programme. However, 0/13 CRC patients with MET amplification derived clinical benefit from this agent [58]. Based upon preclinical work demonstrating synergistic activity in CRC between MEK and MET inhibitors [59], the MErCuRIC1 study is investigating the combination of the MEK inhibitor PD-0325901 and crizotinib in patients with KRAS mutant, or KRAS WT, MET-amplified CRC (Table 1).
ALK/ROS1 Translocations
The EML4-ALK fusion gene is produced by inversion in the short arm of chromosome 2, where anaplastic large-cell lymphoma kinase (ALK) joins echinoderm microtubule-associated protein-like 4(EML4), resulting in a chimeric protein with constitutive ALK activity [60]. ROS1 is an orphan receptor tyrosine kinase phylogenetically related to ALK [61]. These carcinogenic chromosomal translocations are found in 5 % of NSCLC, where the targeted ALK, ROS1 and MET inhibitor crizotinib has been used with up to 74 % response rates [62, 63]. In CRC, ALK or ROS1, gene rearrangements have not been extensively studied. Several papers have detailed a unique subpopulation of between 0.8–2.5 % patients with metastatic CRC (mCRC) with either ALK or ROS1 rearrangement of their tumour [64, 65]. Due to small patient numbers and inconsistent and expensive testing methods for ROS1 translocations, developing a clinical trial of targeted therapy for this patient subset poses considerable challenges.
Defining Molecular Subtypes in CRC: Gene Expression Profiling
Molecular subtyping of CRC has presented challenges, resulting in inconsistencies within the literature with regard to understanding of prognostic and predictive biomarkers. This may reflect the heterogeneity of CRC, possibly due to the lack of exclusivity outlined between the major pathways driving carcinogenesis and disease progression. Using gene expression profiling, the function of multiple genes can be assessed simultaneously to give a clearer picture of the complex characteristics underlying disease and reflecting genetic and epigenetic regulation. This technique may ensure a more informative result in addition to reducing the time for molecular testing and potentially has cost-saving implications for clinical practice.
Early-Stage Disease
A number of gene expression profiling tools have been validated for clinical use to inform prognosis in stage II CRC, where the benefit for chemotherapy remains unclear [66]. Oncotype DX and ColDx were developed using formalin-fixed paraffin-embedded (FFPE) tumours, and ColoPrint used fresh frozen tissue [67–69]. Oncotype DX utilises a 12-gene signature comprising genes associated with recurrence and with clinical benefit to 5FU to stratify tumours into low-, medium- or high-risk for recurrence [67]. The ColDx (now called GeneFX colon) signature comprises a 634-gene signature which differentiates stage II tumours into low- and high-risk for recurrence. The hazard ratio for cancer-related death at 5 years was 2.21, and the test performed better than use of traditional clinicopathological variables [68]. ColoPrint uses an 18-gene signature to classify patients with stage II disease as high- or low-risk of recurrence and improved the accuracy of clinicopathological variables alone for prediction of low-risk CRC [69]. Practical difficulties with obtaining fresh frozen tissue may limit use of this assay in routine clinical practice.
Advanced Disease
Independent investigator groups have used gene expression profiles to identify intrinsic molecular subtypes [8, 10, 70, 71]. Due to poor agreement across the resulting subtypes for several gene expression profiles, members of the Colorectal Cancer Subtyping Consortium agreed to combine their genomic datasets to generate a consensus molecular subtyping strategy (CMS) by applying unsupervised clustering techniques to 4151 samples [72••]. From this, four subsets have been established. CMS1 comprised 14 % of the patients and classified older, female patients with hypermutated MSI tumours demonstrating BRAF mutation and immune activation. This is consistent with the established MSI phenotype. CMS2 was the most common subset comprising 37 %. These MSS tumours displayed CIN, strong WNT/MYC pathway activation, TP53 mutation and EGFR amplification/overexpression. These tumours were associated with a better OS and predominantly originated in the left colon. Tumours clustering as CMS3 were associated with low CIN, moderate WNT/MYC pathway activation. KRAS and PIK3CA mutation was commonly found among this subtype, as was IGFBP2 overexpression. This subset was seen in 13 % and was associated with intermediate survival. Finally, 23 % of the tumours clustered to CMS4. These were CIN/MSI heterogeneous, demonstrating mesenchymal/TGF-beta activation and NOTCH3/VEGFR2 overexpression. These patients were diagnosed at a younger age and had poorer survival outcomes. A subgroup (13 %) that remained unclassified was also identified, with no common features among these tumour tissues, and the authors suggest that these may represent a transitional phenotype or intratumoural heterogeneity. Although not a therapeutic stratifier (as it is not possible to classify individual patient tumours into these subtypes prospectively), this large dataset has facilitated greater understanding of the broad biological groups within CRC (Fig. 1b).
Antiangiogenic Agents
There are now five antiangiogenic agents licensed for use in CRC, but empirical use of these drugs without selection contributes to underwhelming levels of efficacy. There is still no validated biomarker to predict benefit from these agents either as monotherapy or in addition to chemotherapy, despite almost two decades of investigation. A 63-gene signature has recently been shown to predict sensitivity to antiangiogenic therapy in ovarian cancer using FFPE tumour material and is currently being explored in CRC [73].
Innovative Clinical Trial Design
With the development of novel treatment strategies for subgroups of patients with CRC, finding the optimal biomarker and ensuring appropriate targeted therapy without exhausting limited diagnostic tissue is becoming increasingly challenging. A number of trials address this issue using a basket design to stratify patients into the appropriate arm for therapy based upon the molecular characteristics of the tumour at point of testing.
The UK-based FOCUS4 study is an adaptive, biomarker-driven, multiarm, multistage trial designed to enable stratification of therapy for patients with mCRC during first-line chemotherapy (see http://www.focus4trial.org). By prospectively analysing diagnostic tumour samples for predefined biomarkers, patients with responding or stable disease following 16 weeks of first-line chemotherapy can be allocated to an appropriate treatment arm. The aim of this study is to assess response of targeted treatment strategies using novel or repurposed agents in these molecularly enriched cohorts. There is a hierarchy of biomarker-defined cohorts (Table 2) [74••].
MODUL is a randomised, multicentre, controlled, open-label, parallel-group study sponsored by Roche/Genentech that is investigating the efficacy and safety of biomarker-driven maintenance treatment for first-line mCRC after 16 weeks of induction of FOLFOX and bevacizumab [75]. Patients with BRAF mutations are randomised between fluoropyrimidine (+/−bevacizumab) maintenance and the same with addition of BRAF and EGFR inhibitors; BRAF WT patients are randomised between fluoropyrimidine (+/−bevacizumab) maintenance and the same with addition of a PD-L1 inhibitor.
A similar precision medicine approach is planned in the USA in second-line treatment of CRC in the ASSIGN trial [76]. Here, molecularly defined subgroups based on metastatic site biopsies will be randomised between targeted agents and chemotherapy, dependent on the availability of therapies at the time that the study opens. Current plans are to investigate novel EGFR pathway targeting agents in “all WT” CRC, BRAF inhibitors in BRAF-mutant CRC, targeted agents in those with ALK/ROS-1 translocations, PI3K inhibitors in those with PTEN loss, HER-2 inhibitors or TDM-1 in those with HER-2 over-expression and immune checkpoint inhibitors in those with MSI.
Screening Patients for Efficient Clinical Trial Access in advanced colorectal cancer (SPECTAcolor) is an EORTC initiative launched in 2013, comprising a network of collaborating European institutions treating patients with CRC. Tumour tissue from participants is processed for centralised, high-throughput screening for driver mutations including KRAS, NRAS, BRAF and PIK3CA, in addition to immunohistochemical staining for MSI. Next-generation sequencing will also be performed for 360 key cancer genes, with a view towards enrolment in “downstream” targeted multinational clinical trials [77]. These and similar mutational screening platforms are designed to optimise use of potentially small tissue samples in order to provide a variety of appropriately targeted additional treatment options for patients with mCRC.
Tumour Heterogeneity
Decisions regarding therapeutic strategy are currently made using available tissue, most commonly from the primary tumour at the time of diagnosis. During progression of malignant disease, by a process of clonal evolution, molecular differences may occur between primary tumours and metastatic disease. This may render application of a biomarker to a primary tumour sample inaccurate for the purposes of targeted therapy for more advanced disease, with a lack of knowledge of the wider tumour landscape [78••]. For the clinically relevant driver genes, including KRAS, NRAS, BRAF, PIK3CA and TP53, concordance between tumour primary and metastatic disease site has been shown to be >90 % in CRC [79•]. There appears to be less concordance for PTEN expression by IHC, varying from 47 to 98 % between primary and metastatic site, reducing utility of this genomic marker in clinical practice [80]. Less is known about the MSI subgroup where a baseline genomically unstable tumour may become increasingly hypermutated during disease progression.
The liquid biopsy is a minimally invasive sampling technique utilising circulating cell-free (cf) DNA to reflect the dynamic nature of clonal evolution, thereby potentially eliminating inaccuracies in current molecular status of the tumour while minimising discomfort and risk to the patient [81]. Its potential utility in CRC has been demonstrated for KRAS and BRAF mutation identification during treatment [82•].
CRC has been highlighted as one of the tumour types where development of this technology has been identified as a priority. Techniques to optimise laboratory procedures for analysis and interpretation of results are currently being investigated [83]. However, validation of this exciting technology is required prior to routine clinical adoption.
Conclusion
Although unique subtypes within CRC have been identified, critical questions remain regarding the pathogenesis and biology of these tumours, and optimal biomarkers with evidence-based therapeutic strategies remain elusive. The emergence of immunotherapy for treatment of malignancy has widened treatment options for many patients, but trials for patients with CRC are limited and are currently focused largely on those with metastatic MSI disease. The biological features of this subgroup with immune infiltration as a key feature of tumourigenesis would appear to validate this choice. However, the mechanisms underlying poor prognosis for these patients remain incompletely understood. In order to progress management of this disease, it is critical that we understand the resistance mechanisms to therapy and address these in developing the next generation of therapeutic strategies. Optimising use of EGFR inhibitors, improving treatment for those patients with RAS and RAF mutations where no proven targeted therapy exists, and for those with RAS/RAF WT disease resistant to EGFR inhibition, are key areas of unmet clinical need.
Enhancing our understanding of the CRC disease process may result in better use and scheduling of systemic treatment, reducing morbidity and mortality in this disease. Development of robust and validated biomarkers will be at the core of personalised therapy, and the novel trial designs currently underway will aim to rapidly isolate subpopulations for treatment while ensuring efficient use of limited tissue resources. In addition, collaboration between multiple centres will facilitate information sharing and generate more rapid trial accrual to answer these clinical questions. Although biomarkers are validated for prediction of benefit for chemotherapy in the adjuvant setting, targeted therapy has not yet demonstrated any benefit in early disease. Finally, through interrogation of the molecular and immune characteristics driving progression from adenoma to carcinoma, there is also potential for preventative measures to be explored.
Compliance with Ethical Standards
Conflict of Interest
Donna M. Graham has received honoraria from Amgen as compensation for giving a lecture.
Vicky M. Coyle has received non-financial support (travel to educational meetings) from Sanofi and Bristol-Myers Squibb.
Richard D. Kennedy has received compensation from Almac for serving as Medical Director.
Richard H. Wilson has received compensation from Merck Serono and Sanofi for service on advisory boards and has received reimbursement from Merck Serono for travel expenses to educational meetings.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
This article is part of the Topical Collection on Personalized Medicine in Colorectal Cancer
Contributor Information
Donna M. Graham, Phone: +44 (0)2890 972760, Email: dgraham06@qub.ac.uk
Vicky M. Coyle, Phone: +44 (0)2895 048492, Email: v.coyle@qub.ac.uk
Richard D. Kennedy, Phone: +44 (0)2890 972760, Email: r.kennedy@qub.ac.uk
Richard H. Wilson, Phone: +44 (0)2895 048492, Email: r.wilson@qub.ac.uk
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Van Cutsem E, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011–2019. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann V, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1065–1075. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 3.Schwartzberg LS, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32(21):2240–2247. doi: 10.1200/JCO.2013.53.2473. [DOI] [PubMed] [Google Scholar]
- 4.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med. 2009;361(25):2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shepherd FA, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 7.Camidge DR, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadanandam A, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19(5):619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roepman P, et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer. 2014;134(3):552–562. doi: 10.1002/ijc.28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marisa L, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10(5):e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21(6):1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 14.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073–2087. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannavo E, et al. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J Biol Chem. 2007;282(5):2976–2986. doi: 10.1074/jbc.M609989200. [DOI] [PubMed] [Google Scholar]
- 16.Nosho K, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. 2010;222(4):350–366. doi: 10.1002/path.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63(17):5209–5212. [PubMed] [Google Scholar]
- 18.Kakar S, et al. Frequency of loss of hMLH1 expression in colorectal carcinoma increases with advancing age. Cancer. 2003;97(6):1421–1427. doi: 10.1002/cncr.11206. [DOI] [PubMed] [Google Scholar]
- 19.Watson P, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123(2):444–449. doi: 10.1002/ijc.23508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elsayed FA, et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet. 2015;23(8):1080–1084. doi: 10.1038/ejhg.2014.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spier I, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. 2015;137(2):320–331. doi: 10.1002/ijc.29396. [DOI] [PubMed] [Google Scholar]
- 22.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23(3):609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 23.Phipps AI, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148(1):77–87. doi: 10.1053/j.gastro.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jover R, et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006;55(6):848–855. doi: 10.1136/gut.2005.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sinicrope FA, et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Clin Oncol. 2013;31(29):3664–3672. doi: 10.1200/JCO.2013.48.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaanan A, et al. Impact of p53 expression and microsatellite instability on stage III colon cancer disease-free survival in patients treated by 5-fluorouracil and leucovorin with or without oxaliplatin. Ann Oncol. 2010;21(4):772–780. doi: 10.1093/annonc/mdp383. [DOI] [PubMed] [Google Scholar]
- 27.Des Guetz G, et al. Prognostic impact of microsatellite instability in colorectal cancer patients treated with adjuvant FOLFOX. Anticancer Res. 2010;30(10):4297–4301. [PubMed] [Google Scholar]
- 28.Nopel-Dunnebacke S, et al. Prognostic value of microsatellite instability and p53 expression in metastatic colorectal cancer treated with oxaliplatin and fluoropyrimidine-based chemotherapy. Z Gastroenterol. 2014;52(12):1394–1401. doi: 10.1055/s-0034-1366781. [DOI] [PubMed] [Google Scholar]
- 29.Tougeron D, et al. Impact of adjuvant chemotherapy with 5-FU or FOLFOX in colon cancers with microsatellite instability: An AGEO multicenter study. J Clin Oncol. 2014;32:5s. doi: 10.1200/JCO.2013.49.4757. [DOI] [Google Scholar]
- 30.Tran B, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117(20):4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tentori L, et al. Influence of MLH1 on colon cancer sensitivity to poly(ADP-ribose) polymerase inhibitor combined with irinotecan. Int J Oncol. 2013;43(1):210–218. doi: 10.3892/ijo.2013.1932. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi M, et al. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase inhibitor. J Biol Chem. 2011;286(14):12157–12165. doi: 10.1074/jbc.M110.198804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwitalle Y, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134(4):988–997. doi: 10.1053/j.gastro.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 34.•.Llosa NJ, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5(1):43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 36.••.Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 38.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 39.Lievre A, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 40.De Roock W, et al. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12(6):594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 41.Inatomi O, et al. Regulation of amphiregulin and epiregulin expression in human colonic subepithelial fibroblasts. Int J Mol Med. 2006;18(3):497–503. [PubMed] [Google Scholar]
- 42.Khambata-Ford S, et al. Expression of epiregulin and amphiregulin and K-ras status predict disease control in metastatic colorectal cancer patients treated with cetuximab. JCO. 2007;25(22):3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 43.Jacobs B, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. JCO. 2009;27(30):5068–5074. doi: 10.1200/JCO.2008.21.3744. [DOI] [PubMed] [Google Scholar]
- 44.Pentheroudakis G, et al. Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer. 2013;13(49):1–12. doi: 10.1186/1471-2407-13-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Douillard JY, Rong A, Sidhu R. RAS mutations in colorectal cancer. N Engl J Med. 2013;369(22):2159–2160. doi: 10.1056/NEJMc1312697. [DOI] [PubMed] [Google Scholar]
- 46.Segelov E, et al. The AGITG ICECREAM Study: The irinotecan cetuximab evaluation and cetuximab response evaluation amongst patients with a G13D mutation—analysis of outcomes in patients with refractory metastatic colorectal cancer harbouring the KRAS G13D mutation. European Cancer Congress 2015; 32LBA.
- 47.Misale S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siena S, et al. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101(19):1308–1324. doi: 10.1093/jnci/djp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siena S, et al. Trastuzumab and lapatinib in HER2-amplified metastatic colorectal cancer patients: The HERACLES trial. J Clin Oncol 33, 2015 (suppl; abstr 3508).
- 50.Turke AB, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72(13):3228–3237. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Roock W, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 52.Yang H, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012;72(3):779–789. doi: 10.1158/0008-5472.CAN-11-2941. [DOI] [PubMed] [Google Scholar]
- 53.•.Prahallad A, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 54.Liao X, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367(17):1596–1606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liska D, et al. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011;17(3):472–482. doi: 10.1158/1078-0432.CCR-10-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di Renzo MF, et al. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res. 1995;1(2):147–154. [PubMed] [Google Scholar]
- 57.Fumagalli D, et al. A rapid, sensitive, reproducible and cost-effective method for mutation profiling of colon cancer and metastatic lymph nodes. BMC Cancer. 2010;10:101. doi: 10.1186/1471-2407-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vassal G, et al. Biomarker-driven access to crizotinib in ALK, MET or ROS1 positive (+) malignancies in adults and children: the French Mational AcSé Program. Eur J Cancer. 2015;51(S3):S715. doi: 10.1016/S0959-8049(15)30070-8. [DOI] [Google Scholar]
- 59.Van Schaeybroeck S, et al. ADAM17-dependent c-MET-STAT3 signaling mediates resistance to MEK inhibitors in KRAS mutant colorectal cancer. Cell Rep. 2014;7(6):1940–1955. doi: 10.1016/j.celrep.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 60.Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 61.Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta. 2009;1795(1):37–52. doi: 10.1016/j.bbcan.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 62.Solomon BJ, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 63.Shaw AT, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aisner DL, et al. ROS1 and ALK fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol Cancer Res. 2014;12(1):111–118. doi: 10.1158/1541-7786.MCR-13-0479-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lipson D, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18(3):382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Narzo AF, et al. Test of four colon cancer risk-scores in formalin fixed paraffin embedded microarray gene expression data. J Natl Cancer Inst, 2014. 106(10). [DOI] [PubMed]
- 67.Gray RG, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol. 2011;29(35):4611–4619. doi: 10.1200/JCO.2010.32.8732. [DOI] [PubMed] [Google Scholar]
- 68.Kennedy RD, et al. Development and independent validation of a prognostic assay for stage II colon cancer using formalin-fixed paraffin-embedded tissue. J Clin Oncol. 2011;29(35):4620–4626. doi: 10.1200/JCO.2011.35.4498. [DOI] [PubMed] [Google Scholar]
- 69.Salazar R, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011;29(1):17–24. doi: 10.1200/JCO.2010.30.1077. [DOI] [PubMed] [Google Scholar]
- 70.Budinska E, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. 2013;231(1):63–76. doi: 10.1002/path.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schlicker A, et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med Genomics. 2012;5:66. doi: 10.1186/1755-8794-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.••.Guinney J, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gourley C, et al. Molecular subgroup of high-grade serous ovarian cancer (HGSOC) as a predictor of outcome following bevacizumab. J Clin Oncol. 2014;32:5s. doi: 10.1200/JCO.2013.49.4757. [DOI] [Google Scholar]
- 74.••.Kaplan R, et al. Evaluating many treatments and biomarkers in oncology: a new design. J Clin Oncol. 2013;31(36):4562–4568. doi: 10.1200/JCO.2013.50.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmoll H, et al. MODUL—a multicentre randomised clinical trial of biomarker-driven therapy for the 1st-line maintenance treatment of metastatic colorectal cancer: a signal-seeking approach. Ann Oncol. 2014;25(4):209. doi: 10.1007/s00432-018-2632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O’Dwyer P. The next generation of targeted therapy trials in colorectal cancer. Clin. Adv. Hematol. Oncol. 2014;12(12):852–854. [PubMed] [Google Scholar]
- 77.Folprecht G, et al. Improving access to molecularly defined clinical trials for patients with colorectal cancer: The EORTC SPECTAcolor platform., in 2015 Gastrointestinal Cancers Symposium. 2015: San Francisco. abstr 575
- 78.••.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.•.Vakiani E, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol. 2012;30(24):2956–2962. doi: 10.1200/JCO.2011.38.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Molinari F, Frattini M. Functions and regulation of the PTEN gene in colorectal cancer. Front Oncol. 2013;3:326. doi: 10.3389/fonc.2013.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Crowley E, et al. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10(8):472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 82.•.Spindler KLG, et al. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res. 2012;18:1177–1185. doi: 10.1158/1078-0432.CCR-11-0564. [DOI] [PubMed] [Google Scholar]
- 83.Experimental Cancer Medicine Centres (ECMC) Network and National Cancer Research Institute (NCRI) Biomarkers and Imaging Clinical Studies Group Cell-free DNA Consensus Meeting: Meeting report. 24th November, 2014.