

Abstract
Feedforward (FF) inhibition is a common motif in many neural networks. Typically, excitatory inputs drive both principal neurons and interneurons; the interneurons then inhibit the principal neurons, thereby regulating the strength and timing of the FF signal. The interneurons introduce a likely nonlinear processing step that could distort the excitation/inhibition (E/I) ratio in the principal neuron, potentially degrading the reliability of computation in the circuit. In the retina, FF inhibition is an essential feature of the circuitry underlying direction selectivity (DS): glutamatergic bipolar cells (BCs) provide excitatory input to direction-selective ganglion cells (DSGCs) and GABAergic starburst amacrine cells (SACs), and the SACs then provide FF inhibition onto DSGCs. Robust DS computation requires a consistent synaptic E/I ratio in the DSGC in various visual conditions. Here, we show in mouse retina that the E/I ratio is maintained in DSGCs over a wide stimulus contrast range due to compensatory mechanisms in the diverse population of presynaptic BCs. BC inputs to SACs exhibit higher contrast sensitivity, so that the subsequent nonlinear transformation in SACs reduces the contrast sensitivity of FF inhibition to match the sensitivity of direct excitatory inputs onto DSGCs. Measurements of light-evoked responses from individual BC synaptic terminals suggest that the distinct sensitivity of BC inputs reflects different contrast sensitivity between BC subtypes. Numerical simulations suggest that this network arrangement is crucial for reliable DS computation.
SIGNIFICANCE STATEMENT Properly balanced excitation and inhibition are essential for many neuronal computations across brain regions. Feedforward inhibition circuitry, in which a common excitatory source drives both the principal cell and an interneuron, is a typical mechanism by which neural networks maintain this balance. Feedforward circuits may become imbalanced at low stimulation levels, however, if the excitatory drive is too weak to overcome the activation threshold in the interneuron. Here we reveal how excitation and inhibition remain balanced in direction selective ganglion cells in the mouse retina over a wide visual stimulus range.
Keywords: calcium imaging, direction selectivity, feedforward, iGluSnFR, starburst amacrine, synaptic balance
Introduction
Retinal direction-selective ganglion cells (DSGCs) respond best to motion in one “preferred” direction (PD) across their receptive field (Barlow and Hill, 1963; Demb, 2007; Vaney et al., 2012). Direction selectivity (DS) originates in starburst amacrine cell (SAC) dendrites, which operate autonomously and respond preferentially to motion away from their soma (Euler et al., 2002; Lee et al., 2010). SAC dendrites selectively contact DSGCs tuned to the opposite direction, so that postsynaptic inhibition in DSGCs is greater in response to their null direction (ND) stimuli (Fried et al., 2002; Lee et al., 2010; Briggman et al., 2011; Wei et al., 2011; Yonehara et al., 2011). SACs also excite DSGCs via cholinergic inputs that are more directionally symmetric (Kittila and Massey, 1997; Chiao and Masland, 2002; Lee et al., 2010; but see Pei et al., 2015). DSGCs also receive directionally untuned glutamatergic input from bipolar cells (BCs; Cohen and Miller, 1995; Tjepkes and Amthor, 2000; Lee et al., 2010, 2014; Yonehara et al., 2013; Park et al., 2014; Stafford et al., 2014; Pei et al., 2015), which also excite SACs directly (Famiglietti, 1991; Helmstaedter et al., 2013; Duan et al., 2014; Greene et al., 2016), and a weaker glutamatergic input from vGlut3 amacrine cells (Lee et al., 2014). The mammalian retina contains at least 12 different BC subtypes (Ghosh et al., 2004; Helmstaedter et al., 2013; Euler et al., 2014); BC input to SACs and DSGCs originate from two partially overlapping populations (Helmstaedter et al., 2013; Duan et al., 2014; Kim et al., 2014). Although it has not been shown that an individual BC can contact both postsynaptic cells, this may be possible given the cofasciculation of SAC and DSGC dendrites in the inner plexiform layer (Jeon et al., 2002; Helmstaedter et al., 2013).
The general structure of the DS network therefore represents a feedforward (FF) circuit in which BCs provide excitatory drive to both the principal cells (DSGCs) and interneurons (SACs; Fig. 1A). Theoretically, this FF arrangement could compromise reliable DS computation: If the input/output relationship of SACs is not linear, GABAergic input from SACs to DSGCs may not always balance properly the excitatory inputs from BCs, an excitation/inhibition (E/I) mismatch that could degrade directional tuning in DSGCs.
Figure 1.
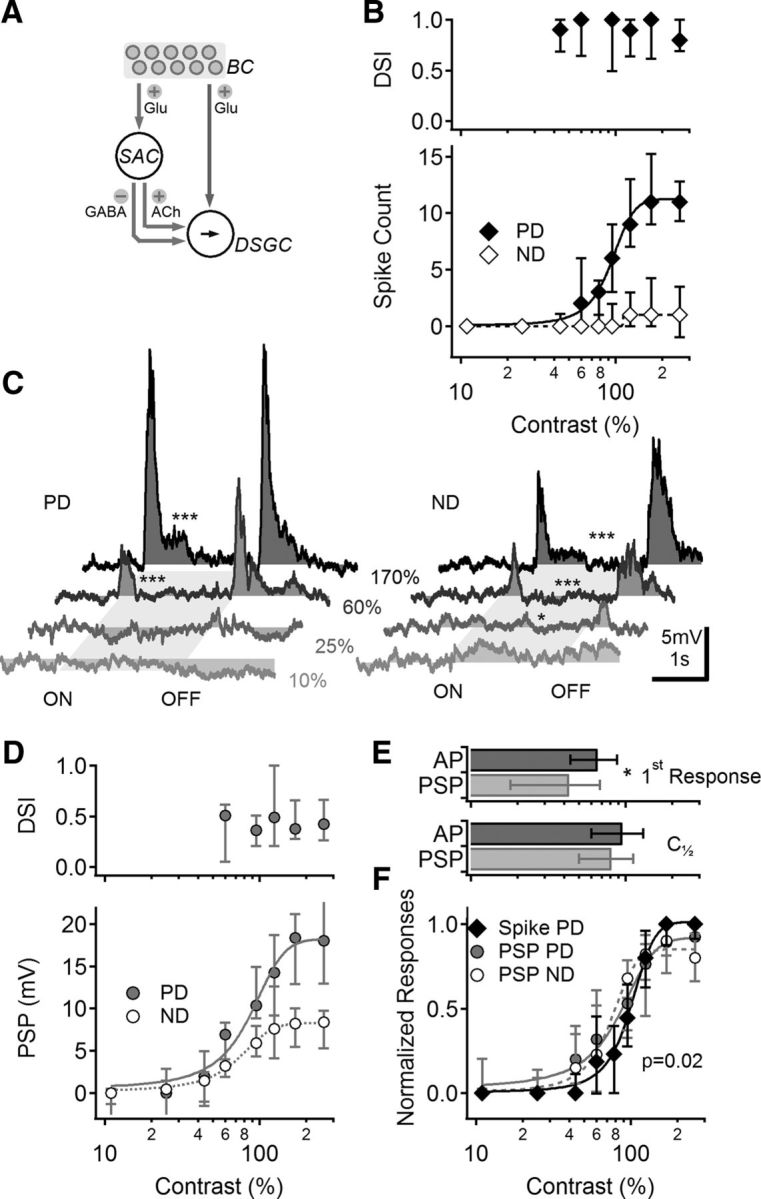
Contrast sensitivity of visual responses in DSGCs. A, Schematic representation of the main components of the retinal DS network. Photoreceptors (data not shown) transduce light stimuli and signal to BCs. BCs activate two pathways: one is a direct connection to DSGCs (right), and another goes via intermediate amacrine cells (left), most notably SACs. SACs then provide directionally tuned inhibition and excitation to DSGCs, forming a FF circuit. B, Median (±quartiles) calculated DSI (top) and spike count for the preferred and null directions (PD, ND; bottom) in DSGCs at different stimulus contrasts (n = 43). C, Representative PSPs evoked by PD (left) and ND (right) stimuli, shade-coded for different contrast levels. The stimulus consisted of a moving bar; the approximate time interval over which the stimulus occupied the cell's receptive is indicated in gray. Asterisks indicate comparison between PSPs and baseline (*p < 0.05, ***p < 0.001, t test). D, Median (±quartiles) calculated DSI values (top) and PSP amplitudes (bottom) for different contrasts, as in B. n = 16. E, Top, Mean (±SD) lowest contrast level that elicited a measurable signal in DSGCs, for suprathreshold (black) and subthreshold (gray) responses (*p = 0.005, t test). Bottom, The contrast level that elicited a half-maximal response was similar between spike (suprathreshold) and PSP (subthreshold) responses. F, Normalized contrast sensitivity curves (median ± quartiles) of the suprathreshold (black) and subthreshold (gray) responses. PSP contrast tuning was similar between PD and ND. For clarity, only PD suprathreshold responses are shown.
We examined contrast sensitivity of light responses in neurons within the DS circuitry to test whether and how excitation balances inhibition in DSGCs. We found that the E/I ratio in DSGCs varied with stimulus direction, as expected. In responses to motion in a particular direction, however, the E/I ratio remained consistent over a wide range of stimulus contrast, despite the fact that excitatory inputs to SACs are more sensitive than GABAergic outputs from SACs to DSGCs. A potential E/I mismatch in DSGCs is avoided because BC inputs to SACs are also more sensitive than BC inputs to DSGCs, so that excitatory and inhibitory inputs to DSGC exhibit similar contrast sensitivity. We show that the distinct contrast sensitivity of BC input reflects differences in contrast tuning between BCs, not from contrast sensitivity variation between synapses within the same BC.
Materials and Methods
Recording procedures.
All animal procedures were conducted in accordance with U.S. National Institutes of Health guidelines, as approved by the National Institute of Neurological Disorders and Stroke Animal Care and Use Committee. Retinas were isolated from eGFP-DRD4/Chat-Cre/TdTomato mice of either sex after eye opening (postnatal days 14–56). Except where noted, all subsequent procedures and recordings were performed in Ames media equilibrated with 95% O2/5% CO2. Mice were not dark-adapted and recordings were performed in ambient light levels to reduce rod-pathway activation. All experiments were performed at ∼35°C.
Recordings from labeled cells were performed in cell-attached and whole-cell patch-clamp configurations with 4–6 MΩ pipets filled the following solutions: Ames (cell attached); 90 mm CsCH3SO4, 20 mm TEA-Cl, 10 mm HEPES, 10 mm EGTA, 10 mm phosphocreatine disodium salt hydrate, 4 mm Mg-ATP, and 0.4 mm Na-GTP, 5 mm N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium bromide (QX-314; whole-cell voltage-clamp); 110 mm K-MeSO4H, 10 mm NaCl, 5 mm HEPES,4 mm Mg-ATP, and 0.4 mm Na-GTP, 5 mm QX-314, 2 mm EGTA (whole-cell current-clamp). For calcium recordings from patched cells, EGTA was replaced with 0.2 mm OGB-1 or 0.2 mm Fluo-4. In some experiments, calcium dyes were introduced with sharp electrodes (resistance 100–300 MΩ, filled with 10 mm OGB-1 or Fluo-4 in water). Access to a cell was gained by positioning the sharp electrode next to the cell membrane and applying a brief (∼1 ms) sinusoidal current pulse. In most cases, the same electrode could be used to label multiple cells.
To record subthreshold responses under current-clamp, sodium channels were blocked with bath applied octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethano-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentol (tetrodotoxin, 1 μm; Ascent Scientific) or N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX 314, 5 mm; Sigma-Aldrich) added to the intracellular patch solution. Recordings were obtained with an Axopatch 700B amplifier (Molecular Devices), low-pass filtered at 4 kHz (Bessel) and digitized at 20–50 kHz with a 16-bit A/D board (Instrutech) controlled by custom acquisition software written in IgorPro (WaveMetrics). NMDARs were blocked with d-(-)-2-amino-5-phosphonopentanoic acid (AP5, 50 μm; Sigma-Aldrich) or 5R,10S)-(-)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cylcohepten-5,10-imine maleate (MK801, 10 μm; Sigma-Aldrich). Both drugs exerted similar effects on light responses and experiments were pooled for analysis. Nicotinic and muscarinic blockers, N,N,N,N′,N′,N′-hexamethylhexamethylenediammonium dibromide (hexamethonium bromide, 100 μm; Sigma-Aldrich) and (RS)-(8-methyl-8-azabicyclo[3.2.1]oct-3-yl) 3-hydroxy-2-phenylpropanoate (atropine, 5 μm; Sigma-Aldrich) were used to block cholinergic transmission.
iGluSnFR expression.
To measure iGluSnFR responses in SACs, ∼1.0 μl of AAV1/AAV9.hsyn.FLEX.iGluSnFR.WPRE.SV40 (FLEX, Cre-dependent; 1.5 × 1012 IU/μl in water; Penn Vector Core) was injected into the vitreous humor of Chat-Cre transgenic mice, so that iGluSnFR was expressed only in SACs. Experiments on retinas from these animals were performed 14–28 d following virus injection. iGluSnFR expression in DSGCs was achieved by injecting Cre-independent versions of the virus (AAV1/AAV9.hsyn.iGluSnFR.WPRE.SV40 1.5 × 1012 IU/μl in water; Penn Vector Core). On-off DSGCs were then identified using directionally tuned light responses in cell-attached configuration. DSGCs were selected for iGluSnFR recordings only if no adjacent SACs were also infected. One SAC was also identified and recorded using the cre-independent virus. SACs and DSGCs were filled with an internal solution containing 0.01 mm AlexaFluor 647 to visualize the dendritic tree and iGluSnFR fluorescence was recorded from the labeled dendrites (see below).
Light stimulation.
Light stimuli were generated with a 405 nm LED collimated and masked by a LCD display (240 × 240 pixels; 4 d systems; refresh rate of 20 Hz) controlled by the acquisition software and focused through a the microscope condenser to illuminate the tissue at the focal plane of the photoreceptors (pixel size ∼10 μm). Photon flux was measured at the focal plane of the photoreceptors using a radiometer (DR-2000, Gamma Scientific) and converted to photoisomerizations per rod assuming a collecting area of 0.5 μm2 (Field and Rieke, 2002); the resulting background light intensity was ∼30,000 R* rod−1 s−1. Stimuli consisted of rectangles (500 × 2000 μm) moving at 1 mm/s along their long axis in each of eight different directions, presented at 10–15 s intervals. The stimuli were restricted to a 2 × 2 mm sized area on the retina centered on the recorded cell. This stimulus was, therefore, sufficient to activate the center and surround portions of DRD4 DSGC receptive fields (Hoggarth et al., 2015).
Imaging.
Calcium imaging was performed with a Zeiss LSM-510 microscope. Two-photon excitation was provided by pulsed laser light (820 nm, ∼1 μW power output at the objective; Chameleon, Coherent). Fluorescence was acquired between 500 and 550 nm with a confocal (descanned) photomultiplier tube (PMT). The confocal pinhole set to full aperture, thereby permitting adequate detection of OGB-1/Fluo-4/iGluSnFR signals while largely preventing stimulus light, which was focused on the photoreceptor layer, from reaching the PMT. Frame scans (10–20 Hz) were acquired in bidirectional scan mode and filtered by a 2–5 Hz low-pass filter off-line. Patched SACs were imaged within the first 20 min after gaining whole-cell access, to minimize rundown of responses due to washout of internal components.
BC labeling and analysis.
We developed a new technique to fill BCs with fluorescent dyes. To stain the cells, the retina was placed photoreceptor side up on a nitrocellulose filter paper, removed from the extracellular solution, and placed in a chamber with concentrated (2 mm) OBG-1 dye dissolved in Ames for a few seconds. The tissue was then returned back to the control solution and rapidly peeled off the filter paper. We found that the resulting bending of the tissue was sufficient to effectively load multiple cells with the fluorescent dye; the degree of tissue bending was the main factor that influenced the number of stained cells and viability of the tissue. The best light responses from BCs were obtained when only a few cells were labeled per retina.
Light response rise time was measured as the time it took for the fluorescent signal to raise from 10% to 90% of the peak response. The transient part of the response was measured at the first second after the estimated entrance of the visual stimulus to the receptive field of the cell. The rest of the response was considered to be sustained and the transient/sustained ratio was computed as the ratio between the peak dF/F values in the two regions.
Following recording of light responses from axon terminals, Z-stack scans were performed to determine the morphology of the BC. Simultaneously corecorded transmitted light image was used to find the boundaries of the IPL. Axonal span in the IPL was measured as the difference between the position in IPL of the highest and lowest locations of axonal terminals. Axonal area was calculated as the area of the smallest ellipse that contained a z-projection of the axonal terminals.
Data analysis.
The PD for each DSGC was determined by recording (in cell-attached mode) spike responses to light onset (ON) of light bars moving in eight different directions. We then computed the angle of the vector of the average response (Taylor and Vaney, 2002), and designated the closest of the eight directions to be the PD. The ND was defined as the direction 180° opposite from the PD. For suprathreshold recordings, we measured the number of action potentials. For subthreshold responses, we measured the mean peak depolarization (current-clamp) or peak current (voltage-clamp) for light onset. DSI was calculated as 1 − Rnull /Rpreferred, where Rnull and Rpreferred are the responses at the null/preferred directions, respectively.
To measure the detectability of responses to visual stimuli we compared the recorded signals during baseline activity to the signals during the visual stimulus presentation. The contrast of the first observable response was defined as the lowest contrast level in which the response to the stimulus was significantly larger (p < 0.025, one-tailed t test) higher than the baseline activity.
Because electrical recordings were digitized at a much higher rate than fluorescence signals, in experiments where we compared electrical recordings with fluorescence signals, we downsampled the recorded membrane potentials to 20 Hz and further filtered by a 5 Hz filter, to match the fluorescence acquisition parameters. This manipulation did not affect the detectability of electrical events in response to the stimulus (data not shown).
The signal-to-noise ratio (SNR) was determined as the ratio between the mean response to the stimulus and the SD of the baseline signals. We estimated the SNR values that would produce a detectable response by simulating sham baseline and stimulus-evoked responses based on the experimental parameters and measuring detectability as described above. Taking into account the number and the distribution of data points in the baseline and the stimulus regions, we found SNR above ∼1.8 to be detectable in >80% of the trials.
Simulation.
The simulation was based on a recent DSGC model (Poleg-Polsky and Diamond, 2016). One recorded DSGC was reconstructed using the ImageJ plugin “Simple Neurite Tracer” and converted into a multicompartmental model (121 ON-; 119 OFF-stratifying dendritic segments). Simulations were run using the NEURON simulation environment (Hines and Carnevale, 1997). The distribution and parameters of the passive and active conductances were set to match the experimentally recorded DSGC behavior: Membrane capacitance was set to 1 μF/cm2, the specific axial resistance was 100 Ωcm, leak current was equal across all compartments, with a conductance of 0.55 mS/cm2 and reversal potential of −60 mV. We matched the firing rate of experimentally recorded somatic current injections with the following distribution of voltage-gated channels at DSGC soma (peak conductance in mS/cm2): sodium (400), fast potassium rectifier (70), delayed rectifier (0.5). The reversal potentials for sodium and potassium were set to +50 and −77 mV, respectively. In experiments, we observed that activation of voltage-gated channels following a step-current injection introduced significant variability in the membrane potential of experimentally recorded DSGCs. To simulate the effect of channel noise in the model, we included stochastic behavior into the description of state transitions of the channels in the model (Linaro et al., 2011). All simulations were 0.5 s long with integration time of 0.1 ms. Ten trials were conducted for each simulated condition.
Simulated light responses.
Each of the 121 simulated DSGC dendritic compartments in the ON layer was innervated by a synaptic input containing a BC- and a SAC-derived component. Presynaptic cells were modeled as point sources located at the middle of the innervated branch. Light activation was simulated by a 1-mm-long rectangular area moving at 1 mm/s over the network. When the leading edge of the stimulus overlapped with the position of a presynaptic cell, the cell became depolarized to a new potential with a rise time of 50 ms and a decay time of 200 ms. The degree of presynaptic depolarization was set according to the contrast sensitivity of the synaptic drive. Three scenarios were modeled: In the first the E/I was balanced (ie, the E/I ratio was the same at all contrast levels), so that the peak “light” induced depolarization was similar between the excitatory and inhibitory drives. In the second and third scenarios, the inhibitory release occurred either at lower or higher contrast levels. Direction selectivity was mediated by the amplitude of the GABAergic drive; at each contrast level, we modeled the null direction responses first. Then the GABAergic drive was reduced threefold and the simulation repeated to model the preferred direction.
Each presynaptic synapse had a readily releasable pool (RRP) of 10 vesicles, which could be released every 1 ms with a probability of release dependent on the membrane potential. Vesicles then could be replenished from an infinite vesicle pool at a rate of 500 ms−1 (Singer and Diamond, 2006). The amplitude of the peak stimulus-induced depolarization was set to deplete on average 30% of the RRP at the highest contrast level.
Simulated synaptic release.
We simulated three distinct classes of postsynaptic receptors: (1) Ohmic excitatory receptors, corresponding to AMPA and nicotinic cholinergic receptors. Ohmic synapses had an instantaneous rise time and a decay time of 2 ms and a unitary conductance of 0.5 nS following simulated transmitter release. (2) NMDARs, which had a rise time of 2 ms, decay time of 60 ms, and a peak unitary conductance of 0.5 nS. The conductance was voltage-dependent as follows: NMDAG = (Jahr and Stevens, 1990), where v is the membrane potential of the postsynaptic dendritic compartment. Excitatory conductances reversed at 0 mV. (3) GABAergic inputs, which were activated with an instantaneous rise time, 30 ms decay time, 0.5 nS conductance, and had a reversal potential of −60 ms (Lee et al., 2010).
Results
Contrast sensitivity of DRD4-DSGCs
Rabbit DSGCs respond to higher contrast stimuli with increased firing but do not change their DS tuning (Grzywacz et al., 1998; but see Lipin et al., 2015). We first tested whether this basic response feature is present in mouse ON–OFF DSGCs fluorescently labeled via the DRD4 promoter (Huberman et al., 2009) and visualized in intact, isolated retinas with a two-photon microscope (see Materials and Methods). Light stimulation consisted of bars moving in any of eight different directions on a dimmer background (30,000 R* rod−1 s−1) that was sufficient to saturate rods and fully engage the cone pathway (Nikonov et al., 2006). We recorded axosomatic action potentials (APs) from DRD4+ DSGCs with somatic patch electrodes in the cell-attached configuration, stimulated the retina with light bars moving in eight different directions, and determined PD and ND stimuli from the AP rate evoked by light onset. We then recorded AP responses to PD and ND moving bars at different contrast levels relative to the background and calculated the direction selectivity index [DSI = (PD response − ND response)/PD response] for each contrast level (Fig. 1B). Next, whole-cell recordings were obtained from the same cells and visual stimulation was repeated in current-clamp mode to record light-evoked postsynaptic potentials (PSPs; Fig. 1C,D). DRD4+ DSGCs exhibited reliable DS over a broad contrast range in both subthreshold and suprathreshold recordings (Fig. 1B–F; Grzywacz et al., 1998). Not all contrast levels generated detectable responses in DSGCs, however (Fig. 1B,D). For each cell, we identified a contrast threshold as the highest contrast level that did not evoke a detectable signal in response to either PD or ND stimulation. The mean (±SD) threshold contrast was lower for PSPs (45 ± 25%; n = 13) than for APs (68 ± 24%; n = 43; Fig. 1E; p = 0.03, Wilcoxon signed rank test), indicating that AP threshold prevented small PSPs, evoked at low contrasts, from effectively triggering APs. The shape of the contrast dependence of suprathreshold and subthreshold responses was compared by normalizing PSPs or APs in each cell by the largest recorded responses (Fig. 1F). The normalized signals exhibited a sigmoidal relationship with respect to contrast level. The spike and PSP contrast tuning curves were significantly different (p = 0.02, two-way ANOVA, PD responses only), mainly due to lack of spiking responses to low contrasts described above. The contrast level that evoked half-maximal responses was similar between the groups (Fig. 1E).
Similar contrast sensitivity of excitatory and inhibitory inputs to DSGCs
The undetectable responses in DSGCs to lower contrast stimuli may reflect the absence of excitatory synaptic drive and/or strong shunting inhibition. We distinguished between the two possibilities by obtaining whole-cell voltage-clamp recordings from DSGCs and examining the contrast sensitivity of EPSCs and IPSCs by clamping the cell at the estimated reversal potential of the alternative synaptic input (−65 and 0 mV; Fried et al., 2002). As with PSP recordings, EPSCs and IPSCs were undetectable below contrast levels of 38 ± 14% and 39 ± 30%, respectively (Fig. 2A,B, left; p = 0.3 between EPSCs and IPSCs, n = 23 cells, Wilcoxon Signed Rank Test). Evoked PSCs grew larger with increasing contrasts and reached a maximal plateau at ∼150% contrast (Fig. 2C). At all contrasts that evoked reliable responses, EPSCs were larger in response to PD stimulation, whereas inhibition was larger in response to ND stimulation, as reported previously (Fried et al., 2002; Taylor and Vaney, 2002; Park et al., 2014; Poleg-Polsky and Diamond, 2016). We attribute the larger EPSCs evoked by PD stimuli to space clamp errors (Poleg-Polsky and Diamond, 2011; Vaney et al., 2012), as glutamate release from BCs is not strongly DS (Yonehara et al., 2013; Park et al., 2014; Pei et al., 2015; Poleg-Polsky and Diamond, 2016). Interestingly, when we normalized the recorded signals by the corresponding peak responses, we found that both EPSCs and IPSCs in PD and ND directions exhibited statistically similar contrast relationships (Fig. 2D; p = 0.4, n = 23, two-way ANOVA) and half-maximal contrasts (Fig. 2B, right; p = 0.4 between EPSCs and IPSCs, n = 23 cells, Wilcoxon signed rank test). Although space clamp errors may have distorted the relative amplitude of PD and ND responses, stimulus contrast did not appear to affect the relative magnitude of EPSCs and IPSCs; the E/I ratio in DSGCs was contrast independent (Fig. 2E).
Figure 2.

Excitatory and inhibitory inputs to DSGCs exhibit balanced contrast sensitivity. A, Representative IPSCs (top; green) and EPSCs (bottom; black) in DSGCs in response to PD (left) and ND (right) stimulation at different contrast levels. Gray shaded areas indicate the approximate time interval over which the moving bar occupied the receptive field of the cell. Asterisks indicate comparison between PSCs and baseline (**p < 0.01, ***p < 0.001, t test). Inset, Schematic showing the primary excitatory and inhibitory inputs to the DSGC. B, Contrast levels that elicited the first observable synaptic current (left) and half-maximal response (right) were similar between inhibitory and excitatory currents (n = 25, individual experiments in gray, population mean ± SD; black). C, Median (± quartile) contrast tuning of EPSCs (black) and IPSCs (green). PD, solid lines; ND, dotted lines. D, Median (± quartile) normalized contrast sensitivity curves were similar between PD and ND responses and between EPSCs and IPSCs (same color scheme as in C). E, EPSC and IPSC amplitudes for each contrast level (mean ± SEM) remained proportional over the entire range of contrast stimuli tested (r = 0.94). Responses are grayscale coded based on contrast level (inset).
Highly contrast-sensitive inhibition required for reliable DS
The E/I ratio is an important parameter in DS computation, as the inhibitory drive largely determines DSGC directional tuning (Demb, 2007; Zhou and Lee, 2008; Trenholm et al., 2011; Vaney et al., 2012). DS would therefore be expected to degrade if the synaptic input to DSGCs did not include a prominent inhibitory component. We examined this prediction with a mathematical model in which we could tune independently the contrast sensitivity of non-DS excitatory and DS inhibitory inputs onto a simulated DSGC (Fig. 3; see Materials and Methods). First, we simulated a perfectly balanced condition in which the contrast sensitivity of the excitatory and the inhibitory drives was identical (Fig. 3Ai). Note that “balance” is not meant to imply that excitatory and inhibitory conductances are equal. Rather, it means that the E/I ratio is consistent over the stimulus contrast range (Fig. 3Aii). When this condition was met in the model, PD and ND PSP amplitudes were poorly separated at low contrasts, due to weak inhibitory drive (Fig. 3Aiii), resulting in lower DSI (Fig. 3D), similar to our experimental findings (Fig. 1). We then estimated how subthreshold DS translated to spike responses by including voltage gated sodium and potassium conductances in the simulation (see Materials and Methods). Because low-contrast PSPs typically did not cross spike threshold, the poorest subthreshold DS was not reflected in action potential responses (Fig. 3Aiv,E). At slightly higher contrasts, only PD responses crossed spike threshold (Fig. 3Aiv), resulting in a DSI near 1 (Fig. 3E). As contrast was increased further, ND responses also began to elicit spikes (Fig. 3Aiv), but by this point subthreshold DS had improved sufficiently to produce high suprathreshold DS, particularly following the sharp nonlinear transformation of PSPs to action potentials (Fig. 3E).
Figure 3.

Simulated responses predict that differences in E/I contrast sensitivity would degrade DS computation. A–C, Top, Schematic illustration of the inputs to the cell and the simulated contrast sensitivities of the excitatory (black) and inhibitory (green) conductances. The inhibitory drive was set to have either similar contrast sensitivity to the excitatory input (A), less sensitivity (B), or greater sensitivity (C) arrows mark the contrast for half-maximal activation. Directional information was provided via a larger ND inhibitory conductance (green, dotted). Middle and Bottom, Simulated PSPs and APs, respectively, for PD and ND stimuli. D, E, DSI for subthreshold (D) and suprathreshold (E) responses for the three simulated scenarios: equal excitation and inhibition (light brown), more sensitive excitation (violet), or more sensitive inhibition (red).
Next, we set the excitatory drive to activate at lower contrasts than the inhibitory drive (Fig. 3Bi). This manipulation caused the E/I ratio to vary over the contrast range (Fig. 3Bii). As expected, weaker inhibition at lower contrasts permitted larger subthreshold PSPs (Fig. 3Biii), increased the simulated suprathreshold ND responses (Fig. 3Biv), and degraded DSI values over a wide contrast range (Fig. 3D,E). DSI measured from spike responses approached values in the balanced condition only for high contrast levels at which the inhibitory drive was finally strong enough to inhibit spiking in response to ND stimulation (Fig. 3E).
Finally, we tested the condition in which inhibitory input is activated at lower contrast levels relative to the excitatory input (Fig. 3C). Due to the decisive role of inhibition in DS computation, larger inhibition at low contrasts forced a greater difference between PD and ND PSPs and a better subthreshold DSI (Fig. 3Ciii,D). Enhanced subthreshold performance did not translate to improved suprathreshold DS compared with the balanced E/I condition (Fig. 3Civ,E), however, because the high inhibitory conductance at low-contrast levels precluded the DSGC from firing in response to low-contrast stimulation, even for contrast levels at which the balanced E/I case conveyed DS information reliably (Fig. 3E). These simulations suggest that balanced E/I input provides the optimal DS signal, at least for the modeled conditions (but see Lipin et al., 2015).
Possible mechanisms of contrast independent E/I ratio in DSGCs
Given the structure of the DS circuitry, the consistent balance between the excitatory and the inhibitory inputs to DSGCs over a range of contrast levels (Fig. 2) is surprising. A significant fraction of excitatory synaptic input to DSCGs comes directly from BCs (Lee et al., 2014), whereas inhibition is mediated exclusively by amacrine cells (predominantly SACs; Demb, 2007; Zhou and Lee, 2008; Vaney et al., 2012). Most neurons exhibit a nonlinear, approximately sigmoidal input/output relationship, such that the weakest synaptic inputs do not exceed a threshold to generate an output. Within the DS circuitry, low-contrast stimuli that evoke only weak release from BCs likely would not elicit GABA release from SACs. If DSGCs received the same BC input as SACs, an E/I imbalance would occur at the lower contrast levels. The fact that we did not observe an E/I imbalance indicates that some mechanism preserves a consistent relative contribution of excitatory and inhibitory inputs to DSGCs across a range of contrasts.
What mechanism mediates this compensation? One possibility is that SACs, which also provide cholinergic excitation to DSGCs (Kittila and Massey, 1997; Chiao and Masland, 2002; Lee et al., 2010), or other amacrine cells that provide excitatory input to DSGCs (Lee et al., 2014) predominate over BC inputs in DRD4+ DSGCs. If SAC-derived excitatory input to DSGCs were substantially larger than that from BCs, and if GABA and ACh release exhibited similar contrast dependence (but see Lee et al., 2010), DSGCs could encounter a similar E/I balance at all contrasts.
Second, if the input/output (I/O) transformation in SACs were actually linear, then any synaptic input, regardless of amplitude, would elicit release to DSGCs. This scenario may be feasible, as both synaptic input and output sites are located on SAC dendrites and action potentials are not required to drive GABA release (Euler et al., 2002; Lee et al., 2010). If true, linear processing in SACs would produce similar contrast sensitivity between SAC inputs and outputs, both of which can be determined experimentally.
A third potential mechanism involves presynaptic BCs. Given that BCs subtypes are heterogeneous and distinct combinations of BC subtypes innervate SACs and DSGCs (Shi et al., 2011; Helmstaedter et al., 2013; Chen et al., 2014; Kim et al., 2014; Greene et al., 2016), BC input to SACs may activate at lower contrasts to compensate for nonlinear I/O behavior in SACs.
AChR and NMDAR blockers exert similar effects on the contrast sensitivity of EPSCs in DSGCs
To test the viability of the first scenario, that amacrine-derived excitatory input is significantly stronger than BC-mediated glutamatergic excitation, we pharmacologically blocked different components of the EPSCs recorded in DSGCs (Fig. 4). The AChR component was blocked with nAChR and mAChR antagonists hexamethonium (100 μm) and atropine (5 μm, n = 10; Fig. 4A); the NMDAR component was blocked with AP5 (50 μm, n = 7; Fig. 4B); the AMPAR component was isolated by combining AChR and NMDAR antagonists (n = 6; Fig. 4C). We focused on PD responses because they were larger and easier to analyze. All blockers reduced light-evoked EPSC amplitudes at all contrast levels measured (Fig. 4A–C), but the contrast sensitivity of EPSCs was not different in the presence of cholinergic or glutamatergic antagonists (Fig. 4D,E). These experiments show that, at all contrast levels studied here, cholinergic and glutamatergic inputs mediate comparable fractions of the excitatory drive to DSGCs. As the relative contribution of BCs and amacrine cells to the glutamatergic input onto DSGCs cannot be determined with current recordings alone (Lee et al., 2014), at this point we were unable to conclude whether the contrast sensitivity of the BC signal to DSGCs differs from the sensitivity of the excitatory input from glutamatergic amacrine cells (Fig. 1A; Lee et al., 2014). Experiments described below, conducted on SACs, address this question while simultaneously addressing alternative mechanisms for E/I compensation.
Figure 4.
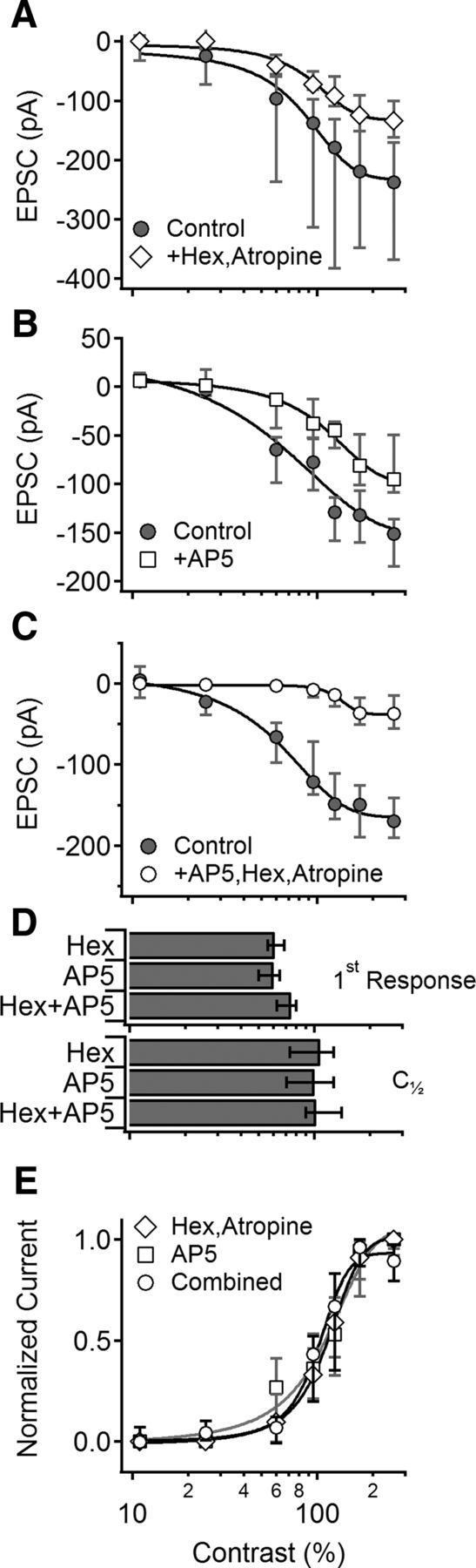
Cholinergic and glutamatergic inputs to DSGCs exhibit similar contrast sensitivity. A, Median (±quartile) contrast sensitivity of EPSCs before (circles) and after (diamonds) cholinergic receptor blockade by hexamethonium and atropine. Cholinergic blockade eliminated SAC-mediated excitatory input, leaving only the BC-mediated glutamatergic component. B, As in A for NMDAR blockade (+AP5, squares). C, As in A following combined application of cholinergic and NMDAR blockers to isolate the AMPA component (open circles). D, Mean (±SD) lowest contrast level that evoked measurable EPSCs (top) and half-maximal contrast levels (C[1/2], bottom) were similar in all conditions (p > 0.2, t test). E, Median (±quartile) normalized contrast tuning curves in the different conditions were similar (p > 0.5, two-way ANOVA), indicating that glutamatergic and cholinergic excitatory inputs to DSGCs exhibited similar contrast tuning.
EPSCs in SACs reveal sensitivity to low contrasts
To test the second possibility, that the SAC I/O relationship is linear, we first examined the contrast dependence of excitatory input to SACs with whole-cell voltage-clamp recordings from ON SACs (Fig. 5). EPSCs, elicited with the same moving bar stimuli used for DSGCs, exhibited a sharp initial component followed by a more sustained component (Fig. 5A). The contrast sensitivity of the two components was slightly different: the transient component was first reliably observed at 15.2 ± 8.5% contrast, and its half-maximal contrast was just 32.5.2 ± 13.5% (Fig. 5B,C; n = 13 cells), whereas the half-maximal contrast of activation of the sustained component was 50 ± 27% (Fig. 5B,C; p = 0.02, paired t test, n = 13). Despite this difference, SAC EPSCs overall were significantly more sensitive than EPSCs recorded in DSGCs (Fig. 5C). The contrast sensitivity of SACs and DSGCs was not correlated with postnatal age (Fig. 5D). As BCs provide the primary source of excitatory input to SACs after eye opening (Famiglietti, 1991; Wei et al., 2011; Yonehara et al., 2011; Helmstaedter et al., 2013), these data indicate that excitatory input from BCs to SACs exhibits relatively high contrast sensitivity.
Figure 5.
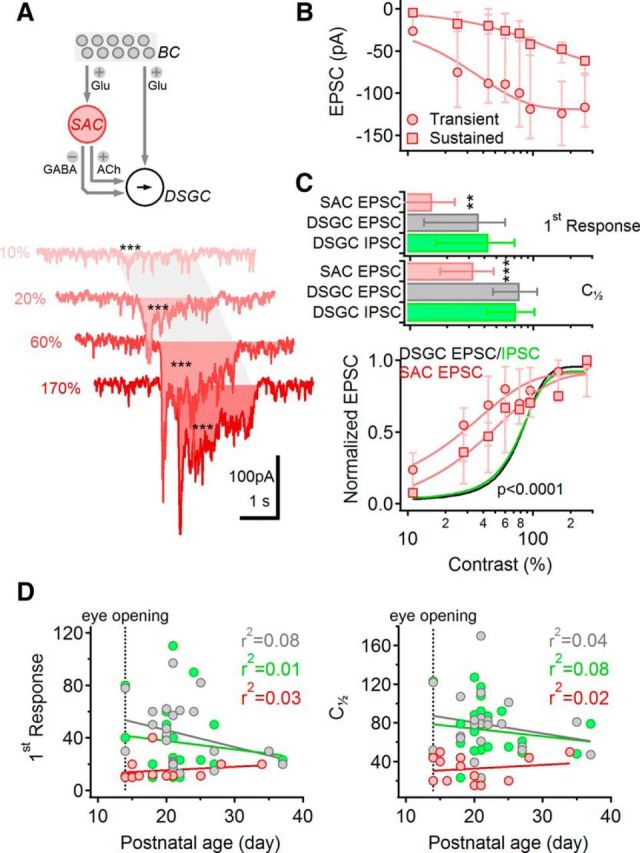
Excitatory input to SACs exhibits high contrast sensitivity. A, Top, Schematic highlighting SACs in the DS network. Bottom, EPSCs from a representative SAC in response to moving bar stimulation, color-coded according to the contrast level. The shaded area indicates the approximate time interval over which the stimulus occupied the receptive field of the cell. Asterisks indicate comparison between stimulus response and baseline (***p < 0.001). Note that EPSCs were evident at the lowest contrast level tested. B, Median (±quartile) amplitudes of the sustained (squares) and the transient (circles) components of the EPSC evoked by different contrast levels (n = 13 cells). C, Top, Lowest detectable (mean ± SD; top) and half-maximal (bottom) contrast levels were significantly lower for SAC EPSCs (measured at the peak; red) compared with either EPSCs (gray) or IPSCs (green) recorded in DSGCs; **p < 0.01; ***p < 0.001 (one-way ANOVA). Bottom, Data from (B) normalized by the largest response in each cell (red). The contrast tuning of the sustained SAC-EPSC was significantly different from that of the transient SAC response (p < 0.00001, two-way ANOVA). Normalized contrast tuning curves of DSGC EPSCs (black) and IPSCs (green) are shown for comparison. D, Contrast sensitivity [measured as first observable response (left) and half-maximal contrast levels (right)] was not correlated with postnatal age. Color coding as in C. Responses were fitted with a linear regression.
SACs exhibit a nonlinear I/O relationship
The results thus far indicate that excitatory synaptic inputs to SACs, measured as SAC EPSCs, exhibit higher contrast sensitivity than SAC outputs, measured as IPSCs in DSGCs (Fig. 5C). Several potential mechanisms could underlie this observation. First, if calcium channels in SAC dendrites respond nonlinearly to postsynaptic depolarization, small PSPs elicited by lower contrasts may not evoke calcium influx. Consistent with this idea, activation of calcium channels expressed in SACs (N, P/Q; Cohen, 2001; Kaneda et al., 2007; Lee et al., 2010) requires strong depolarization. An alternative, not mutually exclusive explanation is that a nonlinear (power) relationship between calcium and GABA release may preferentially limit release at low contrasts, when the calcium current is expected to be lower than in response to stronger stimuli.
To distinguish these two possibilities, we patched SACs with electrodes containing OBG-1 to measure the change in dendritic calcium during visual stimulation (Euler et al., 2002) while simultaneously recording somatic PSPs in current-clamp mode (Fig. 6A,B). We first compared the contrast sensitivity curve of the somatically recorded PSP signals in the absence and presence of the imaging laser light and confirmed that the laser illumination did not influence light-evoked signals in SACs (Fig. 6C, top; p > 0.1, two-way ANOVA, n = 8 SACs). The contrast sensitivity of SAC PSPs was similar to that of SAC-EPSCs (compare Figs. 6B,C, bottom, and 5C), but it was significantly different from that of the dendritic calcium signals (Fig. 6B,D–F): calcium influx was not detected in response to low-contrast stimuli up to 38 ± 32% (Fig. 6D–F; p = 0.01, paired t test vs PSPs, n = 11 cells), and the half-maximal activation contrast was significantly higher for calcium signals (26 ± 17% vs 66 ± 35% for PSPs vs OGB-1 fluorescence, respectively, p = 0.0005, paired t test; Fig. 6E, right).
Figure 6.
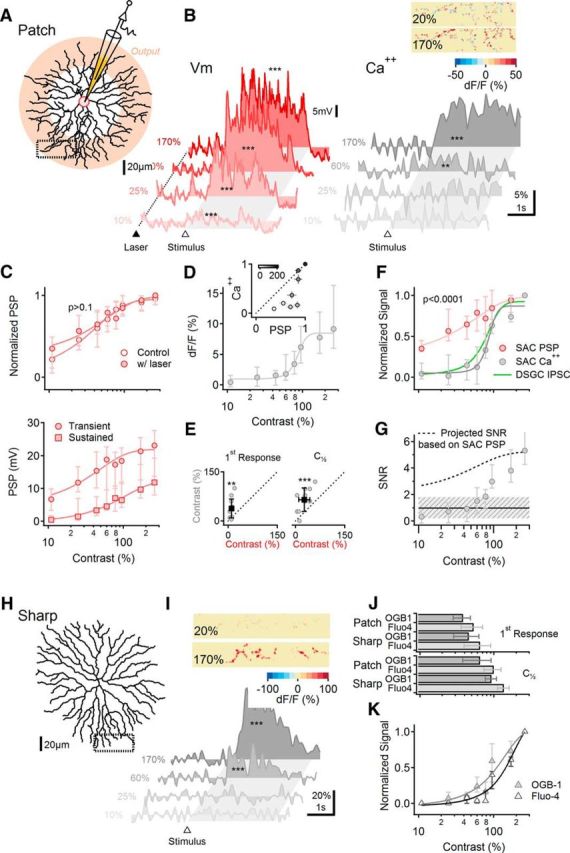
SACs exhibit nonlinear I/O behavior. A–G, Somatic electrical and dendritic calcium signals exhibit different contrast dependence. A, Schematic of experimental design. SACs were patched and filled with a calcium indicator (OGB-1 or Fluo-4). Pink shading indicates approximately the area of the dendrites from which GABA and ACh release occurs. Visual stimulation consisted of a bar moving in the centrifugal direction (ie, from the soma to the recorded dendritic location). B, Representative somatic PSPs (left) and corresponding dendritic calcium transients were visualized using OGB-1 from the rectangular ROI in A (right). The dotted line and filled triangle mark the beginning of 2P laser scanning. The open triangle and the shaded area indicate the approximate entrance and the time interval over which the stimulus occupied the cell's receptive field. Asterisks indicate comparison between responses and baseline (**p < 0.01, ***p < 0.001, t test). Inset, Heat map showing the spatial distribution of dendritic calcium signals from the dotted region in A for two stimulus contrast levels, color-coded by the peak dF/F values. C, Top, Illumination from the imaging laser did not affect the contrast tuning of the somatic PSPs (two-way ANOVA). Bottom, Contrast sensitivity of the transient (circles) and the average (squares) somatic PSPs (n = 12 cells). D, Contrast tuning of dendritic calcium signals recorded from the same cells as in C. Inset, Comparison between normalized contrast responses of calcium and voltage signals (mean ± SEM; greyscale coded as in Fig. 2E) reveals strong deviation from unity (dotted line) at low to intermediate contrast levels. E, Left, The lowest contrast level that elicited a measurable response was significantly (**p < 0.01, paired t test) higher for the calcium (vertical axis) than the voltage (horizontal axis) signals (n = 12). Right, Similar analysis of the contrast that elicited a half-maximal response (C[1/2]; ***p < 0.001, paired t test). F, Normalized contrast sensitivity curves for SAC PSPs and calcium signals (p < 0.0001, two-way ANOVA). Contrast sensitivity of DSGC-recorded IPSCs (green) is shown for comparison. G, Signal-to-noise ratio of dendritic calcium responses. Dotted curve indicates the estimated SNR if calcium signals followed the contrast sensitivity of the somatic membrane potentials. Dashed region indicates the estimated range of SNR over which the responses to the stimulus would not be reliably distinguished from baseline noise. H–K, Calcium contrast dependence is similar in SACs that were filled via patch and sharp electrodes. H, Reconstructed morphology of a SAC impaled and filled with OGB-1 via sharp electrode. I, Similar to B (right) for the cell shown in H. J, Similar first observable (top) and half-maximal responses (C[1/2]; bottom) between cells patched and filled with OGB-1 (n = 12) or Fluo-4 (n = 5) and cells in which dyes were introduced via sharp electrode (OGB-1, n = 7; Fluo-4, n = 5). K, Normalized contrast sensitivity curves for calcium signals from SACs filled by sharp electrode. Unless noted otherwise, error bars represent first and third quartiles.
Could the difference in contrast sensitivity between electrical and calcium signals reflect a detection bias? The recorded fluorescence had higher baseline variability and was acquired at a lower sampling rate than electrical signals (Fig. 6G; Materials and Methods). This can limit the detectability of the responses to the stimulus over the background activity, especially at low-contrast stimulation trials, where the expected change in fluorescence is small. To address this issue, we computed the ratio between the mean responses during visual stimulation to the variability of the baseline fluorescence signals. The resulting SNRs varied as a function of contrast: at the highest contrast level measured (170%), the median SNR was 5.3 and the calcium responses were readily detected (Fig. 6G). SNR values got progressively smaller at lower contrasts. We estimate that calcium responses are poorly distinguishable from baseline for SNRs <1.8 (Fig. 6G, shaded region; see Materials and Methods; Vlasits et al., 2016).
Next we estimated the fluorescence SNR that we would observe if dendritic calcium signals exhibited the same contrast sensitivity as somatic PSPs. The projected SNR values (calculated from the peak calcium signals) in this scenario followed the dotted curve in Figure 6G. Critically, the projected SNR exceeded 2 for all contrast levels, indicating that our recordings of OGB-1 fluorescence were sufficiently sensitive to detect responses to visual stimulation at even the lowest contrast levels. The fact that no responses were detected at low contrasts (Fig. 6E,F) therefore reflects true differences in contrast sensitivity between dendritic calcium signals and somatic PSPs. In fact, calcium responses in SACs exhibited contrast sensitivity that was more similar to that of IPCSs in DSCGs than PSPs in SACs (Fig. 6F). These data suggest that the nonlinear voltage dependence of calcium channels in SAC dendrites likely underlies the nonlinear I/O behavior of SAC dendrites and reduces the contrast sensitivity of SAC outputs relative to SAC inputs.
SAC calcium signals may run down during whole-cell recordings (Hausselt et al., 2007; Vlasits et al., 2016) or be distorted by saturation of OGB-1 at high contrasts. We addressed the first concern by filling SACs with OGB-1 via sharp electrodes (Fig. 6H–K). Following sharp-electrode filling, calcium responses in SACs remained stable for hours. To ensure cell longevity, the pipette was extracted immediately following dye injection, precluding somatic potential recordings. We did not observe significant differences with patch and sharp electrodes in either the contrast that elicited the first observable light response or the half-maximal contrast level (Fig. 6J). These results indicate that the difference between electrical responses recorded from SAC somata and calcium responses recorded in SAC dendrites was not due to artifacts of whole-cell recordings. To address the second concern about calcium indicator saturation, we repeated the experiments with a lower affinity dye, Fluo-4 (Fig. 6J–K). As expected from the distinct binding kinetics of the dyes, we detected a statistically significant difference in the contrast dependence of OBG-1 and Fluo-4 signals (Fig. 6J; p = 0.03 for both the first observable and half-amplitude responses). Despite the approximately twofold difference in affinity, however, the contrast dependence curves of the two calcium indicators were relatively similar compared with the contrast dependence of the somatic voltage signals (Fig. 6F,K).
Different contrast sensitivity of glutamate release onto SACs and DSGCs
The observed difference in contrast tuning of EPSCs in DSGCs and SACs could reflect differences in presynaptic BC characteristics or differences in sensitivity of the postsynaptic cells to synaptically released glutamate. To distinguish between these possibilities, we expressed iGluSnFR, a genetically encoded glutamate sensor (Marvin et al., 2013), in either SACs or DSCGs (see Materials and Methods; Fig. 7A–D). If BC inputs to DSGCs and SACs exhibited distinct contrast sensitivities, any differences would be evident in the iGluSnFR signals recorded in the two cell types. If the difference between SAC and DSGC contrast sensitivity were instead due to distinct postsynaptic sensitivities to glutamate, the contrast sensitivity of iGluSnFR signals should be similar in the two cell types. iGluSnFR fluorescence was measured in eight SACs and nine DSGCs; in a subset of experiments we also obtained voltage recordings to directly compare the contrast sensitivity of the different signals and to ensure that the scanning laser light did not evoke light responses (Fig. 7B,D). iGluSnFR fluorescence was detected at significantly lower contrast levels in SACs compared with DSGCs (Fig. 7B–F). The lowest contrast that evoked a detectable iGluSnFR response was 16.2 ± 8% in SACs versus 65.5 ± 17% in DSGCs (Fig. 7E, top; p < 0.00001, t test). We did not compare the half-activation contrast between cell types because we were unable to confirm the synapse specificity of iGluSnFR signals at very high contrast levels, conditions under which iGluSnFR may be more likely to respond to glutamate spillover between neighboring synapses.
Figure 7.

Glutamatergic inputs to SACs and DSGCs exhibit distinct contrast sensitivity. A, DSGCs were patched and filled with AlexaFluor-647 for electrical recordings and morphological reconstruction. The morphology of the DSGC (left) from which voltage and iGluSnFR signals shown in B were recorded. Right, Top, Magnified view of DSGC dendrite (red) from the dotted box in A overlaid with the mean iGluSnFR background fluorescence (teal). Middle and Bottom, Heat map of the peak change in iGluSnFR signals for two contrast levels during stimulation with a moving bar. B, Representative somatic PSPs (left) and the corresponding dendritic iGluSnFR transients (right) from the DSGC in A. The dotted line and a filled triangle mark the beginning of 2P laser scanning. The open triangle and the shaded area indicate the approximate time interval over which the stimulus entered and occupied the cell's receptive field. C, D, Similar to A and B for a SAC. E, Top, Lowest contrast that evoked detectable EPSCs and iGluSnFR signals (mean ± SD) were significantly lower in SACs (n = 11 cells) compared with DSGCs (p = 0.00001, n = 8 cells, t test). Within each cell type, the somatic voltage responses and iGluSnFR fluorescence exhibited similar contrast activation thresholds. Bottom, Contrast sensitivity of the median (±quartile) iGluSnFR signal in DSGC and SAC dendrites. F, The data in E (bottom) normalized to the largest response in each cell. Error bars indicate first and third quartiles.
These results indicate that glutamatergic inputs to SACs and DSGCs exhibit distinct contrast tuning, suggesting that BC inputs to DS circuitry exhibit a range of contrast sensitivity and that SACs receive, on average, more highly contrast-sensitive inputs than do DSGCs. Recent anatomical and functional evidence suggests that SACs and DSGCs receive inputs from distinct but overlapping populations of BCs, mostly cone bipolar type 5 and type 7 cells (Shi et al., 2011; Helmstaedter et al., 2013; Duan et al., 2014; Greene et al., 2016). In addition, BC axon terminals are morphologically diverse and highly branched; individual terminal processes contain several ribbon synapses and receive distinct inhibitory inputs (Euler et al., 2014), suggesting that single BCs may mediate multiple signals with different contrast tuning.
Axon terminals within individual bipolar cells exhibit similar contrast sensitivity
We examined the contrast tuning of light-evoked calcium signals in axon terminals of BCs using a newly developed technique to stain cells in the retina with OGB-1 (Poleg-Polsky A, Diamond JS, unpublished observations). In brief, gentle mechanical manipulation of intact retina in solution containing OGB-1 causes a small fraction of neurons to take up the dye, and those that do are brightly labeled (Fig. 8A). This manipulation preserves light responses, enabling us to activate BCs with the same type of physiological stimuli used for SACs and DSGCs. Calcium signals in responsive cells were robust and stable for 1–2 h, indicating that they were not injured significantly by the labeling procedure.
Figure 8.
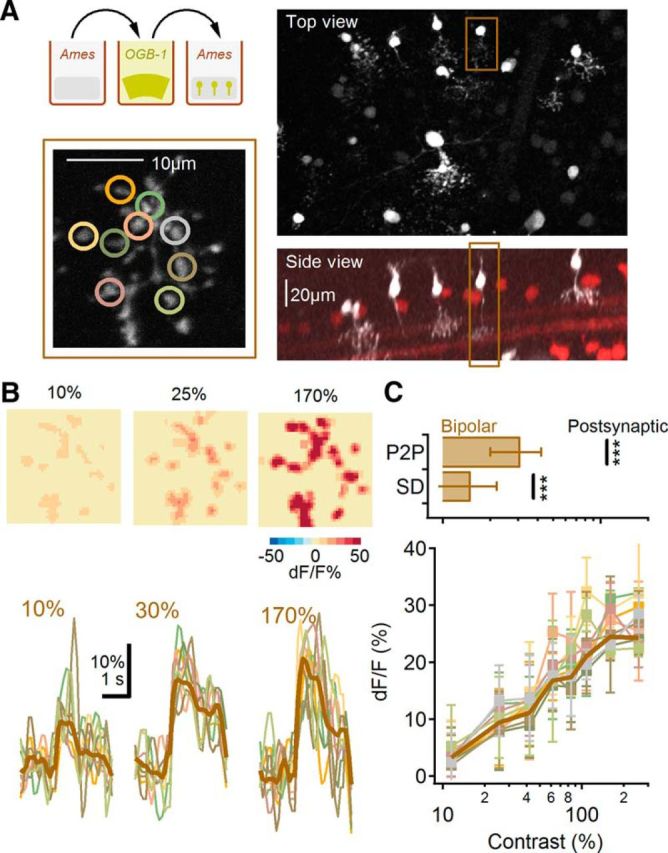
Similar contrast sensitivity in different axonal boutons of a single BC. A, Experimental protocol. ON BCs were filled with OGB-1 in whole-mount retina with a newly developed method of labeling multiple neural types by gentle bending of the retinal tissue in a concentrated indicator solution (top, left). Right, Top (top) and side (bottom) views of brightly and sparsely labeled ON-BCs. Brown frame marks the ON-BC chosen for axon terminal recording (bottom, left; B). B, Representative calcium transients from individual terminals (bottom; color-coded to match the locations in A, bottom, left) and the spatial distribution of the peak dF/F levels (top) for three contrast levels. Thick brown line indicates the average response of the entire axonal field-of-view. Responses were elicited by the same moving bar stimulus used elsewhere. C, Top, The range of contrast sensitivity in different axonal boutons from individual cells quantified as P2P or SD of the half-maximal response was significantly smaller than the differences between SAC and DSGC responses (black lines; ***p < 0.001, t test). Bottom, Fluorescence signals (mean ± SD) from all the recorded axon boutons color coded as in B, bottom.
We first tested the possibility that different parts of the axon terminals of individual BC cells exhibit distinct contrast sensitivities. To compare contrast sensitivity between regions within a single axon terminal, we first determined the contrast level that elicited a half-maximal calcium signal in each of 5–10 different regions within the terminal (Fig. 8B). Next, we calculated the SD and the difference between the largest to the smallest [peak to peak (P2P)] contrasts that elicited half-maximal responses in all the regions within the terminal and then compared them to similar metrics of contrast sensitivity distributions between SACs and DSGCs (Fig. 8C). Light-evoked calcium signals in different regions of the same terminal had relatively similar contrast dependence (n = 13; P2P = 33 ± 14%, SD = 12 ± 5%; Fig. 8C) especially when compared with the difference within the postsynaptic half-maximal activation between postsynaptic cells (Fig. 8C; p < 0.001; Mann–Whitney U test).
Axon terminals of different ON CBs exhibit different contrast sensitivity
The results in Figure 8 indicate that regions within individual ON BC axon terminals respond over a similar contrast range. Next, we compared contrast sensitivity between ON BCs. We recorded calcium signals from the entire axon terminals of 30 ON BCs and compared their contrast sensitivities with one another and with those of SAC and DSGC EPSCs (Fig. 9). As a group, ON BCs exhibited a broad range of contrast tuning; the contrast sensitivity of many individual cells overlapped with either the highly sensitive SAC EPSCs or the less-sensitive DSGC EPSCs (Fig. 9A; two examples shown in B,C).
Figure 9.
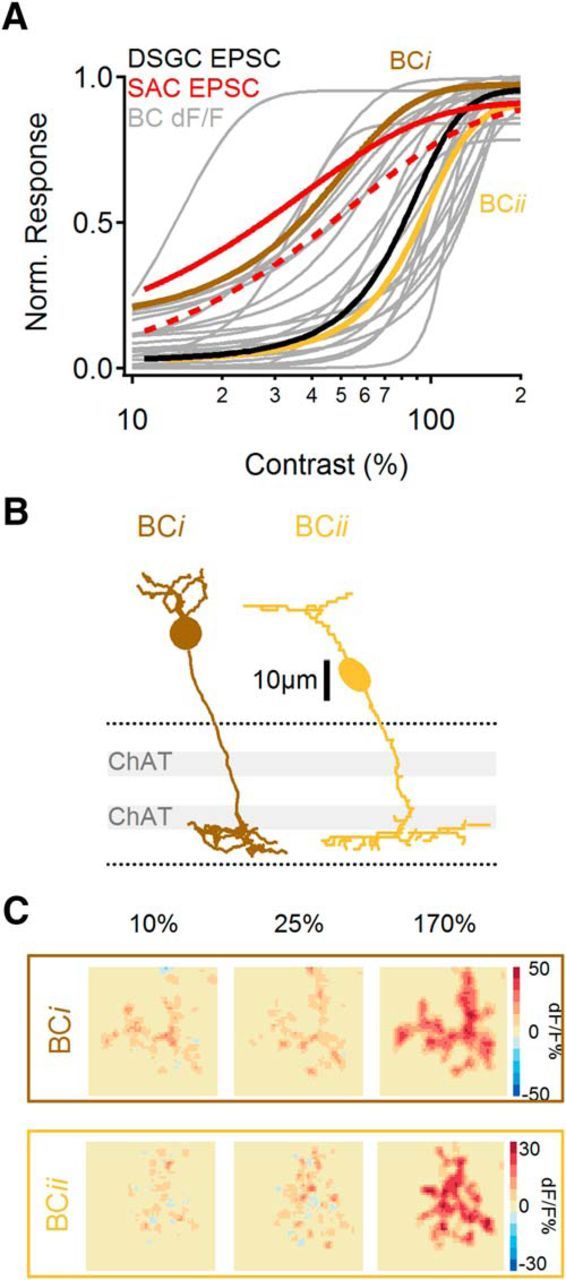
Individual BCs exhibit distinct contrast tuning. A, Contrast sensitivity of normalized dF/F responses of all recorded BCs (gray). Brown and yellow traces indicate tuning of individual cells shown in B and C. Contrast tuning curves of EPSCs in SACs (peak EPSC, solid red; mean EPSC, dotted red) and DSGCs (black) are shown for comparison. B, Representative morphologies of two BCs with contrast sensitivities similar to the mean contrast sensitivities recorded in SACs (BCi) and in DSGCs (BCii). Dotted lines indicate limits of the IPL, gray bars represent ChAT bands formed by SAC dendrites. C, Spatial distribution of peak dF/F signals at different contrast levels for the two BCs shown in B.
Next, we tested whether cell morphology, which varied substantially between BCs (Figs. 8A, 9B), was correlated with contrast sensitivity (Ichinose et al., 2014). Previous studies have identified specific ON BC subtypes using molecular/genetic markers (Chen et al., 2014; Ichinose et al., 2014) or by comparing all ON BCs in a dense reconstruction of a circumscribed region of a single retina (Helmstaedter et al., 2013; Greene et al., 2016). Here, because we examined one cell at a time in the absence of specific markers, we were unable to unambiguously assign individual ON BCs to specific cell classes. Instead, we split the recorded BCs into two groups based upon whether their contrast sensitivity (as indicated by the contrast that evoked a half-maximal response) was closer to that of SACs (n = 12 BCs) or DSGCs (n = 18 BCs) and then tested whether the two groups were morphologically distinguishable (Fig. 10). We compared morphological characteristics of the axon terminals; mean depth in the IPL, (z-axis) span in IPL depth, and lateral (x–y) area (Fig. 10A). These characteristics, as well as calcium response time course and contrast sensitivity, are shown for each BC in both groups (Fig. 10B,C). This analysis showed that the axon terminals of BCs with higher (SAC-like) contrast sensitivity ramified higher and more narrowly in the IPL than did less-sensitive BCs (Fig. 10D1,D2), whereas no differences in lateral area were observed between the two groups (Fig. 10D3). These data suggest that the range of contrast sensitivity in BCs likely reflects physiological differences between morphologically/genetically defined subtypes (Ichinose et al., 2014). The substantial overlap between the two groups may indicate that multiple BC subtypes populate each group and/or that individual subtypes may exhibit broad contrast tuning. No correlation was observed between contrast tuning and other calcium response characteristics measured (rise time, peak amplitude, transience; Fig. 10D4–6). Differential connectivity between ON BC subtypes and SACs and DSGCs may therefore explain the distinct contrast tuning of the postsynaptic cells. Together, our results suggest that SACs receive some of their input from more contrast-sensitive BCs than do DSGCs.
Figure 10.

Contrast sensitivity in BCs is correlated with morphology and but not with the shape of the calcium response. A, Schematic of the morphological (top) and functional (calcium transients; bottom) parameters analyzed in BCs. B, C, BCs were divided into two classes based on the similarity of their half peak amplitude response to the responses in SACs (B; 12 BCs) or in DRGCs (C; 18 BCs). Left, The mean stratification level of the BC axonal tree in the IPL. Vertical bars = axonal field area (x, y); horizontal bars = (z-axis) span in IPL of the axonal terminals, grayscale coded by the contrast level of the half-peak amplitude response. Middle, Calcium responses in BCs (black, mean; gray, SD) evoked by a moving bar stimulus (170% contrast). Gray bar indicates the approximate time interval over which the stimulus occupied the cells' receptive fields. Error bars, 20% dF/F, 500 ms. Right, Normalized contrast sensitivity (mean ± SD) for each BC. D, Left, Correlation between morphological and functional parameters to the half-maximal contrast level (C[1/2]), color coded by the two BC groups and fitted by a liner regression (note the logarithmic scale of the bottom axis). Right, Between-class comparison (mean ± SD; *p < 0.05, t test with Bonferroni correction for multiple comparisons). The following parameters were compared: mean stratification level of the BC axonal tree in the IPL (D1), z-axis span of the axonal terminals in the IPL (D2), x–y-axis area of the axonal tree (D3), rise time of the calcium response (D4), peak change in fluorescence (recorded at 170% contrast; D5), and ratio of the transient and sustained component of the calcium response waveform (D6).
Discussion
Here we have compared the contrast tuning of different elements within the DS circuitry of the mouse retina. We found that excitatory and inhibitory synaptic inputs to mouse DSGCs remain balanced over a broad stimulus range (Fig. 2), thereby enabling the circuit to compute motion independently of contrast (Figs. 1, 3). This balance occurs because BC inputs to SACs are more sensitive than those onto DSGCs (Figs. 6, 7), effectively compensating for the nonlinear I/O characteristics of SACs (Fig. 6) so that GABAergic and glutamatergic inputs to DSGCs exhibit similar contrast tuning. The varied contrast sensitivity of BC inputs likely reflects the contribution of multiple ON BC subtypes to DS signaling (Figs. 8–10; Helmstaedter et al., 2013; Greene et al., 2016).
Responses to visual motion stimuli were recorded using several different approaches, each with their own caveats. Thin processes and remote synaptic inputs likely compromise spatial control of SAC and DSGC dendrites during voltage-clamp recordings (Poleg-Polsky and Diamond, 2011; Vaney et al., 2012), and key features of visual signaling in SAC dendrites are often obscured in somatic responses (Euler et al., 2002). In addition, calcium responses depend critically on the kinetics and affinity of the indicator dyes, and iGluSnFR recordings may present uncertainties about synapse specificity. In several cases, however, different approaches corroborated each other. For example, the difference in contrast sensitivity of iGluSnFR responses in SACs and DSGCs (Fig. 7F) was analogous to that observed in EPSCs (Fig. 5C), and calcium signals in SAC dendrites exhibited similar contrast tuning to IPSCs recorded in DSGCs (Fig. 6F). Similarly, the contrast sensitivity of calcium signals in a population of BC terminals spanned the range between the distinct tuning of SAC and DSGC EPSCs (Fig. 9A). Together then, these results constitute a cohesive explanation for how the retina computes DS robustly over a wide contrast range.
Other features of the data suggest interesting complexities that, for the time being, remain obscure. For example, light-evoked responses in SAC somata exhibit a pronounced slower component (Figs. 5, 6) that is less apparent in the time course of dendritic calcium signals or GABA release. Future experiments may reveal whether this component contributes to some other aspect of SAC signaling.
The DS network is a feedforward circuit
The DS circuitry is a well studied example of FF inhibition, a common circuit motif throughout the brain (Lawrence and McBain, 2003; Alonso and Swadlow, 2005; Silberberg, 2008; Ehrlich et al., 2009; Jörntell et al., 2010; Bruno, 2011). In the DS FF circuit, presynaptic neurons (BCs) excite interneurons (SACs) and principal neurons (DSGCs), and SACs convey FF inhibitory signals to DSGCs. Computation in many FF inhibitory circuits depends on E/I balance (Anderson et al., 2000; Shu et al., 2003; Wehr and Zador, 2003; Zhang et al., 2003; Haider et al., 2006; Poo and Isaacson, 2009; Isaacson and Scanziani, 2011; Xue et al., 2014), similar to the processing in retinal DSGCs (Fried et al., 2002; Vaney et al., 2012). As interneurons typically filter the presynaptic signal nonlinearly, for example, by introducing a threshold, the E/I ratio in principal cells may vary with stimulus intensity. Preferential recruitment of interneurons at low stimulation levels is a general feature of FF inhibition (Hull et al., 2009; Pouille et al., 2009; Atallah et al., 2012), as reflected within the DS circuitry by the increased contrast sensitivity of SACs relative to DSGCs.
The importance of E/I balance
The present results suggest that balanced synaptic inputs are important for shaping DS responses in DSGCs. Our model, built upon the assumption that GABAergic input from SACs provides the sole source of DS tuning, revealed that balanced E/I drive outperforms other scenarios. Specifically, when the contrast sensitivity of inhibitory inputs was reduced, DS was diminished over a wide contrast range (Fig. 3): because inhibition is not present at low contrast levels, no directional information is provided to the DSGC. Interestingly, shifting inhibition to be more contrast-sensitive promoted strong DS in subthreshold DSGC responses, but suprathreshold responses underperformed compared with the balanced E/I case because the pronounced inhibitory drive prevented firing at low contrasts (Fig. 3).
Our interpretation of these simulations is tempered by the recognition that, although SAC-derived GABAergic inhibition is an important source of DS tuning, it may not be the only one. A recent study suggested that SACs also provide DS cholinergic input to DSGCs (Pei et al., 2015). An excitatory DS signal would reduce the requirement for precise E/I balance, as DS excitation could compensate for a mismatched inhibitory drive. Interestingly, in rabbit release of ACh and GABA from SACs is mediated by distinct calcium channel types (Lee et al., 2010) and may exhibit distinct contrast sensitivity (Lipin et al., 2015). We did not detect differences between the contrast sensitivity of IPSCs and the various EPSC components in DSGCs (Fig. 4), a discrepancy that may reflect interspecies variation or different experimental conditions.
Mechanisms to maintain E/I balance
Theoretically, several different compensatory mechanisms could maintain a consistent E/I balance. In somatosensory cortex, for example, principal cells and interneurons express distinct glutamate receptors and receive different numbers of synaptic inputs, causing them to respond distinctly to thalamocortical input and possibly compensating for nonlinear processing in interneurons (Bruno and Simons, 2002; Gabernet et al., 2005; Cruikshank et al., 2007; Hull et al., 2009). Our results suggest that, although such differences may exist between SACs and DSGCs, a major feature that distinguishes FF excitation and inhibition in DS circuitry is contrast sensitivity. If the differences in SAC and DSGC activation were due primarily to postsynaptic mechanisms or the number of synaptic inputs, one would expect both cell types to respond over a similar stimulus range. Instead, we observed no detectable responses in DSGCs over a low-contrast range that consistently evoked responses in SACs (Figs. 2, 5).
Alternatively, the E/I balance would be conserved if both excitation and inhibition were mediated by the FF arms of the circuit. In the case of DSGCs, excitation comes from multiple presynaptic sources, including BCs, SACs, and vGluT3 amacrine cells (Lee et al., 2014). Theoretically, if DSGCs did not receive direct excitation from BCs, all remaining synaptic inputs would originate in amacrine cells and potentially exhibit comparable contrast sensitivity. The fact that BCs do provide a sizable portion of excitatory input (Lee et al., 2014) indicates an important role for BCs in DS signaling. We recently showed that glutamatergic and GABAergic inputs to DSGCs interact multiplicatively, thereby increasing excitatory gain while preserving DS and enhancing motion discrimination in noisy visual conditions (Poleg-Polsky and Diamond, 2016). We speculate that BC inputs exhibit different noise characteristics compared with amacrine cell-mediated inputs, enabling the system to reliably extract correlated signals in these two parallel pathways (Cafaro and Rieke, 2010).
The role of BCs in DS computation
Recent anatomical data indicate that DSGCs sample from CB3-5 cells, whereas SACs receive inputs from CB1-7 (Helmstaedter et al., 2013; Greene et al., 2016). Functional mapping of presynaptic partners confirmed CB5 input to ON DSGCs (Yonehara et al., 2013), although a different study suggested that CB7 also contributes (Shi et al., 2011), potentially conflicting findings that may reflect DSGC diversity (Vaney et al., 2012). We found that axon terminals within an individual BC exhibit similar contrast sensitivity (Fig. 8). Thus, if an individual BC contacted both SACs and DSGCs, it should provide similar contrast information to both cell types. We predict, therefore, that at least some of the BC input is not shared between SACs and DSGCs.
Consistent with SACs sampling from multiple BC subtypes, we observed two contrast tuning channels in SACs: the transient component of EPSCs and PSPs was more sensitive and easily saturated than the slower, more sustained component (Fig. 5). This tuning may indicate that SACs receive input from kinetically distinct BC subtypes (Kim et al., 2014; Greene et al., 2016), although we observed no correlation between BC response kinetics and contrast sensitivity (Fig. 10). Distinct BC contrast tuning likely contributes to the wider contrast tuning range in SACs (Figs. 5, 6).
Contrast sensitivity of BCs
What mechanisms could underlie differential contrast tuning between BC subtypes? Although all ON BCs detect photoreceptor signals via the same metabotropic glutamate receptor (mGluR6), different ON BC subtypes exhibit distinct signaling dynamics due to differences in post-receptor signaling, voltage-gated channel expression, and feedback inhibition in the IPL (Euler and Masland, 2000; Protti et al., 2000; Xu et al., 2008; Baden et al., 2011; Dreosti et al., 2011; Odermatt et al., 2012; Baden et al., 2013b; Euler et al., 2014). Future experiments will determine how each of these mechanisms contributes to BC contrast sensitivity.
A wide range in contrast sensitivity between different cell types in the retina likely enriches the neural representation of the visual world (Odermatt et al., 2012; Baden et al., 2013a; Ichinose et al., 2014), Here we propose an additional functional role for differential BC tuning in DS signaling. Given that many ganglion cells receive FF inhibitory input, differential contrast tuning between BC subtypes may compensate for nonlinear processing in amacrine cells in other circuits within the retina.
Footnotes
This work was supported by the NINDS Intramural Research Program (NS003145). We thank Hua Tian for mouse husbandry and genotyping support, and Huayu Ding and Dr Cole Graydon for discussions.
The authors declare no competing financial interests.
References
- Alonso JM, Swadlow HA. Thalamocortical specificity and the synthesis of sensory cortical receptive fields. J Neurophysiol. 2005;94:26–32. doi: 10.1152/jn.01281.2004. [DOI] [PubMed] [Google Scholar]
- Anderson JS, Carandini M, Ferster D. Orientation tuning of input conductance, excitation, and inhibition in cat primary visual cortex. J Neurophysiol. 2000;84:909–926. doi: 10.1152/jn.2000.84.2.909. [DOI] [PubMed] [Google Scholar]
- Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. 2012;73:159–170. doi: 10.1016/j.neuron.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden T, Esposti F, Nikolaev A, Lagnado L. Spikes in retinal bipolar cells phase-lock to visual stimuli with millisecond precision. Curr Biol. 2011;21:1859–1869. doi: 10.1016/j.cub.2011.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden T, Euler T, Weckström M, Lagnado L. Spikes and ribbon synapses in early vision. Trends Neurosci. 2013a;36:480–488. doi: 10.1016/j.tins.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Baden T, Berens P, Bethge M, Euler T. Spikes in mammalian bipolar cells support temporal layering of the inner retina. Curr Biol. 2013b;23:48–52. doi: 10.1016/j.cub.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Barlow HB, Hill RM. Selective sensitivity to direction of movement in ganglion cells of the rabbit retina. Science. 1963;139:412–414. doi: 10.1126/science.139.3553.412. [DOI] [PubMed] [Google Scholar]
- Briggman KL, Helmstaedter M, Denk W. Wiring specificity in the direction-selectivity circuit of the retina. Nature. 2011;471:183–188. doi: 10.1038/nature09818. [DOI] [PubMed] [Google Scholar]
- Bruno RM. Synchrony in sensation. Curr Opin Neurobiol. 2011;21:701–708. doi: 10.1016/j.conb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno RM, Simons DJ. Feedforward mechanisms of excitatory and inhibitory cortical receptive fields. J Neurosci. 2002;22:10966–10975. doi: 10.1523/JNEUROSCI.22-24-10966.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafaro J, Rieke F. Noise correlations improve response fidelity and stimulus encoding. Nature. 2010;468:964–967. doi: 10.1038/nature09570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Lee S, Park SJ, Looger LL, Zhou ZJ. Receptive field properties of bipolar cell axon terminals in the direction-selective sublaminas of the mouse retina. J Neurophysiol. 2014;112:1950–1962. doi: 10.1152/jn.00283.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiao CC, Masland RH. Starburst cells nondirectionally facilitate the responses of direction-selective retinal ganglion cells. J Neurosci. 2002;22:10509–10513. doi: 10.1523/JNEUROSCI.22-24-10509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ED. Voltage-gated calcium and sodium currents of starburst amacrine cells in the rabbit retina. Vis Neurosci. 2001;18:799–809. doi: 10.1017/S0952523801185135. [DOI] [PubMed] [Google Scholar]
- Cohen ED, Miller RF. Quinoxalines block the mechanism of directional selectivity in ganglion cells of the rabbit retina. Proc Natl Acad Sci U S A. 1995;92:1127–1131. doi: 10.1073/pnas.92.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Demb JB. Cellular mechanisms for direction selectivity in the retina. Neuron. 2007;55:179–186. doi: 10.1016/j.neuron.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Dreosti E, Esposti F, Baden T, Lagnado L. In vivo evidence that retinal bipolar cells generate spikes modulated by light. Nat Neurosci. 2011;14:951–952. doi: 10.1038/nn.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Krishnaswamy A, De la Huerta I, Sanes JR. Type II cadherins guide assembly of a direction-selective retinal circuit. Cell. 2014;158:793–807. doi: 10.1016/j.cell.2014.06.047. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Euler T, Masland RH. Light-evoked responses of bipolar cells in a mammalian retina. J Neurophysiol. 2000;83:1817–1829. doi: 10.1152/jn.2000.83.4.1817. [DOI] [PubMed] [Google Scholar]
- Euler T, Detwiler PB, Denk W. Directionally selective calcium signals in dendrites of starburst amacrine cells. Nature. 2002;418:845–852. doi: 10.1038/nature00931. [DOI] [PubMed] [Google Scholar]
- Euler T, Haverkamp S, Schubert T, Baden T. Retinal bipolar cells: elementary building blocks of vision. Nat Rev Neurosci. 2014;15:507–519. doi: 10.1038/nrn3783. [DOI] [PubMed] [Google Scholar]
- Famiglietti EV. Synaptic organization of starburst amacrine cells in rabbit retina: analysis of serial thin sections by electron microscopy and graphic reconstruction. J Comp Neurol. 1991;309:40–70. doi: 10.1002/cne.903090105. [DOI] [PubMed] [Google Scholar]
- Field GD, Rieke F. Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron. 2002;34:773–785. doi: 10.1016/S0896-6273(02)00700-6. [DOI] [PubMed] [Google Scholar]
- Fried SI, Münch TA, Werblin FS. Mechanisms and circuitry underlying directional selectivity in the retina. Nature. 2002;420:411–414. doi: 10.1038/nature01179. [DOI] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, Scanziani M. Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron. 2005;48:315–327. doi: 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Ghosh KK, Bujan S, Haverkamp S, Feigenspan A, Wässle H. Types of bipolar cells in the mouse retina. J Comp Neurol. 2004;469:70–82. doi: 10.1002/cne.10985. [DOI] [PubMed] [Google Scholar]
- Greene MJ, Kim JS, Seung HS, EyeWirers Analogous convergence of sustained and transient inputs in parallel on and off pathways for retinal motion computation. Cell Rep. 2016;14:1892–1900. doi: 10.1016/j.celrep.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzywacz NM, Merwine DK, Amthor FR. Complementary roles of two excitatory pathways in retinal directional selectivity. Vis Neurosci. 1998;15:1119–1127. doi: 10.1017/s0952523898156109. [DOI] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausselt SE, Euler T, Detwiler PB, Denk W. A dendrite-autonomous mechanism for direction selectivity in retinal starburst amacrine cells. PLoS Biol. 2007;5:e185. doi: 10.1371/journal.pbio.0050185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter M, Briggman KL, Turaga SC, Jain V, Seung HS, Denk W. Connectomic reconstruction of the inner plexiform layer in the mouse retina. Nature. 2013;500:168–174. doi: 10.1038/nature12346. [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997;9:1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- Hoggarth A, McLaughlin AJ, Ronellenfitch K, Trenholm S, Vasandani R, Sethuramanujam S, Schwab D, Briggman KL, Awatramani GB. Specific wiring of distinct amacrine cells in the directionally selective retinal circuit permits independent coding of direction and size. Neuron. 2015;86:276–291. doi: 10.1016/j.neuron.2015.02.035. [DOI] [PubMed] [Google Scholar]
- Huberman AD, Wei W, Elstrott J, Stafford BK, Feller MB, Barres BA. Genetic identification of an on–off direction-selective retinal ganglion cell subtype reveals a layer-specific subcortical map of posterior motion. Neuron. 2009;62:327–334. doi: 10.1016/j.neuron.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Isaacson JS, Scanziani M. Postsynaptic mechanisms govern the differential excitation of cortical neurons by thalamic inputs. J Neurosci. 2009;29:9127–9136. doi: 10.1523/JNEUROSCI.5971-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose T, Fyk-Kolodziej B, Cohn J. Roles of on cone bipolar cell subtypes in temporal coding in the mouse retina. J Neurosci. 2014;34:8761–8771. doi: 10.1523/JNEUROSCI.3965-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahr CE, Stevens CF. Voltage dependence of NMDA-activated macroscopic conductances predicted by single-channel kinetics. J Neurosci. 1990;10:3178–3182. doi: 10.1523/JNEUROSCI.10-09-03178.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon CJ, Kong JH, Strettoi E, Rockhill R, Stasheff SF, Masland RH. Pattern of synaptic excitation and inhibition upon direction-selective retinal ganglion cells. J Comp Neurol. 2002;449:195–205. doi: 10.1002/cne.10288. [DOI] [PubMed] [Google Scholar]
- Jörntell H, Bengtsson F, Schonewille M, De Zeeuw CI. Cerebellar molecular layer interneurons: computational properties and roles in learning. Trends Neurosci. 2010;33:524–532. doi: 10.1016/j.tins.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Kaneda M, Ito K, Morishima Y, Shigematsu Y, Shimoda Y. Characterization of voltage-gated ionic channels in cholinergic amacrine cells in the mouse retina. J Neurophysiol. 2007;97:4225–4234. doi: 10.1152/jn.01022.2006. [DOI] [PubMed] [Google Scholar]
- Kim JS, Greene MJ, Zlateski A, Lee K, Richardson M, Turaga SC, Purcaro M, Balkam M, Robinson A, Behabadi BF, Campos M, Denk W, Seung HS, EyeWirers Space-time wiring specificity supports direction selectivity in the retina. Nature. 2014;509:331–336. doi: 10.1038/nature13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittila CA, Massey SC. Pharmacology of directionally selective ganglion cells in the rabbit retina. J Neurophysiol. 1997;77:675–689. doi: 10.1152/jn.1997.77.2.675. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, McBain CJ. Interneuron diversity series: containing the detonation–feedforward inhibition in the CA3 hippocampus. Trends Neurosci. 2003;26:631–640. doi: 10.1016/j.tins.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Lee S, Kim K, Zhou ZJ. Role of ACh-GABA cotransmission in detecting image motion and motion direction. Neuron. 2010;68:1159–1172. doi: 10.1016/j.neuron.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Chen L, Chen M, Ye M, Seal RP, Zhou ZJ. An unconventional glutamatergic circuit in the retina formed by vGluT3 amacrine cells. Neuron. 2014;84:708–715. doi: 10.1016/j.neuron.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linaro D, Storace M, Giugliano M. Accurate and fast simulation of channel noise in conductance-based model neurons by diffusion approximation. PLoS Comput Biol. 2011;7:e1001102. doi: 10.1371/journal.pcbi.1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipin MY, Taylor WR, Smith RG. Inhibitory input to the direction selective ganglion cells is saturated at low contrast. J Neurophysiol. 2015;114:927–941. doi: 10.1152/jn.00413.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen TW, Bargmann CI, Orger MB, Schreiter ER, Demb JB, Gan WB, Hires SA, Looger LL. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods. 2013;10:162–170. doi: 10.1038/nmeth.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov SS, Kholodenko R, Lem J, Pugh EN., Jr Physiological features of the S- and M-cone photoreceptors of wild-type mice from single-cell recordings. J Gen Physiol. 2006;127:359–374. doi: 10.1085/jgp.200609490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odermatt B, Nikolaev A, Lagnado L. Encoding of luminance and contrast by linear and nonlinear synapses in the retina. Neuron. 2012;73:758–773. doi: 10.1016/j.neuron.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Kim IJ, Looger LL, Demb JB, Borghuis BG. Excitatory synaptic inputs to mouse on–off direction-selective retinal ganglion cells lack direction tuning. J Neurosci. 2014;34:3976–3981. doi: 10.1523/JNEUROSCI.5017-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Z, Chen Q, Koren D, Giammarinaro B, Acaron Ledesma H, Wei W. Conditional knock-out of vesicular GABA transporter gene from starburst amacrine cells reveals the contributions of multiple synaptic mechanisms underlying direction selectivity in the retina. J Neurosci. 2015;35:13219–13232. doi: 10.1523/JNEUROSCI.0933-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleg-Polsky A, Diamond JS. Imperfect space clamp permits electrotonic interactions between inhibitory and excitatory synaptic conductances, distorting voltage clamp recordings. PLoS One. 2011;6:e19463. doi: 10.1371/journal.pone.0019463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleg-Polsky A, Diamond JS. NMDA receptors multiplicatively scale visual signals and enhance directional motion discrimination in retinal ganglion cells. Neuron. 2016;89:1277–1290. doi: 10.1016/j.neuron.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo C, Isaacson JS. Odor representations in olfactory cortex: “sparse” coding, global inhibition, and oscillations. Neuron. 2009;62:850–861. doi: 10.1016/j.neuron.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Marin-Burgin A, Adesnik H, Atallah BV, Scanziani M. Input normalization by global feedforward inhibition expands cortical dynamic range. Nat Neurosci. 2009;12:1577–1585. doi: 10.1038/nn.2441. [DOI] [PubMed] [Google Scholar]
- Protti DA, Flores-Herr N, von Gersdorff H. Light evokes Ca2+ spikes in the axon terminal of a retinal bipolar cell. Neuron. 2000;25:215–227. doi: 10.1016/S0896-6273(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Shi Z, Trenholm S, Zhu M, Buddingh S, Star EN, Awatramani GB, Chow RL. Vsx1 regulates terminal differentiation of type 7 ON bipolar cells. J Neurosci. 2011;31:13118–13127. doi: 10.1523/JNEUROSCI.2331-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Silberberg G. Polysynaptic subcircuits in the neocortex: spatial and temporal diversity. Curr Opin Neurobiol. 2008;18:332–337. doi: 10.1016/j.conb.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Vesicle depletion and synaptic depression at a mammalian ribbon synapse. J Neurophysiol. 2006;95:3191–3198. doi: 10.1152/jn.01309.2005. [DOI] [PubMed] [Google Scholar]
- Stafford BK, Park SJ, Wong KY, Demb JB. Developmental changes in NMDA receptor subunit composition at ON and OFF bipolar cell synapses onto direction-selective retinal ganglion cells. J Neurosci. 2014;34:1942–1948. doi: 10.1523/JNEUROSCI.4461-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WR, Vaney DI. Diverse synaptic mechanisms generate direction selectivity in the rabbit retina. J Neurosci. 2002;22:7712–7720. doi: 10.1523/JNEUROSCI.22-17-07712.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjepkes DS, Amthor FR. The role of NMDA channels in rabbit retinal directional selectivity. Vis Neurosci. 2000;17:291–302. doi: 10.1017/s0952523800172128. [DOI] [PubMed] [Google Scholar]
- Trenholm S, Johnson K, Li X, Smith RG, Awatramani GB. Parallel mechanisms encode direction in the retina. Neuron. 2011;71:683–694. doi: 10.1016/j.neuron.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaney DI, Sivyer B, Taylor WR. Direction selectivity in the retina: symmetry and asymmetry in structure and function. Nat Rev Neurosci. 2012;13:194–208. doi: 10.1038/nrn3165. [DOI] [PubMed] [Google Scholar]
- Vlasits AL, Morrie RD, Tran-Van-Minh A, Bleckert A, Gainer CF, DiGregorio DA, Feller MB. A role for synaptic input distribution in a dendritic computation of motion direction in the retina. Neuron. 2016;89:1317–1330. doi: 10.1016/j.neuron.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–446. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- Wei W, Hamby AM, Zhou K, Feller MB. Development of asymmetric inhibition underlying direction selectivity in the retina. Nature. 2011;469:402–406. doi: 10.1038/nature09600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Sulaiman P, Feddersen RM, Liu J, Smith RG, Vardi N. Retinal ON bipolar cells express a new PCP2 splice variant that accelerates the light response. J Neurosci. 2008;28:8873–8884. doi: 10.1523/JNEUROSCI.0812-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Atallah BV, Scanziani M. Equalizing excitation–inhibition ratios across visual cortical neurons. Nature. 2014;511:596–600. doi: 10.1038/nature13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonehara K, Balint K, Noda M, Nagel G, Bamberg E, Roska B. Spatially asymmetric reorganization of inhibition establishes a motion-sensitive circuit. Nature. 2011;469:407–410. doi: 10.1038/nature09711. [DOI] [PubMed] [Google Scholar]
- Yonehara K, Farrow K, Ghanem A, Hillier D, Balint K, Teixeira M, Jüttner J, Noda M, Neve RL, Conzelmann KK, Roska B. The first stage of cardinal direction selectivity is localized to the dendrites of retinal ganglion cells. Neuron. 2013;79:1078–1085. doi: 10.1016/j.neuron.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Tan AY, Schreiner CE, Merzenich MM. Topography and synaptic shaping of direction selectivity in primary auditory cortex. Nature. 2003;424:201–205. doi: 10.1038/nature01796. [DOI] [PubMed] [Google Scholar]
- Zhou ZJ, Lee S. Synaptic physiology of direction selectivity in the retina. J Physiol. 2008;586:4371–4376. doi: 10.1113/jphysiol.2008.159020. [DOI] [PMC free article] [PubMed] [Google Scholar]