

Abstract
The brainstem contains diverse neuronal populations that regulate a wide range of processes vital to the organism. Proper cell-fate specification decisions are critical to achieve neuronal diversity in the CNS, but the mechanisms regulating cell-fate specification in the developing brainstem are poorly understood. Previously, it has been shown that basic helix-loop-helix transcription factor Ptf1a is required for the differentiation and survival of neurons of the inferior olivary and cochlear brainstem nuclei, which contribute to motor coordination and sound processing, respectively. In this study, we show that the loss of Ptf1a compromises the development of the nucleus of the solitary tract, which processes viscerosensory information, and the spinal and principal trigeminal nuclei, which integrate somatosensory information of the face. Combining genetic fate-mapping, birth-dating, and gene expression studies, we found that at least a subset of brainstem abnormalities in Ptf1a−/− mice are mediated by a dramatic cell-fate misspecification in rhombomeres 2–7, which results in the production of supernumerary viscerosensory and somatosensory neurons of the Lmx1b lineage at the expense of Pax2+ GABAergic viscerosensory and somatosensory neurons, and inferior olivary neurons. Our data identify Ptf1a as a major regulator of cell-fate specification decisions in the developing brainstem, and as a previously unrecognized developmental regulator of both viscerosensory and somatosensory brainstem nuclei.
SIGNIFICANCE STATEMENT Cell-fate specification decisions are critical for normal CNS development. Although extensively studied in the cerebellum and spinal cord, the mechanisms mediating cell-fate decisions in the brainstem, which regulates a wide range of processes vital to the organism, remain largely unknown. Here we identified mouse Ptf1a as a novel regulator of cell-fate decisions during both early and late brainstem neurogenesis, which are critical for proper development of several major classes of brainstem cells, including neurons of the somatosensory and viscerosensory nuclei. Since loss-of-function PTF1A mutations were described in human patients, we suggest Ptf1a-dependent cell-fate misspecification as a novel mechanism of human brainstem pathology.
Keywords: cell-fate specification, hindbrain, human mid-hindbrain malformation disorders, mouse, neuronal progenitors
Introduction
Brainstem neurons control several vital body functions. In particular, neurons of the nucleus of the solitary tract (nTs) integrate viscerocensory information regulating the activity of the respiratory, cardiovascular, and digestive organs; principal sensory trigeminal nuclei (PrV) and spinal trigeminal nuclei (SpV) process and relay somatosensory information of the face; inferior olivary nuclei (ION) contribute to motor coordination; and cochlear nuclei process sounds (Woolsey, 1990; Ruigrok and Cella, 1995; Blessing, 1997; Pickles, 2015). Previous studies revealed that at least a subset of nTs, PrV, SpV, ION, and cochlear neurons arise from ventricular zone (VZ) progenitors in the alar plate of early rhombomeres (rh) 2–7, and as development proceeds, migrate to their final destinations (Farago et al., 2006; Oury et al., 2006; Yamada et al., 2007; Fujiyama et al., 2009; D'Autréaux et al., 2011; Gray, 2013). The mechanisms regulating the development of dorsally derived brainstem neurons, including the mechanisms of their cell-fate specification, however, remain poorly understood.
Based on the expression of transcription factors, eight newborn neuronal populations, named dA1–4 and dB1–4, were described in dorsal mouse rh2–7 at embryonic days (E) 10.5–11.5, at the beginning of neurogenesis in the dorsal hindbrain (Fig. 1A–D; Sieber et al., 2007). Some of these populations, such as dA4 and dB1, extend through rh2–7, while others, such as dA3, are present only in some rhombomeres (Sieber et al., 2007; Storm et al., 2009). In contrast to rh2–6, where dA/dB neurons arise throughout neurogenesis, in rh7, dBLa and dBLb neurons arise in a salt-and-pepper pattern from the dorsal VZ after E12 (Fig. 1E; Sieber et al., 2007). Fate-mapping and gene expression studies identified dA3 cells as differentiating Lmx1b+/Phox2b+ neurons of the nTS, dA4 cells of the caudal brainstem as newborn ION neurons, and dBLb cells as Lmx1b+ SpV neurons (Sieber et al., 2007; Storm et al., 2009; D'Autréaux et al., 2011), while the eventual fates of most other dA/dB populations, as well as dBLa cells, remain unknown.
Figure 1.

Summary of neuronal subtypes arising during early and late waves of neurogenesis in rh2–7. A, Side view of E10.5–E11.5 hindbrain with dA and dB neuronal populations extending through rh2–7. Specific markers for some of these populations are shown based on the data in studies by Sieber et al., 2007 and Storm et al., 2009. The dorsal–ventral (D–V) and anterior–posterior (A–P) axes are labeled. B–D, Schematics of transverse sections of E10.5–E11.5 hindbrain taken at the level of rh2–3 (B), rh4–6 (C), and rh 7 (D). The dorsal part of each hemisection is shown. Ptf1a expression domain in the VZ, located adjacent to dA4 and dB1 neurons, is shown as a hatched area, based on the study by Sieber et al. (2007) and Storm et al. (2009). The D–V axis is labeled. E, Schematic of transverse section of E12.5 rh7, showing dBLa and dBLb neurons arising in a salt-and-pepper pattern from a broad domain of the dorsal VZ during late neurogenesis.
Recently, several regulators of hindbrain cell-fate decisions were identified, including homeodomain and basic helix-loop-helix (bHLH) transcription factors Phox2b, Olig3, and Lbx1, which are necessary for the specification of dA3 Lmx1b+/Phox2b+ nTs neurons, dA4 ION neurons, and dBLb Lmx1b+ SpV neurons, respectively (Sieber et al., 2007; Storm et al., 2009; D'Autréaux et al., 2011). Another bHLH transcription factor, Ptf1a, is transiently expressed in a restricted domain of the dorsal VZ, which extends through rh2–7 (Fig. 1B–D; Yamada et al., 2007, Fujiyama et al., 2009; Storm et al., 2009), and also in the cerebellum, which arises from dorsal rh1 (Hoshino et al., 2005). Loss of Ptf1a leads to cerebellar agenesis and prevents formation of the ION and dorsal cochlear nuclei. In Ptf1a−/− mice, an increased number of apoptotic cells was detected, suggesting that excessive cell death contributes to the development of hindbrain pathology in the absence of Ptf1a (Hoshino et al., 2005; Yamada et al., 2007; Fujiyama et al., 2009). More recently, however, we and others (Pascual et al., 2007; Millen et al., 2014; Yamada et al., 2014) found that Ptf1a−/− cerebellar agenesis is largely caused by a cell-fate misspecification in dorsal rh1. Currently, it remains poorly understood whether, in the hindbrain, Ptf1a regulates cell-fate decisions beyond rh1, and it is unknown whether this gene is required for the development of brainstem cells other than neurons of the ION and cochlear nuclei.
In this study, by performing genetic fate-mapping in mice, we identify Ptf1a as a previously unrecognized developmental regulator of both somatosensory (SpV and PrV) and viscerosensory (nTs) nuclei. Our analysis revealed that an early and widespread cell-fate misspecification in rh2–7 underlies at least some brainstem abnormalities in Ptf1a−/− mice. Since loss-of-function PTF1A mutations were described in human patients with hindbrain malformation disorders (Sellick et al., 2004), our data also suggest misspecification of neurons originating from the rh2–7 VZ as a novel mechanism of human brainstem pathology.
Materials and Methods
Animals.
We used Ptf1aCre (Kawaguchi et al., 2002), Ptf1aYFP (Burlison et al., 2008), ROSA26-LacZ (Soriano, 1999), ROSA26-YFP (Srinivas et al., 2001), Tau-nLacZ reporter [which labels neuronal progeny of Cre-expressing cells by nuclear β-galactosidase (β-gal) expression, referred to as nLacZ reporter throughout this article; Hippenmeyer et al., 2005], and Gad67-GFP (Chattopadhyaya et al., 2004) alleles. Because the Ptf1a protein-coding sequence is replaced by Cre or YFP in Ptf1aCre and Ptf1aYFP alleles, respectively (Kawaguchi et al., 2002; Burlison et al., 2008), they are Ptf1a-null alleles. In this study, we used Ptf1aCre/Cre and Ptf1aCre/YFP embryos for the analysis of hindbrain development and fate mapping of the progeny of Ptf1a-expressing cells in the absence of Ptf1a function. All mice used in this study were maintained on a mixed genetic background and were genotyped as previously described (Soriano, 1999; Srinivas et al., 2001; Kawaguchi et al., 2002; Hippenmeyer et al., 2005; Burlison et al., 2008). Noon of the day of the plug was designated as E0.5. For birth-dating experiments, pregnant mice were intraperitoneally injected with BrdU at a concentration of 50 mg/kg. Both male and female embryos were studied. All animal experiments were approved by the Institutional Animal Care and Use Committee.
Immunohistochemistry.
Immunohistochemistry was performed as previously described (Swanson et al., 2010; Millen et al., 2014). Briefly, embryos were fixed in 4% paraformaldehyde for 2–12 h at 4°C, sunk in 30% sucrose, embedded in optimum cutting temperature compound, and sectioned on a cryostat. Cryosections were washed in PBS, blocked in PBS containing 1% normal goat serum and 0.1% Triton X-100, and incubated with primary antibodies overnight at 4°C. Then, sections were washed in PBS and incubated with secondary antibodies for 1 h at room temperature. The following primary antibodies were used: rabbit anti-Pax2 (Zymed), anti-Phox2b (Pattyn et al., 1997), anti-Ptf1a (H. Edlund, University of Umea, Umea, Sweden), anti-activated caspase 3 (Promega), anti-β-galactosidase (M.P. Chapel), and anti-Tlx3 antibodies (M. Goulding, Salk Institute, La Jolla, CA), mouse anti-Lhx1/5 antibody (Developmental Studies Hybridoma Bank), goat anti-Lmx1a antibody (Santa Cruz Biotechnology), guinea pig anti-Lmx1b antibody (E. Hedlund and T. Perlmann, Karolinska Institute, Stockholm, Sweden), chick anti-β-galactosidase (Abcam), and anti-GFP antibody, which also recognizes yellow fluorescent protein (YFP; Abcam). Species-appropriate fluorescent dye-conjugated secondary antibodies were purchased from Life Technologies and Jackson ImmunoResearch.
BrdU-labeled cells were detected by immunohistochemistry with primary rat anti-BrdU (Abcam) and secondary goat anti-rat fluorescent dye-conjugated antibodies (Life Technologies). For BrdU detection, embryos were processed and sections were treated exactly as described for immunohistochemistry with other antibodies above, except before blocking in PBS with normal goat serum and Triton, sections were incubated in 2N HCl for 20 min at 37°C and rinsed in 0.1 m borate buffer, pH 8.5. Double or triple labeling using anti-BrdU, anti-β-galactosidase, and anti-Lmx1b or anti-Pax2 antibodies was performed sequentially. Sections were first incubated with anti-β-galactosidase and anti-Lmx1b or anti-Pax2 primary antibodies and their secondary antibodies. Then, sections were treated with HCl, and anti-BrdU antibodies were applied. Some sections were counterstained with DAPI (Sigma-Aldrich) to visualize cell nuclei. Images were taken using a Zeiss AxioImager Z1 microscope equipped with an AxioCam MRc camera, and figure panels were assembled in Photoshop.
Anatomy, measurements, and statistical analysis.
At least three embryos of each genotype at each developmental stage were analyzed. For each embryo, the entire hindbrain was tangentially serially sectioned at 12 μm, and all sections were evaluated under a microscope. Similar numbers of sections were placed on each slide to more easily identify the position of each section along the anterior–posterior axis of the hindbrain. Rhombomeric units were identified using morphological landmarks such as exit points of cranial nerves (Cordes, 2001) and hindbrain nuclei, as previously described (Sieber et al., 2007; Storm et al., 2009). In addition, molecular markers were used to confirm the rhombomeric identity of E11.5 sections. At E11.5, rh7 dorsal VZ contains Ngn1+ cells (dA2 progenitors) that are not present in more anterior rhombomeres (Landsberg et al., 2005, Yamada et al., 2007; Storm et al., 2009). E11.5 rh1 contains Lmx1a+ c3 cells that are not present in more posterior rhombomeres (Chizhikov et al., 2006). By staining every fifth serial section of caudal hindbrain with an anti-Ngn1 antibody and every fifth serial section of rostral hindbrain with an anti-Lmx1a antibody, we confirmed the rhombomeric identity of rh7 and rh2 sections selected for molecular analysis (data not shown). At E14.5, rh7 was defined as a hindbrain segment caudal to the facial nucleus, as previously described (Sieber et al., 2007). In Ptf1a−/− embryos, the position and extent of this nucleus along the anterior–posterior axis of the hindbrain was not affected (data not shown). To evaluate the positions of specific brainstem nuclei, every fifth serial section of the hindbrain was stained with cresyl violet (Schambra, 2008). Then, the remaining sections, spanning the entire brainstem along its anterior–posterior axis, were colabeled with antibodies to more specifically identify and analyze the PrV, nTs, and SpV nuclei (Sieber et al., 2007; Dai et al., 2008; Storm et al., 2009; Xiang et al., 2012), as described in the Results section. In Ptf1a mutants, the PrV, nTs, and SpV nuclei formed at appropriate positions in the brainstem, but, as described in the Results section, their neuronal composition was affected. For cell counts, sections from control and Ptf1a−/− littermates were carefully selected to ensure that they were taken from comparable anterior–posterior levels. For consistency, cell counts were performed on three sections per embryo, separated by 48 μm from each other. In most cases, we quantified the cells of interest using immunohistochemistry against nuclear markers (e.g., Lmx1b, Phox2b, Pax2, and nuclear β-gal), and cell numbers were determined by counting positive nuclei. To evaluate apoptosis, we quantified Casp3+ cells. All raw cell counts were corrected by multiplying the raw values by the Abercrombie (1946) correction factor, determined as T/T + h, where T is section thickness and h is mean nuclear diameter (or mean diameter of Casp3+ cell bodies). For each cell type, h was determined by measuring 30 cells from three different embryos of each genotype at each developmental stage, as previously described (Jahagirdar and Wagner, 2010). For clarity, cell counts in Ptf1a−/− embryos were normalized to the counts in control embryos, which were set as 1, as previously described (Huang et al., 2010; Sudarov et al., 2011; Millen et al., 2014). Quantitative data are expressed as the mean ± SD. Statistical significance was determined by two-tailed t test. p < 0.05 was considered to be statistically significant.
Results
In early Ptf1a−/− rh7, Ptf1a-expressing progenitors produce dA3 and dB3 neurons instead of dA4 and dB1 neurons
To investigate whether Ptf1a regulates cell-fate decisions in the hindbrain beyond rh1, we analyzed rh2–7 of Ptf1a−/− embryos beginning at E11.5, when early-born neurons are generated and were described as dA/dB populations (Fig. 1A–D; Sieber et al., 2007; Storm et al., 2009). Since cells arising from Ptf1a-expressing progenitors turn Ptf1a expression off once they initiate neuronal differentiation and exit the VZ (Yamada et al., 2007; Fujiyama et al., 2009; Storm et al., 2009), to visualize the Ptf1a lineage, we used Ptf1aCre/+;ROSA-YFP (control) and Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) embryos, in which Ptf1a-expressing progenitors in the hindbrain VZ and their progeny are permanently labeled with YFP expression from the ROSA-YFP allele (Srinivas et al., 2001).
In rh7 of control E11.5 Ptf1aCre/+;ROSA-YFP embryos, Lhx1/5+/Pax2− dA4 and Lhx1/5+/Pax2+ dB1 neurons were YFP+, supporting previous studies suggesting that these populations originate from Ptf1a-expressing progenitors (Fig. 2A,C; Storm et al., 2009). The vast majority of Lmx1b+/Phox2b+ dA3 neurons, which dorsally flank dA4 neurons, and Lmx1b+/Phox2b− dB3 neurons, which ventrally flank dB1 neurons (Figs. 1D, 2A,F; Sieber et al., 2007; Storm et al., 2009), were YFP− in control rh7 (Fig. 2J,k,m). Although in Ptf1aCre/+;ROSA-YFP rh7, we occasionally observed Lmx1b+/Phox2b+/YFP+ cells, the number of these cells was very small (a few cells per section; Fig. 2k–l″). Thus, during early neurogenesis, in wild-type rh7, Ptf1a-expressing progenitors mostly produce dA4 and dB1 neurons.
Figure 2.
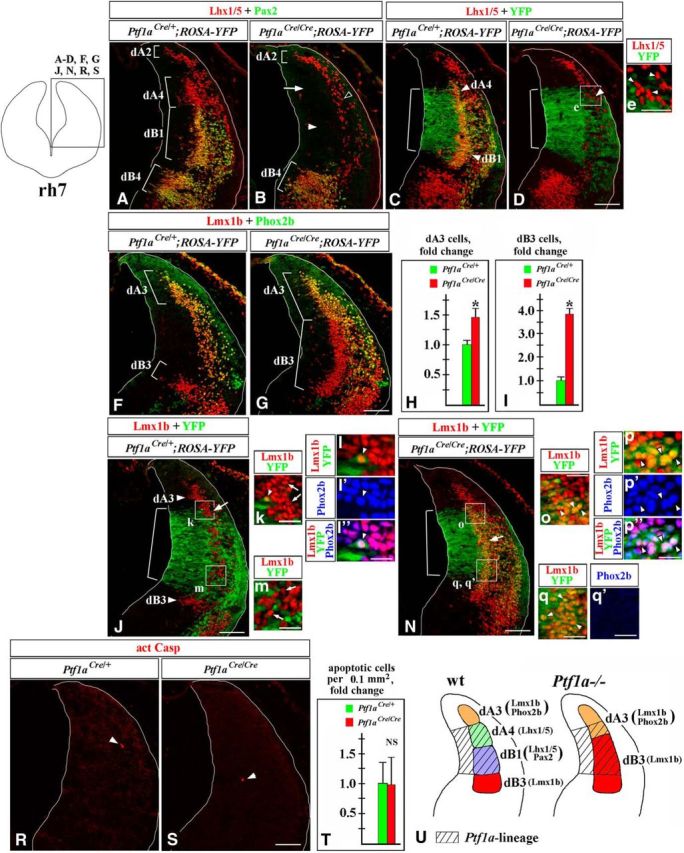
In E11.5 Ptf1a−/− rh7, Ptf1a-expressing progenitors produce dA3 and dB3 neurons instead of dA4 and dB1 neurons. Transverse sections of E11.5 rh7 with genotypes and antibody markers indicated. Low-magnification panels correspond to the region boxed in the rh7 schematic in the top left corner. Magnified regions (e, k, m, o, q, q′) correspond to areas boxed in related data panels. l–l″ and p–p″ show higher magnification of cells, pointed out by arrowheads in k and o, respectively. Brackets in A, B, F, and G indicate the positions along the dorsal–ventral axis at which specific neuronal populations emerge. Brackets in C, D, J, and N indicate the dorsal–ventral extent of YFP expression in the VZ, which was not significantly different between Ptf1aCre/+;ROSA-YFP and Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) embryos. A–D, Lhx1/5+/Pax2− dA4 and Lhx1/5+/Pax2+ dB1 cells (A) were YFP+ in control Ptf1aCre/+;ROSA-YFP embryos (C), and, therefore, originate from Ptf1a-expressing progenitors. B, In Ptf1a−/− rh7, neither dA4 (arrow) nor dB1 (arrowhead) are present. A small group of Lhx1/5+ cells present in lateral Ptf1a−/− rh7 (B, open arrowhead, D, arrowhead) was YFP− in Ptf1aCre/Cre;ROSA-YFP embryos (e, arrowheads), indicating that these cells do not originate from Ptf1a-expressing progenitors and, therefore, are likely ventrally migrated dA2 cells rather than dA4 cells. F, G, In Ptf1a−/− rh7, dA3 neurons expanded ventrally, while dB3 expanded dorsally. H, I, Quantification of dA3 and dB3 cells in Ptf1a−/− rh7 revealed a significant increase in the number of dA3 cells (p = 0.0016; H), and dB3 cells (p = 8.2 × 10−5; I) in Ptf1a−/− embryos relative to control littermates. J–l″, In Ptf1aCre/+;ROSA-YFP embryos, virtually all Lmx1b+ cells, including both dA3 and dB3 cells, were YFP−, indicating that a majority of these cells do not normally originate from Ptf1a-expressing progenitors (J, k, m, arrows). Rare Lmx1b+/YFP+ cells found in Ptf1aCre/+;ROSA-YFP control embryos (k, arrowhead) coexpressed Phox2b (l–l″) and, therefore, were dA3 cells. N–q′, In Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) embryos, many Lmx1b+ cells were YFP+ (N, arrow, o, q, arrowheads), indicating that they originated from Ptf1a-expressing progenitors. Some of these Lmx1b+/YFP+ cells, particularly those located adjacent to a dorsal segment of the YFP+ VZ, coexpressed Phox2b (p–p″, arrowheads), suggesting that they were dA3 cells. Lmx1b+/YFP+ cells, located adjacent to a ventral segment of the YFP+ VZ were Phox2b− (q, q′), adopting an expression profile of dB3 cells. R–T, Arrowheads point to Casp3+ cells. Quantification of apoptotic cells in the rh7 alar plate, where Ptf1a+ VZ progenitors and their newly generated progeny are located, revealed no significant difference between control and Ptf1a−/− embryos (cell counts were normalized to controls; NS, nonsignificant). U, Summary of cell-fate misspecification in early Ptf1a−/− rh7. In the absence of Ptf1a, Ptf1a-expressing progenitors in the VZ rather than producing dA4 and dB1 neurons, produce dA3 and dB3 neurons, which normally arise adjacent to cells belonging to the Ptf1a lineage. Scale bars: A–D, F, G, J, N, R, S, 100 μm; e, 40 μm; k, m, o, q, q′, 30 μm; l–l″, p–p″, 50 μm.
In contrast to wild-type embryos, Lhx1/5/Pax2 immunostaining did not reveal any dA4 or dB1 neurons in Ptf1a−/− rh7 (Fig. 2A,B, white arrowhead and arrow). Non-numerous Lhx1/5+ cells found in lateral rh7 of E11.5 Ptf1a−/− mutants (Fig. 2B, open arrowhead, D, arrowhead) were YFP− in Ptf1aCre/Cre ;ROSA-YFP embryos (Fig. 2e, arrowheads) and, therefore, likely, were ventrally migrated dA2 cells (which originate from Ptf1a− progenitors located dorsal to the Ptf1a-expressing VZ; Storm et al., 2009) rather than dA4 cells. Strikingly, in Ptf1a−/− rh7, dA3 cells expanded ventrally, while dB3 cells expanded dorsally (Fig. 2F,G), populating areas occupied by dA4 and dB1 neurons in control rh7 (Fig. 2A). Cell counts revealed a >3.5-fold increase in the number of dB3 cells (p = 8.2 × 10−5) and almost a 50% increase in the number of dA3 cells (p = 0.0016) in Ptf1a−/− embryos compared with control littermates (Fig. 2H,I). In contrast to control rh7, many Lmx1b+ cells were YFP+ in Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) embryos, arguing that in Ptf1a−/− mutants, excessive Lmx1b+ neurons originate from Ptf1a-expressing progenitors (Fig. 2J,N, arrow, o,q, arrowheads). In Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) embryos, some Lmx1b+/YFP+ cells, in particular those located within a dorsal part of the YFP+ domain, coexpressed Phox2b, and, therefore, resembled dA3 cells (Fig. 2N–p″). Many other Lmx1b+/YFP+ cells, specifically those located within a ventral part of the YFP+ domain, were Phox2b−, resembling dB3 cells (Fig. 2N,q,q′).
Previously, it has been shown that the loss of Ptf1a does not affect proliferation in the E11.5 rh7 VZ (Yamada et al., 2007). We found that apoptosis was not significantly increased in the Ptf1a−/− rh7 alar plate (Fig. 2R–T). Therefore, our data argue that in early Ptf1a−/− rh7, rather than producing dA4 and dB1 neurons, Ptf1a-expressing progenitors in the rh7 VZ produce cells molecularly resembling dA3 and dB3 neurons. In E11.5 Ptf1aCre/Cre;ROSA-YFP mutants, the ectopic Lmx1b+/Phox2b+/YPF+ cells were located adjacent to a dorsal segment of the Ptf1a+/YFP+ VZ (Fig. 2N–q′), which normally gives rise to dA4 neurons (Fig. 2A,C; Sieber et al., 2007, Storm et al., 2009). Thus, it is likely that in the absence of Ptf1a at least some dorsally located Ptf1a-expressing VZ progenitors produce dA3-like cells instead of dA4 neurons, while more ventrally located Ptf1a-expressing progenitors aberrantly generate Lmx1b+/Phox2b− dB3-like neurons (summarized in Fig. 2U).
Cell-fate misspecification in early Ptf1a−/− rh2–6
Next, we analyzed rh2–6 at E11.5. In rh4, Lhx1/5+ dA4 and Lhx1/5+/Pax2+ dB1 cells, normally arising from Ptf1a-expressing progenitors, did not appear in Ptf1a−/− mutants (Fig. 3A–E). Instead, as in Ptf1a−/− rh7, in Ptf1a−/− rh4, Lmx1b+/Phox2b+ dA3 cells expanded ventrally and were increased in number (by ∼70%; p = 0.003; Fig. 3F–H). Compared with rh7, in rh4–6 of wild-type E11.5 embryos, an additional population, Phox2b+/Lmx1b− dB2 cells, arises between dB1 and dB3 neurons (Figs. 1C, 3F; Sieber et al., 2007, 2009). Thus, in wild-type rh4, dB1 cells are ventrally flanked by dB2 rather dB3 cells. Unexpectedly, in Ptf1a−/− rh4, Phox2b+ dB2 cells did not expand dorsally (Fig. 3F–G). Instead, ectopic dB3-like Lmx1b+/Phox2b− cells appeared dorsal to the band of dB2 cells (Fig. 3G, arrow). Analysis of embryos carrying Ptf1aCre/ROSA-YFP fate-mapping alleles revealed that in contrast to control rh4, where very few YFP+/Lmx1b+ cells were detected (Fig. 3I–l), in Ptf1a−/− rh4, many YFP+ cells coexpressed Lmx1b (Fig. 3M, arrow, n). Some of these Lmx1b+/YFP+ cells, in particular those located within a dorsal part of the YFP+ domain, coexpressed Phox2b, adopting a gene expression profile of dA3 cells (Fig. 3M, n–p, arrowheads). Other Lmx1b+/YFP+ cells were Phox2b−, corresponding to ectopic dB3-like cells [Fig. 3M, n-p, arrows (summarized in Fig. 3Q)].
Figure 3.

Similar to rh7, in E11.5 Ptf1a−/− rh4, early-born dA4 and dB1 neurons transfate to dA3 and dB3 neurons. Transverse sections of E11.5 hindbrain taken at the level of rh4 with genotypes and antibody markers indicated. B–G, I, and M correspond to the region boxed in rh4 schematic in A. Brackets in B, C, F, and G indicate the positions along the dorsal–ventral axis at which specific neuronal populations emerge. Brackets in D, E, I, and M indicate the dorsal–ventral extent of YFP expression in the VZ, which was not dramatically different between Ptf1aCre/+;ROSA-YFP and Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) embryos. B–E, Lhx1/5+/Pax2− dA4 and Lhx1/5+/Pax2+ dB1 cells (B) were YFP+ in control Ptf1aCre/+;ROSA-YFP embryos (D), and, therefore, originate from Ptf1a-expressing progenitors. C, In Ptf1a−/− rh4, neither dA4 (arrow) nor dB1 (arrowhead) are present. F–H, In Ptf1a−/− rh4, dA3 neurons expanded ventrally. Quantification of dA3 cells in Ptf1a−/− rh4 revealed a significant increase in the number of dA3 cells relative to control littermates (p = 0.003; H). dB2 cells did not expand dorsally. Instead, in Ptf1a−/− rh4, dB3-like Lmx1b+ cells appeared at ectopic positions dorsal to dB2 domain (labeled as dB3* in G). I–p, rh4 of control and Ptf1a−/− embryos carrying Ptf1a-Cre/ROSA-YFP fate-mapping alleles. High-magnification insets (j–l and n–p) correspond to regions boxed in I and M. I–I, In Ptf1aCre/+;ROSA-YFP (control) embryos, virtually all Lmx1b+ cells, including both dA3 and dB3 cells were YFP−, indicating that the majority of these cells do not normally originate from Ptf1a-expressing progenitors. j–l, Rare Lmx1b+/YFP+ cells found in Ptf1aCre/+;ROSA-YFP control embryos (arrowhead) coexpressed Phox2b and, therefore, were dA3 cells. M–p, In Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) embryos, many Lmx1b+ cells were YFP+ (M, arrow, n–p, arrowheads and arrows), indicating that they originated from Ptf1a-expressing progenitors. Some of these Lmx1b+/YFP+ cells, particularly those located adjacent to a dorsal segment of the YFP+ VZ, coexpressed Phox2b (n–p, arrowheads), suggesting that they adopted the fate of dA3 cells. Lmx1b+/YFP+ cells, located adjacent to a ventral segment of the YFP+ VZ, were Phox2b− (n–p), adopting an expression profile of dB3 cells. While in Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) mutants, the majority of YFP+ cells were clearly Lmx1b+, some YFP+ cells appeared Lmx1b− (M, arrowhead). The identity of these YFP+/Lmx1b− cells is unknown. Q, Summary of cell-fate misspecification in early Ptf1a−/− rh4. In the absence of Ptf1a, Ptf1a-expressing progenitors in the VZ rather than producing dA4 and dB1 neurons, produce dA3- and dB3-like neurons. Scale bars: B–G, I, M, 100 μm; j–p, 25 μm.
In wild-type rh2–3, Lmx1b+/Phox2b+ dA3 cells are not produced (Figs. 1B, 4F; Sieber et al., 2007; Storm et al., 2009). In the absence of Ptf1a, in rh2, Ptf1a-expressing cells switched from producing dA4 and dB1 cells to generating Lmx1b+/Phox2b− (dB3-like) neurons (Fig. 4). Therefore, similar to rh7, in the absence of Ptf1a, during early (E10.5–E11.5) neurogenesis, Ptf1a-expressing progenitors in both rh4–6 and rh2–3 produce Lmx1b+ neurons (either Lmx1b+/Phox2b+ dA3-like neurons or Lmx1b+/Phox2b− neurons) rather than dA4/dB1 neurons (summarized in Fig. 5).
Figure 4.

In E11.5 Ptf1a−/− rh2, early-born dA4 and dB1 neurons transfate to dB3 neurons. Transverse sections of E11.5 hindbrain taken at the level of rh2, with genotypes and antibody markers indicated. B–J, The region boxed in rh2 schematic in A. Brackets in B, C, F, and G indicate the positions along the dorsal–ventral axis at which specific neuronal populations emerge. Brackets in D, E, H, I, and J indicate dorsal–ventral extent of YFP expression in the VZ, which was not significantly different between Ptf1aCre/+;ROSA-YFP and Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) embryos. B–E, Lhx1/5+/Pax2+ dB1 and Lhx1/5+/Pax2− dA4 neurons (B) were YFP+ in Ptf1aCre/+;ROSA-YFP embryos (D), and, thus, originate from Ptf1a-expressing progenitors. In Ptf1a−/− rh2, neither dA4 nor dB1 are present (C, arrow, E). F, G, In Ptf1a−/− rh2, dB2 cells did not expand dorsally. Instead, dB3-like Lmx1b+ cells appeared at ectopic positions dorsal to dB2 domain (labeled as dB3* in G). H–J, In control Ptf1aCre/+;ROSA-YFP rh2, Ptf1a-expressing progenitors are not a significant source of either Lmx1b+ dB3 (H) or Phox2b+ dB2 (J) neurons. In Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) rh2, ectopic Lmx1b+ dB3-like cells were YFP+ (I, arrow, i′, arrowheads), indicating that they originate from Ptf1a-expressing progenitors. K, Summary of cell-fate misspecification in early Ptf1a−/− rh2. In the absence of Ptf1a, Ptf1a-expressing progenitors do not produce dA4 and dB1 neurons. Rather than transfating to adjacent dB2 neurons, cells belonging to the Ptf1a lineage initiate the expression of Lmx1b, a marker of dB3 neurons. Scale bars: B–J, 100 μm; j′, 25 μm.
Figure 5.

Summary of neuronal subtypes in wild-type and Ptf1a−/− rh2–7 at E11.5. A, B, Side view of E11.5 wild-type (A) and Ptf1a−/− (B) hindbrain with dA and dB neuronal populations extending through rhombomeres. In Ptf1a−/− mutants, dA4 and dB1 neurons are not generated through rh2–7, and, depending on the anterior–posterior level, their positions are occupied by excessive dA3 and/or dB3 neurons.
During late neurogenesis, in Ptf1a−/− rh7, Ptf1a-expressing progenitors excessively produce dBLb neurons at the expense of dBLa neurons
In contrast to rh2–6, where dA/dB neurons arise throughout neurogenesis, after E12, Pax2+ dBLa and Lmx1b+ dBLb neurons arise in a salt-and-pepper pattern from a broad dorsal segment of the wild-type rh7 VZ (Figs. 1E, 6E–i′; Sieber et al., 2007). Because the pattern of neuronal formation in late (after E12) rh7 is clearly different from that in early (E10.5–E11.5) rh7 as well as early and late rh2–6, we specifically investigated the role of Ptf1a during late rh7 neurogenesis.
Figure 6.
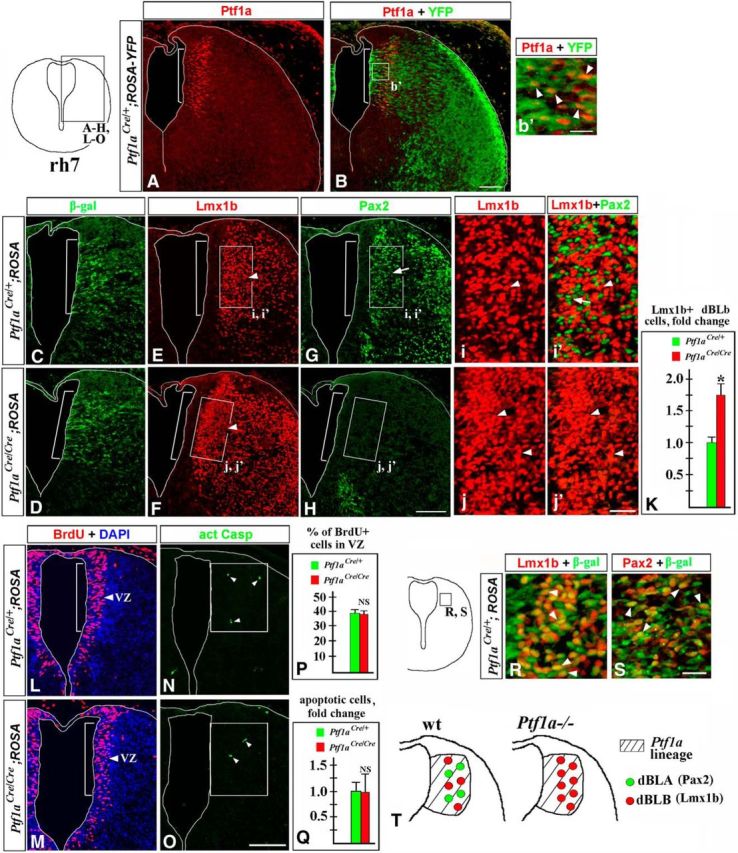
In the absence of Ptf1a, excessive late-born Lmx1b+ dBLb neurons are produced in E12.5 rh7 instead of Pax2+ dBLa neurons. Transverse sections of E12.5 rh7 with genotypes and antibody markers indicated. Low-magnification panels correspond to the region boxed in the rh7 schematic in the top left corner. High-magnification panels correspond to regions boxed in related data panels. A–b′, In wild-type E12.5 rh7, Ptf1a is specifically expressed in the dorsal VZ (A, B, bracket), and Ptf1a-expressing cells (b′, arrowheads) and their progeny are labeled by YFP expression in E12.5 Ptf1aCre/+;ROSA-YFP embryos. C–j′, In Ptf1aCre/+;ROSA-LacZ (control) embryos, both Lmx1b+ dBLb (E, i, i′, arrowheads) and Pax2+ dBLa cells (G, i′, arrows) are located adjacent to the Ptf1a-expressing VZ zone (E, G, bracket), which was identified based on β-gal staining of an adjacent section (C). In Ptf1aCre/Cre;ROSA-LacZ (Ptf1a−/−) rh7, only Lmx1b+ dBLb (F, j, j′, arrowheads) but not Pax2+ dBLa (H, j′) were found. Cell counts in a 100-μm-wide box located adjacent to the Ptf1a-expressing VZ (the region boxed in E–H) revealed a 1.8-fold increase in the number of Lmx1b+ dBLb cells in Ptf1a−/− embryos compared with controls (p = 0.0011). L, M, P, Analysis of proliferation in the Ptf1a-expressing VZ (labeled by bracket and identified based on β-gal staining of an adjacent section). Mice were injected with BrdU and killed 1 h after the BrdU pulse. Approximately the same fraction of cells was BrdU+ in the Ptf1a-epressing VZ in control and Ptf1a−/− embryos [P value is nonsignificant (NS)]. N, O, Q, The number of apoptotic cells was not increased in a 150-μm-wide domain, containing Ptf1a-expressing progenitors and newly generated dBLa and dBLb neurons (boxes in N and O) in Ptf1a−/− mutants compared with controls (Q; NS). Arrowheads point to Casp3+ apoptotic cells (N, O). R, S, Panels from Ptf1aCre/+;ROSA-LacZ (control) embryos corresponding to the region boxed in adjacent rh7 schematic. R, S, Both Lmx1b+ dBLb (R, arrowheads) and Pax2+ dBLa P (S, arowheads) were β-gal+ and, therefore, originate from Ptf1a-expressing progenitors. T, Summary of late-born neurons in rh7 of Ptf1a−/− embryos. Rather than producing dBLa and dBLb neurons, in the absence of Ptf1a, Ptf1a-expressing progenitors produce supernumerary dBLb neurons. Scale bars: A–H, L–O, 100 μm; b′, 15 μm; i–j′, 30 μm; R, S, 20 μm.
We found that in control E12.5 rh7, Ptf1a is specifically expressed in a dorsal segment of the VZ, which, as expected, was YFP+ in Ptf1aCre/+;ROSA-YFP embryos (or β-gal+ in Ptf1aCre/+;ROSA-LacZ embryos; Fig. 6A–C, bracket). The Ptf1a-expressing VZ was located directly adjacent to a more lateral area populated by Pax2+ dBLa and Lmx1b+ dBLb neurons (Fig. 6C,E,G,i,i′). Surprisingly, both Pax2+ dBLa and Lmx1b+ dBLb neurons were β-gal+ in E12.5 Ptf1aCre/+;ROSA-LacZ rh7 (Fig. 6R,S), although in early rh7 (Fig. 2A,C,J) as well as in the spinal cord (Glasgow et al., 2005; Hori et al., 2008; Borromeo et al., 2014) and cerebellum (Millen et al., 2014) Ptf1a-expressing progenitors produce Pax2+ neurons but are not a significant source of Lmx1b+ neurons. In Ptf1a−/− rh7, Pax2+ dBLa neurons were absent, while dBLb neurons significantly increased in number (∼1.8-fold compared with controls, p = 0.0011; Fig. 6E–K). Similar to control littermates, in rh7 of Ptf1aCre/Cre;ROSA-LacZ embryos, Lmx1b dBLb neurons were β-gal+ (data not shown), indicating that in Ptf1a−/− mutants, excessive dBLb neurons originate from Ptf1a-expressing progenitors.
Analysis of proliferation in dorsal rh7 VZ with a 1 h BrdU pulse did not reveal a difference between control and Ptf1a−/− littermates at E12.5 (Fig. 6L,M,P). Similarly, comparable number of Casp3+ apoptotic cells were detected within a domain populated by Ptf1a-expressing progenitors and newly generated dBLa/dBLb neurons in E12.5 Ptf1a−/− and control littermates (Fig. 6N,O,Q). Together, our data suggest that although in E12.5 rh7 dBLa and dBLb neurons are generated from Ptf1a-expressing progenitors, in the absence of Ptf1a these progenitors produce excessive Lmx1b+ dBLb neurons at the expense of Pax2+ dBLa neurons (summarized in Fig. 6T).
In contrast with control embryos, in E14.5 Ptf1a−/− rh7, most cells derived from Ptf1a-expressing progenitors express Lmx1b
Since we observed a dramatic cell-fate misspecification in the caudal hindbrain of Ptf1a mutants during both early and late waves of neurogenesis, we evaluated cellular composition of their rh7 at E14.5, when neurogenesis is essentially complete. Similar to earlier stages, in wild-type E14.5 rh7, Pax2 and Lmx1b continue labeling distinct sets of cells (Fig. 7A,B). In the cerebellum, spinal cord, and other regions of the CNS, Pax2 is expressed in a subset of GABAergic inhibitory neurons, while Lmx1b is expressed in excitatory neurons (Maricich and Herrup, 1999; Cheng et al., 2004; Glasgow et al., 2005; Dai et al., 2008; Xiang et al., 2012). To label GABAergic neurons, we used Gad67-GFP mice (Chattopadhyaya et al., 2004), in which GFP expression is driven by the regulatory elements of the Gad67 (also known as Gad1) gene (Rasmussen et al., 2007). Gad67, a key enzyme for GABA synthesis, is a marker of GABAergic cells (Cheng et al., 2004; Glasgow et al., 2005; Storm et al., 2009), and GFP expression in Gad67-GFP mice reliably labels many types of GABAergic neurons, including those in the developing hindbrain (Schubert et al., 2010; Waite et al., 2012; Gray, 2013). In E14.5 Gad67-GFP embryos, Pax2+ neurons coexpressed GFP, while Lmx1b+ neurons did not (Fig. 7C,D), arguing that, similar to other CNS regions, in rh7, Pax2, but not Lmx1b, is expressed in GABAergic neurons.
Figure 7.
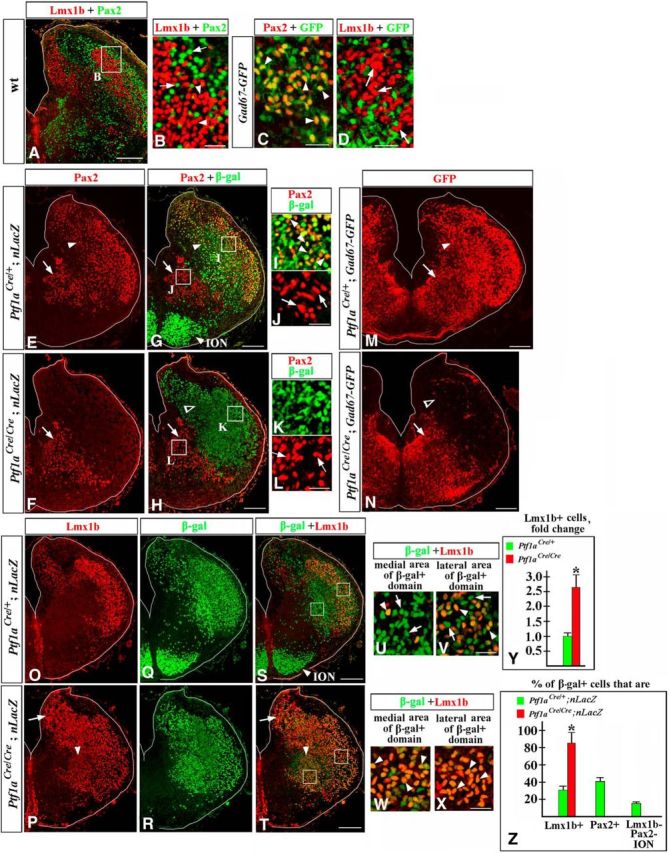
Misspecified neurons in E14.5 Ptf1a−/− rh7. Transverse sections of E14.5 rh7 with genotypes and antibody markers indicated. A–D, Lmx1b and Pax2 are expressed in different sets of cells. B–D show higher magnification of cells located in dorsolateral rh7 (e.g., area indicated by a box in a lower-magnification panel, A). B, Arrowheads point to Lmx1b+ cells, arrows point to Pax2+ cells. C, D, In Gad67-GFP rh7, Pax2+ cells express GFP (C, arrowheads), while Lmx1b+ cells do not (D, arrows), suggesting that Pax2+, but not Lmx1b+, cells are GABAergic. E–L, Ptf1aCre/+;nLacZ (E, G, I, J) and Ptf1aCre/Cre;nLacZ (F, H, K, L) rh7 immunostained against Pax2 and β-gal. Magnified regions (I–L) correspond to areas boxed in G and H. E, G, I, J, In wild-type rh7, Pax2+ dorsolateral cells are β-gal+ (E, G, I, arrowheads) and thus, originate from Ptf1a-expressing progenitors. Pax2+ ventromedial cells are β-gal- (E, G, J, arrows) and originate from progenitors that do not express Ptf1a. F, H, K, L, In Ptf1aCre/Cre;nLacZ (Ptf1a−/−) embryos, β-gal+ cells, populating dorsolateral rh7, do not express Pax2 (F, H, open arrowhead, K), while the ventromedial group of Pax2+ cells is not affected (F, H, L, arrows). M, N, In Ptf1a−/− Gad67-GFP mutants, GFP staining is lost in dorsolateral (N, open arrowhead) but is preserved in ventromedial (arrow) rh7. O–X, Low (O–T) and high (U–X) magnification of Ptf1aCre/+;nLacZ (O, Q, S, U, V) and Ptf1aCre/Cre;nLacZ (P, R, T, W, X) rh7 immunostained against Lmx1b and β-gal. U and W show cells located in ventromedial rh7 (such as area indicated by left box in S and T). V and X show cells located in dorsolateral rh7 (such as area indicated by right box in S and T). O–Z, In Ptf1aCre/+;nLacZ control embryos, a fraction of β-gal+ cells (arrowheads) coexpress Lmx1b (O, Q, S, yellow cells). Approximately a half of β-gal+ cells were Lmx1b+ in lateral rh7 (V, arrowheads), while in more medial rh7, virtually all β-gal+ cells were Lmx1b− (U, arrows). In Ptf1aCre/Cre;nLacZ (Ptf1a−/−) embryos, a vast majority of β-gal+ cells coexpress Lmx1b (P, R, T), including those populating both medial and lateral rh7 (W, X, arrowheads). Y, Cell counts revealed a dramatic increase in the number of Lmx1b+ cells in E14.5 Ptf1a−/− rh7 compared with control littermates (p = 0.006). Excessive Lmx1b+ cells were particularly obvious in dorsomedial (P, T, arrow) and ventromedial (P, T, arrowhead) areas of rh7. Z, Quantification of the fractions of β-gal+ cells expressing specific markers in Ptf1a−/− and control littermates. Approximately 85% of β-gal+ cells coexpressed Lmx1b in Ptf1aCre/Cre/;nLacZ (Ptf1a−/−) rh7 vs 32% of β-gal+ cells in Ptf1aCre/+;nLacZ (control) rh7 (p = 0.001). In Ptf1aCre/+;nLacZ (control) rh7, 41% of β-gal+ cells expressed Pax2, and ∼16% of β-gal+ cells were located in the ION and did not express either Pax2 or Lmx1b. In Ptf1aCre/Cre//nLacZ (Ptf1a−/−) mutants, β-gal+ cells were not present in the ION, and none of the β-gal+ cells expressed Pax2. Scale bars: A, E, G, F, H, M–T, 200 μm; B–D, I–L, U–X, 30 μm.
To label neurons of the Ptf1a lineage in E14.5 embryos, we used the Tau-nLacZ reporter (referred to as nLacZ throughout this study). The nLacZ reporter, which contains a LoxP-Stop-LoxP-nLacZ cassette under the control of a ubiquitous neuronal Tau promoter (Hippenmeyer et al., 2005), was used instead of ROSA26 reporter because it labels neuronal progeny of Cre-expressing cells with nuclear β-gal, helping to more precisely determine the identity of β-gal+ cells by colabeling them with nuclear markers. By analyzing Ptf1aCre/+;nLacZ (control) embryos, we found that, in E14.5 rh7, Pax2+ neurons were divided into two groups based on their origin. Dorsolateral Pax2+ cells were β-gal+ and, therefore, originated from Ptf1a-expressing progenitors (Fig. 7E,G,I, arrowheads). Ventromedial Pax2+ cells were β-gal− (Fig. 7E,G,J, arrows) and, therefore, originated from VZ progenitors that did not express Ptf1a. In E14.5 Ptf1a−/− rh7, the dorsolateral group of Pax2+ cells was absent but the ventrolateral Pax2+ cells were not affected (Fig. 7F,H,K,L). Similarly, in Ptf1a−/− Gad67-GFP embryos, GFP expression was not detected in dorsolateral rh7, while ventromedial GFP signal was still present (Fig. 7M,N). Numerous β-gal+ cells still populated dorsolateral rh7 in Ptf1aCre/Cre;nLacZ (Ptf1a−/−) embryos (Fig. 7H,K, open arrowhead), suggesting that, in the absence of Ptf1a, neurons originating from Ptf1a-expressing progenitors do not die, but rather lose Pax2 expression and activity of the Gad67 promoter, suggesting a cell-fate misspecification.
Opposite to the loss of Pax2 expression in dorsolateral cells, the number of Lmx1b+ cells was 2.6-fold higher in Ptf1a−/− rh7 compared with controls (Fig. 7O,P,Y; p = 0.006), and excessive Lmx1b+ cells were particularly obvious in dorsomedial (Fig. 7P,T, arrow) and ventromedial (Fig. 7P,T, arrowhead) areas of rh7. Furthermore, in control E14.5 Ptf1aCre/+;nLacZ rh7, only 32% of β-gal+ cells expressed Lmx1b (Fig. 7O,Q,S,U,V), while 41% of β-gal+ cells expressed Pax2 (Fig. 7G,I), and 16% of β-gal+ cells were located in the ION and expressed neither Pax2 nor Lmx1b [Fig. 7G,S (quantified in Fig. 7Z)]. In contrast, in Ptf1aCre/Cre;nLacZ (Ptf1a−/−) rh7, ∼85% of β-gal+ cells expressed Lmx1b (Fig. 7P,R,T,W,X; p = 0.001 compared with controls), and none of the β-gal+ cells expressed Pax2 (Fig. 7H,K) or populated the ION [Fig. 7H,T (quantified in Fig. 7Z)]. Together, these data strongly suggest that E14.5 Ptf1a−/− rh7 contains a large population of misspecified neurons of the Ptf1a lineage.
Increased number of Lmx1b+ neurons in the nTs and SpV nuclei in Ptf1a−/− mice
To determine the eventual fates of misspecified hindbrain neurons in Ptf1a−/− mice, we analyzed these mutants at E18.5, the last viable stage. In E18.5 embryos, we focused on the nTs and SpV nuclei, since in earlier Ptf1a mutants we observed an increased number of dA3 cells (Fig. 2F–H), previously described as differentiating Lmx1b+/Phox2b+ neurons destined to form the nTs (Qian et al., 2001; Dauger et al., 2003; Sieber et al., 2007; D'Autréaux et al., 2011), and dBLb cells (Fig. 6E,F,i–K), proposed to contain differentiating Lmx1b+ neurons destined to form the SpV nuclei (Sieber et al., 2007). To identify these nuclei, transverse sections spanning the anterior–posterior axis of E18.5 hindbrain were colabeled with anti-Lmx1b and Phox2b antibodies. The nTs nuclei were identified as dorsomedial medulla areas densely populated by Lmx1b+/Phox2b+ cells, located directly dorsal to Phox2b+/Lmx1b− vagal motoneurons, while the SpV nuclei were identified on the same sections as lateral medulla areas densely populated by Lmx1b+/Phox2b− cells (Fig. 8A), as previously described (Sieber et al., 2007). In E18.5 Ptf1a−/− mutants, both Lmx1b+/Phox2b+ nTs and Lmx1b+/Phox2b− SpV neurons were present at appropriate positions in the brainstem (Fig. 8A,B). However, there was a significant increase in the number of Lmx1b+ neurons in both of these nuclei (an ∼1.7-fold increase in the number of Lmx1b+ nTs neurons, p = 0.0068, and a 1.5-fold increase in the number of Lmx1b+ SpV neurons, p = 0.0043, compared with control littermates; Fig. 8A–D). Thus, loss of Ptf1a compromises the development of the nTs and SpV nuclei. A detailed analysis of these nuclei in Ptf1a mutants is described below.
Figure 8.

nTs and SpV abnormalities in E18.5 Ptf1a−/− embryos. Transverse sections of caudal hindbrain with genotypes and antibody markers indicated. A–D, An increased number of Lmx1b+/Phox2b+ nTs neurons (A–C, p = 0.0068) and Lmx1b+/Phox2b− SpV neurons (A, B, D, p = 0.0043) were detected in Ptf1a−/− embryos compared with control littermates. Arrows point to vagal motoneurons located below the nTs (A, B). E–L, An nTs region boxed in the adjacent diagram. E, F, Both Lmx1b+ (E, arrowheads) and Pax2+ (E, arrows) cells were detected in control nTs, but only Lmx1b+ nTs neurons (F, arrowheads) were present in Ptf1a−/− mutants. G, H, In the nTs of Gad67-GFP mice, Pax2+ cells coexpressed GFP (G, arrowheads), indicating that they were GABAergic neurons. In Ptf1a−/− Gad67-GFP mutants, GFP signal was absent (H). I–L, In Ptf1aCre/+;nLacZ (control) nTs, β-gal+ cells coexpressed Pax2+ (I, arrowheads), but very few β-gal+ cells were Lmx1b+ (K, arrowhead). Arrows in K point to β-gal+/Lmx1b− nTs cells. In Ptf1aCre/YFP;nLacZ (Ptf1a−/−) mutants, β-gal+ nTs cells did not express Pax2 (J, arrows) but expressed Lmx1b (L, arrowheads), suggesting their misspecification. High-magnification images to the right of L correspond to the region boxed in L. In Ptf1aCre/YFP;nLacZ mutants, Lmx1b+/β-gal+ nTs cells coexpressed Phox2b (arrowheads), showing that they adopted the fate of Lmx1b+/Phox2b+ nTs neurons. M, Quantification revealed that in Ptf1aCre/YFP;nLacZ mutants, >35% of Lmx1b+ nTs neurons originate from Ptf1a-expressing progenitors compared with only ∼5% of those cells in Ptf1aCre/+;nLacZ control littermates (p = 0.0064). Scale bars: A, B, 200 μm; E–L, 40 μm; L, inset, 20 μm.
nTs neurogenesis in the absence of Ptf1a
Immunohistochemical analysis revealed that, in addition to Lmx1b+/Phox2b+ cells (Fig. 8E, arrowheads), the nTs of E18.5 control embryos contain Pax2+ cells (Fig. 8E, arrows; Sieber et al., 2007). In contrast to Lmx1b+/Phox2b+ nTs neurons, some of which are noradrenergic (Qian et al., 2001; Sieber et al., 2007), Pax2+ nTs cells were GFP+ in Gad67-GFP mice (Fig. 8G, arrowheads), indicating that they are GABAergic neurons. In E18.5 Ptf1aCre/+;nLacZ (control) embryos, Pax2+ nTs neurons were β-gal+ (Fig. 8I, arrowheads). In contrast, most Lmx1b+ nTs neurons were β-gal− in E18.5 Ptf1aCre/+;nLacZ embryos (Fig. 8K), which is consistent with only a small fraction of newborn Lmx1b+/Phox2b+ nTs neurons (dA3 cells) labeled by YFP in E11.5 Ptf1aCre/+;ROSA-YFP embryos (Fig. 2J,k–l″, 3I–l). Thus, normally Pax2+ nTs neurons originate from Ptf1a-expressing progenitors, while the majority of Lmx1b+ nTs neurons originate from progenitors that do express Ptf1a.
In E18.5 Ptf1a−/− mice, Pax2+ neurons were not detected in the nTs, and there was no GFP expression in the nTs of Ptf1a−/− Gad67-GFP mice (Fig. 8E–H). To investigate whether Ptf1a−/− nTs contain misspecified neurons, we analyzed E18.5 Ptf1aCre/YFP;nLacZ (Ptf1a−/−) mutants, which, similar to Ptf1aCre/+ controls, contained one Cre copy in their genome. In these mutants, numerous β-gal+ cells were detected in the nTs, arguing that, even in the absence of Ptf1a, cells of the Ptf1a lineage contribute to the nTs. However, in contrast to β-gal+ cells in the nTs of Ptf1aCre/+;nLacZ control mice, in Ptf1aCre/YFP;nLacZ (Ptf1a−/−) embryos virtually all β-gal+ nTs cells expressed Lmx1b rather than Pax2 (Fig. 8I–L). Cell counts revealed that in Ptf1aCre/YFP ;nLacZ (Ptf1a−/−) mutants, >35% of Lmx1b+ nTs neurons were β-gal+, compared with only ∼5% of Lmx1b+ neurons that expressed β-gal in the nTs of Ptf1aCre/+;nLacZ control littermates (p = 0.0064; Fig. 8M). Similar to Lmx1b+ nTs neurons originating from Ptf1a− progenitors, in Ptf1aCre/YFP;nLacZ (Ptf1a−/−) mutants, Lmx1b+/β-gal+ nTs neurons coexpressed Phox2b (Fig. 8L, insets). Thus, in the absence of Ptf1a, Pax2+ GABAergic nTs neurons are absent, and the nTs nuclei become populated by supernumerary Lmx1b+/Phox2b+ neurons, some of which aberrantly arise from Ptf1a-expressing progenitors.
We next performed birth-dating experiments to directly identify when misspecified nTs neurons are produced in Ptf1a−/− mutants. Ptf1aCre/+;nLacZ (control) and Ptf1aCre/YFP;nLacZ (Ptf1a−/−) mice were injected with BrdU at E11, during an early phase of hindbrain neurogenesis, or at E12.25, during a late phase of neurogenesis, and were analyzed at E18.5, when the nTs is formed. BrdU is incorporated into DNA during the S phase of the cell cycle, and cells that exit their last cell cycle and differentiate shortly after the BrdU injection become heavily labeled by BrdU (Rash et al., 2013).
In control embryos, a fraction of Pax2+ nTs neurons were BrdU+ when BrdU was injected at both E11 (Fig. 9A,a, arrowheads) and E12.25 (Fig. 9D,d, arrowheads), suggesting that these neurons arise during an extended time period. Colabeling with β-gal in Ptf1aCre/+;nLacZ (control) embryos confirmed that during both early and late neurogenesis Pax2+ nTs neurons arise from Ptf1a-expressing progenitors (Fig. 9a–a″,d–d″). In E18.5 control embryos, a fraction of Lmx1b+ nTs neurons were BrdU+ when BrdU was injected at E11 (Fig. 9B,b, arrowheads), but virtually no Lmx1b+/BrdU+ cells were detected when BrdU was injected at E12.25 (Fig. 9E), confirming previous observations that Lmx1b+ nTs neurons are predominantly generated during an early phase of hindbrain neurogenesis (Qian et al., 2001; Sieber et al., 2007).
Figure 9.

Birth-dating analysis of nTs neurons in Ptf1a−/− mice. Transverse sections through the nTs with genotypes and antibody markers indicated. A–F, Mice were injected with BrdU at E11 (A–C) or E12.25 (D–F) and were analyzed at E18.5. A–F show a dorsal area, including the nTs, boxed in the schematic. The nTs is outlined by a dashed line. Magnified regions in a–a″, b, c–c″, d–d″ correspond to areas boxed in adjacent data panels. In Ptf1aCre/+;nLacZ (control) embryos some Pax2+ cells were labeled by BrdU, when BrdU was injected at both E11 and E12.25 (A, D, arrowheads). In both cases, Pax2+/BrdU+ cells coexpressed β-gal (a–a″, d–d″, arrowheads), indicating that these cells originate from Ptf1a-expressing progenitors. In both Ptf1aCre/+;nLacZ (control) and Ptf1aCre/YFP;nLacZ (Ptf1a−/−) embryos, a fraction of Lmx1b+ cells were labeled by BrdU, when BrdU was injected at E11 (B, C, arrowheads) but not at E12.25 (E, F). Some Lmx1b+/β-gal+ cells in Ptf1aCre/YFP;nLacZ (Ptf1a−/−) embryos, injected with BrdU at E11, coexpressed β-gal (c–c″, arrowheads). Since in control embryos, virtually all Lmx1b+ nTs neurons originate from Ptf1a− progenitors, these Lmx1b+/BrdU+/β-gal+ cells in Ptf1aCre/YFP;nLacZ (Ptf1a−/−) embryos are misspecified cells that arose from Ptf1a-expressing progenitors. Scale bars: A–F, 100 μm; a–a″, b, c–c″, d–d″, 25 μm.
In E18.5 Ptf1a−/− mutants, similar to control embryos, a fraction of Lmx1b+ nTs neurons were BrdU+ when BrdU was injected at E11 but not at E12.25 (Fig. 9C,c, arrowheads, F). In Ptf1aCre/YFP;nLacZ embryos injected at E11, some BrdU+/Lmx1b+ cells were also β-gal+ (Fig. 9c–c″, arrowheads), revealing that in Ptf1a−/− mutants, at least some misspecified Lmx1b+ nTs neurons arise from Ptf1a-expressing progenitors during early neurogenesis. Since in wild-type embryos Lmx1b+/Phox2b+ nTs neurons are generated during early but not late neurogenesis, it is likely that excessive Lmx1b+ neurons arising in Ptf1a−/− rh7 during late neurogenesis (Fig. 6E–K) are not competent to populate the nTs. Notably, although we did not find a difference in the number of Casp3+ apoptotic cells in the nTs of control and Ptf1a−/− mice at E18.5 (Fig. 10A–C), the number of apoptotic cells was increased in the Ptf1a−/− nTs at E14.5 (p = 0.03; Fig. 10D–J), with some Casp3+ cells coexpressing Lmx1b and Phox2b (Fig. 10G–i). Thus, it is possible that in Ptf1a mutants some late-born Ptf1a lineage neurons also aberrantly adopt the fate of Lmx1b+ nTs neurons but do not survive until E18.5.
Figure 10.

Increased apoptosis in the nTs of E14.5 but not E18.5 Ptf1a−/− embryos. Transverse sections through the nTs with genotypes and antibody markers indicated. Images in data panels correspond to area boxed in adjacent schematics. A–J, More Casp3+ apoptotic cells (arrowheads) were present in the nTs of Ptf1a−/− mutants relative to controls at E14.5 (D–J, p = 0.03) but not at E18.5 (A–C; NS, nonsignificant). In E14.5 Ptf1a−/− mutants, some apoptotic cells coexpressed Lmx1b and Phox2b (h, i, white cells pointed out by arrowheads), indicating that they were nTs neurons belonging to the Lmx1b lineage. Scale bars: A, B, D–G, 100 μm; h, i, 30 μm.
SpV neurogenesis in the absence of Ptf1a
Immunohistochemical analysis revealed that in addition to Lmx1b+ neurons (Fig. 11A,B,D, arrowheads), E18.5 wild-type SpV nuclei contain numerous Pax2+ cells (Fig. 11B,D, arrows). Based on the coexpression of Tlx3, a marker and determinant of excitatory cell fate in the CNS (Qian et al., 2001; Cheng et al., 2004; Glasgow et al., 2005; Xiang et al., 2012), Lmx1b+ SpV neurons were previously proposed to be excitatory neurons (Dai et al., 2008). In the SpV nuclei of E18.5 Gad67-GFP embryos, Pax2+ (Fig. 11H, arrowheads), but not Lmx1b+ (data not shown), cells were GFP+, indicating that Pax2+ cells in the SpV nuclei are GABAergic neurons. In the SpV nuclei of E18.5 Ptf1aCre/+;nLacZ (control) embryos, both Pax2+ and Lmx1b+ neurons were labeled by β-gal (Fig. 11J,L), arguing that both these populations originate from Ptf1a-expressing progenitors. β-gal labeling of Lmx1b+ SpV neurons in E18.5 Ptf1aCre/+;nLacZ embryos is consistent with β-gal labeling of dBLb cells in E12.5 Ptf1aCre/+;ROSA-LacZ embryos (Fig. 6R), the cellular population proposed to contain differentiating Lmx1b+ neurons of the SpV (Sieber et al., 2007).
Figure 11.

Lack of Pax2+ GABAergic neurons in the SpV nuclei of E18.5 Ptf1a−/− embryos. Transverse sections of caudal hindbrain with genotypes and antibody markers indicated. Images in B, C, and N–Q correspond to the area boxed in the schematic A. High-magnification images correspond to regions boxed in adjacent data panels. B–E, Both Lmx1b+ (B, D, arrowheads) and Pax2+ (B, D, arrows) SpV neurons were detected in controls, but only Lmx1b+ SpV neurons (C, E, arrowheads) were present in Ptf1a−/− mutants. F, G, Lmx1b+ SpV neurons coexpressed Tlx3 in both controls and Ptf1a−/− mutants (arrowheads). H, I, In the SpV nuclei of Gad67-GFP mice, Pax2+ cells coexpressed GFP (H, arrowheads), suggesting that they were GABAergic neurons. I, In Ptf1a−/− Gad67-GFP mutants, GFP signal was absent. J–M, In the Ptf1aCre/+;nLacZ (control) SpV nuclei, some β-gal+ cells coexpress Pax2 (J, arrowheads), while others coexpress Lmx1b (L, arrowheads). In Ptf1aCre/YFP;nLacZ (Ptf1a−/−) mutants, β-gal+ cells do not express Pax2 (K), but the vast majority of them express Lmx1b (M, arrowheads). N–T, More Casp3+ apoptotic cells (arrowheads) were present in the SpV region of Ptf1a−/− mutants relative to controls (T, p = 0.003). In Ptf1a−/− mutants, some apoptotic cells coexpress Lmx1b (P, Q, r, s, arrowheads). Scale bars: B, C, N–Q, 100 μm; D–M, 40 μm; r, s, 20 μm.
In E18.5 Ptf1a−/− embryos, no Pax2+ cells were detected in the SpV nuclei (Fig. 11C,E), and there was no GFP signal in the SpV nuclei of Ptf1a−/− Gad67-GFP mice (Fig. 11I). At the same time point, similar to wild-type mice, Lmx1b+ SpV neurons in Ptf1a−/− mice expressed their appropriate cell type-specific marker Tlx3 (Fig. 11F,G). In Ptf1aCre/YFP;nLacZ (Ptf1a−/−) embryos, Lmx1b+ SpV neurons were β-gal+, indicating that, as in wild-type mice, they were generated from Ptf1a-expressing progenitors (Fig. 11L,M). In contrast to control E18.5 Ptf1aCre/+;nLacZ embryos, where approximately one-half of β-gal+ cells in the SpV expressed Lmx1b, while the other half expressed Pax2, in Ptf1aCre/YFP;nLacZ mutants the vast majority of β-gal+ SpV cells were Lmx1b+ (Fig. 11J–M). There were an increased number of Casp3+ apoptotic cells in the SpV nuclei of Ptf1a−/− mutants (p = 0.003 compared with controls; Fig. 11N–T). In Ptf1a−/− mutants, some of these Casp3+ cells were Lmx1b+ (Fig. 11P–s), showing that, in the absence of Ptf1a, Lmx1b+ SpV neurons excessively die. Together, our data indicate that loss of Ptf1a prevents populating the SpV nuclei with Pax2+ GABAergic neurons. In contrast, although Lmx1b+ SpV neurons originate from Ptf1a-expressing progenitors, the loss of Ptf1a does not prevent the generation of these neurons. Furthermore, since Lmx1b+ neurons were increased in the number in Ptf1a−/− SpV nuclei (Fig. 8A,B,D), Ptf1a prevents their excessive production. At the same time, since we observed an increased number of apoptotic Lmx1b+ neurons in the Ptf1a−/− SpV nuclei, Ptf1a may be necessary for the survival of Lmx1b+ SpV neurons, although excessive apoptosis of these cells in Ptf1a−/− mutants may be a secondary response to a lack of Pax2+ GABAergic SpV neurons.
To directly analyze when SpV neurons are generated in Ptf1a−/− mutants, we performed BrdU birth-dating experiments. Ptf1aCre/+;nLacZ (control) and Ptf1aCre/YFP;nLacZ (Ptf1a−/−) mice were injected with BrdU at E11, during an early phase of hindbrain neurogenesis, or at E12.25, during a late phase of neurogenesis, and were analyzed at E18.5, when the SpV nuclei are already formed. In wild-type embryos, a fraction of Pax2+ SpV neurons were BrdU+ when BrdU was injected at both E11 (Fig. 12A,b, arrowheads) and E12.25 (Fig. 12O,p, arrowheads), suggesting that these neurons arise during an extended time period. β-gal colabeling in Ptf1aCre/+;nLacZ (control) embryos confirmed that during both early and late neurogenesis, Pax2+ SpV neurons arise from Ptf1a-expressing progenitors (Fig. 12b–d,p–r). Many Lmx1b+ neurons were BrdU+ when control mice were injected with BrdU at E12.25 (Fig. 12S,t). These Lmx1b+/BrdU+ cells were β-gal+ in Ptf1aCre/+;nLacZ (control) embryos (Fig. 12t–v), confirming that they originate from Ptf1a-expressing progenitors. Few Lmx1b+/BrdU+ SpV cells were observed when Ptf1aCre/+;nLacZ mice were injected with BrdU at E11 (Fig. 12E). Furthermore, rather than being heavily labeled with BrdU, a majority of these Lmx1b+/BrdU+/β-gal+ cells contained only one to three small BrdU+ dots (Fig. 12E–i), suggesting that after BrdU incorporation at E11, these cells underwent one or two additional divisions before finally exiting the cell cycle (Rash et al., 2013). Thus, our data indicate that in wild-type embryos most Lmx1b+ SpV neurons arise during late neurogenesis, consistent with previous studies, which proposed late-born dBLb cells as newly generated SpV Lmx1b+ neurons (Sieber et al., 2007).
Figure 12.

Birth-dating analysis of SpV neurons in Ptf1a−/− mice. A–z, Transverse sections through the SpV nucleus with genotypes and antibody markers indicated. Mice were injected with BrdU at E11 (A–n) or E12.25 (O–z) and were analyzed at E18.5. Low-magnification panels correspond to the region boxed in the schematic. High-magnification panels correspond to areas boxed in adjacent data panels. In Ptf1aCre/+;nLacZ (control) embryos, some Pax2+ SpV cells were labeled by BrdU, when BrdU was injected at both E11 and E12.25 (A, O, arrowheads). In both cases, Pax2+/BrdU+ cells coexpressed β-gal (b–d, p–r, arrowheads), indicating that these cells originate from Ptf1a-expressing progenitors. E–i, Few Lmx1b+/BrdU+ cells were detected in Ptf1aCre/+;nLacZ (control) embryos injected with BrdU at E11 (E). Most Lmx1b+/BrdU+ SpV cells found in these embryos, contained only one to three small BrdU dots (E, f–i, arrowheads), suggesting that since the time of BrdU incorporation they underwent one or two additional divisions before exiting the cell cycle. J–n, Many Lmx1b+ cells were heavily labeled with BrdU, when Ptf1aCre/YFP;nLacZ (Ptf1a−/−) embryos were injected with BrdU at E11 (J, arrowheads). These cells coexpressed β-gal (k–n, arrowheads), indicating that they originated from Ptf1a-expressing progenitors. Thus, in contrast to control embryos, in Ptf1a−/− mutants, many cells arising from Ptf1a expressing progenitors at E11 adopt the fate of Lmx1b+ SpV neurons. S–z, In both Ptf1aCre/+;nLacZ (control) and Ptf1aCre/YFP;nLacZ (Ptf1a−/−) embryos, some Lmx1b+ cells were heavily labeled with BrdU, when BrdU was injected at E12.25 (S, W, arrowheads). In both cases, Lmx1b+/BrdU+ SpV cells coexpressed β-gal (t–v, x–z, arrowheads), indicating that they originate from Ptf1a-expressing progenitors. A1, Cell counts revealed an increased number of Lmx1b+/BrdU+ SpV cells in Ptf1a−/− mutants injected with BrdU at E12.25 relative to control littermates (p = 0.0083), indicating that more Lmx1b+ SpV neurons arise in Ptf1a−/− mutants than in controls at E12.25. Scale bars: A, E, J, O, S, W, 75 μm; b–d, f–i, k–n, p–r, t–v, x–z, 25 μm.
In E18.5 Ptf1aCre/YFP/nLacZ (Ptf1a−/−) mutants, similar to control embryos, a fraction of Lmx1b+ SpV neurons were BrdU+ when BrdU was injected at E12.25 (Fig. 12W,x, arrowheads). Cell counts revealed an increased number of Lmx1b+/BrdU+ SpV cells in these mutants relative to control littermates (p = 0.0083; Fig. 12A1), arguing that an increased number of Lmx1b+ SpV neurons in E18.5 Ptf1a mutants are at least partially emerging from their excessive production during late neurogenesis. In addition, and in contrast to wild-type embryos, many Lmx1b+ SpV cells were heavily labeled with BrdU when BrdU was injected at E11 (Fig. 12E,J,k,l,n). Similar to Lmx1b+ SpV neurons labeled by BrdU injection at E12.25, many Lmx1b+ SpV neurons labeled by BrdU injection at E11 were clearly β-gal+ in Ptf1aCre/YFP;nLacZ (Ptf1a−/−) embryos, indicating that they originate from Ptf1a-expressing progenitors (Fig. 12k–n,x–z). Thus, based on our birth-dating studies, in the absence of Ptf1a, more Lmx1b+ SpV neurons are produced during late neurogenesis. Furthermore, in contrast to wild-type embryos, in Ptf1a−/− mutants, a significant fraction of Lmx1b+ SpV neurons arise not only during a late phase of neurogenesis, but also during an early phase.
Loss of Ptf1a compromises neuronal composition of the PrV nuclei
In addition to the SpV nuclei, the PrV nuclei is another critical center of somatosensory information integration (Woolsey, 1990). Mouse genetic fate mapping revealed that, in contrast to the SpV nuclei that develop in caudal brainstem, PrV neurons arise in rh2–3 (Oury et al., 2006). The PrV nuclei contain both Lmx1b+ cells (Fig. 13A,B,D, arrowheads), which are glutamatergic excitatory neurons (Xiang et al., 2012), and Pax2+ cells (Fig. 13B,D, arrows), which are GABAergic inhibitory neurons (Fig. 13F; Xiang et al., 2012). In E18.5 Ptf1aCre/+;nLacZ embryos, virtually all Pax2+ PrV neurons were β-gal+, but the vast majority of Lmx1b+ PrV neurons were β-gal−, indicating that Pax2+, but not Lmx1b+, PrV neurons normally originate from Ptf1a-expressing progenitors (Fig. 13H,J). In E18.5 Ptf1a−/− embryos, Pax2+ neurons appeared reduced in number but were not completely lost (Fig. 13B–E). Furthermore, similar to control mice, some Pax2+ PrV neurons in Ptf1a−/− Gad67-GFP mutants were GFP+, arguing that they maintain the identity of GABAergic inhibitory neurons (Fig. 13F,G). Interestingly, and in contrast to Ptf1aCre/+;nLacZ control embryos, in E18.5 Ptf1aCre/YFP;nLacZ (Ptf1a−/−) mutants, none of the β-gal+ cells populating the PrV nuclei expressed Pax2 (Fig. 13H,I). Instead, virtually all of these β-gal+ cells expressed Lmx1b (Fig. 13K). As a result, in the PrV nuclei of E18.5 Ptf1aCre/YFP;nLacZ mutants, ∼25% of Lmx1b+ neurons were β-gal+ (compared with <3% of Lmx1b+ PrV neurons in control littermates, p = 0.0006; Fig. 13J–L). Thus, rather than producing Pax2+ PrV neurons, in the absence of Ptf1a, Ptf1a-expressing progenitors produce Lmx1b+ PrV neurons. Considering that Pax2+ PrV cells were β-gal+ in Ptf1aCre/+;nLacZ embryos, but were β-gal− in Ptf1aCre/YFP;nLacZ embryos, it is possible that in Ptf1a mutants the loss of Pax2+ PrV cells belonging to the Ptf1a lineage is partially compensated by Pax2+ neurons originating from a Ptf1a− lineage.
Figure 13.

In Ptf1a−/− mutants, cell-fate misspecification leads to a reduction in but not a complete loss of Pax2+ PrV neurons. Transverse sections through the PrV nucleus with genotypes and antibody markers indicated. All images are from E18.5 embryos. B and C correspond to the region boxed in the schematic A. The PrV nuclei are outlined by a dashed line. High-magnification images in D–K correspond to the areas boxed in B and C. B–E, Both Lmx1b+ (arrowheads) and Pax2+ (arrows) PrV neurons were detected in control and Ptf1a−/− mutants, although in Ptf1a mutants Pax2+ cells were reduced in number. F–G, In both control and Ptf1a−/− Gad67-GFP mice, Pax2+ PrV neurons coexpressed GFP (arrowheads), indicating that they were GABAergic neurons. H–K, In the Ptf1aCre/+;nLacZ (control) PrV nuclei, most β-gal+ cells coexpressed Pax2+ (H, arrowheads) but not Lmx1b+ (J, arrow), indicating that Pax2+ PrV neurons originate from Ptf1a-expressing progenitors, while most Lmx1b+ PrV neurons do not. In the Ptf1aCre/YFP;nLacZ (Ptf1a−/−) PrV nuclei, β-gal+ cells coexpress Lmx1b (K, arrowheads) but not Pax2 (I, arrow), suggesting a cell-fate misspecification. L, Quantification revealed that in Ptf1aCre/YFP;nLacZ (Ptf1a−/−) mutants ∼25% of Lmx1b+ PrV neurons originate from Ptf1a-expressing progenitors compared with <3% of Lmx1b+ PrV cells in Ptf1aCre/+;nLacZ control littermates (p = 0.0006). Scale bars: B, C, 100 μm; D–K, 40 μm.
Discussion
In the brainstem, Ptf1a was implicated in the differentiation and survival of ION and cochlear neurons (Yamada et al., 2007, Fujiyama et al., 2009). Here, we show that the loss of Ptf1a prevents the development of GABAergic neurons populating viscerosensory and somatosensory brainstem nuclei. We find that the formation of 3 (dA4, dB1, and dBLa) of the 10 dA/dB and dBLa/b populations in the dorsal hindbrain depends on Ptf1a. Our data argue that in the brainstem Ptf1a acts largely as a regulator of cell-fate specification decisions, suppressing an Lmx1b+ fate in subsets of differentiating cells.
Ptf1a is necessary for development of viscerosensory and somatosensory GABAergic brainstem neurons
Both viscerosensory (nTs) and somatosensory (SpV and PrV) nuclei contain neurons of the Lmx1b lineage. Lmx1b+ viscerosensory and somatosensory neurons share a similar gene expression profile, suggesting common developmental and evolutionary origin of viscerosensory and somatosensory brainstem nuclei (Qian et al., 2002; D'Autréaux et al., 2011; Nomaksteinsky et al., 2013). We show that, in addition to neurons of the Lmx1b lineage, another common feature of the nTs, SpV, and PrV nuclei, is that each contain Pax2+ GABAergic neurons that originate from Ptf1a-expressing progenitors and require Ptf1a for development. While several genes, including Lbx1, Phox2b, Ascl1, and Tlx1/3, are known to regulate the development of Lmx1b+ brainstem neurons (Qian et al., 2001, 2002; Sieber et al., 2007; D'Autréaux et al., 2011), to our knowledge, Ptf1a is the first gene required for specification of Pax2+ GABAergic viscerosensory and somatosensory brainstem neurons.
In the developing brainstem, Ptf1a primarily acts as a regulator of cell-fate specification
Our data support the conclusion that in Ptf1a−/− mutants, the loss of GABAergic nTs and SpV neurons is caused by a cell-fate misspecification during both early (E11–E11.5) and late (E12.5) neurogenesis, which affects not only viscerosensory and somatosensory neurons, but at least one additional population, the climbing fiber neurons of the ION.
Previously, 10 newborn neuronal populations, dA1–4/dB1–4 and dBLa/b, were described in E11–E13 dorsal brainstem (Sieber et al., 2007). dA3 and dA4 cells were identified as newborn Lmx1b+/Phox2b+ nTs and ION neurons, respectively. dBLb cells in E12.5 rh7 were described as Lmx1b+ SpV neurons (Sieber et al., 2007; Storm et al., 2009). Although the eventual fates of dB1 and dBLa cells were unknown, our data suggest that these two populations in caudal brainstem contain differentiating Pax2+ GABAergic neurons of the nTs and SpV. Indeed, by combining birth-dating and fate-mapping studies, we find that Pax2+ nTs and SpV neurons arise from Ptf1a+ progenitors during both early (E11) and late (E12.25) neurogenic phases, when the only Pax2+ populations arising from Ptf1a+ progenitors are dB1 and dBLa cells.
In E11.5 Ptf1a−/− mutants, we detected neither dB1 nor dA4 neurons throughout rh2–7. Instead, rather than producing dA4/dB1 neurons, in these mutants, Ptf1a-expressing progenitors generate Lmx1b+/Phox2b+ and Lmx1b+/Phox2b− neurons (Figs. 2U, 3Q, 4K). In Ptf1aCre/Cre;ROSA-YFP (Ptf1a−/−) caudal brainstem, ectopic Lmx1b+/Phox2b+/YFP+ dA3-like cells occupied the area normally populated by newborn dA4 ION neurons, suggesting that, in the absence of Ptf1a, dorsally located Ptf1a-expressing progenitors generate dA3 cells instead of dA4 ION neurons (Figs. 2U, 3Q). Thus, it is likely that previously reported ION loss in Ptf1a−/− mice (Yamada et al., 2007) is caused by an early misspecification of dA4 ION neurons into dA3 Lmx1b+/Phox2b+ cells. Consistent with transfating of dA4 ION neurons into Lmx1b+/Phox2b+ dA3 cells at E11.5, in E18.5 Ptf1a−/− embryos we observed excessive numbers of Lmx1b+/Phox2b+ nTs neurons, which, based on birth-dating analysis, aberrantly arose from Ptf1a-expressing progenitors during early neurogenesis. Lmx1b+/Phox2b− neurons aberrantly originating from Ptf1a-expressing progenitors in E11.5 Ptf1a−/− embryos, had the gene expression profile of somatosensory neurons. In E18.5 Ptf1a−/− mutants, we detected excessive Lmx1b+ SpV neurons, some of which, based on birth-dating analysis, arose from the Ptf1a lineage during early neurogenesis. Thus, combining E18.5 and E11.5 analyses, we conclude that during early (E11.5) brainstem neurogenesis Ptf1a regulates the production of Pax2+ viscerosensory and somatosensory neurons and ION neurons by segregating these lineages from Lmx1b+ viscerosensory and somatosensory fates.
In contrast to wild-type E12.5 rh7, where dBLa/dBLb neurons arise in a salt-and-pepper pattern (Sieber et al., 2007), in Ptf1a−/− E12.5 rh7, Pax2+ dBLa cells were absent while Lmx1b+ dBLb neurons increased in number. A lack of changes in proliferation and apoptosis strongly suggests that the production of dBLa/dBLb cells in Ptf1a−/− rh7 was affected by a cell-fate misspecification. Since both dBLa and dBLb cells normally arise from Ptf1a-expresing progenitors, we could not use Ptf1aCre/ROSA fate mapping to directly demonstrate transfating of dBLa to dBLb cells. Birth-dating experiments revealed that in Ptf1a−/− mutants, excessive Lmx1b+ neurons born at E12.25, contribute to the SpV. Thus, our data support the conclusion that during late neurogenesis Ptf1a promotes the production of Pax2+ viscerosensory and somatosensory neurons, and limits the excessive generation of Lmx1b+ SpV neurons from Ptf1a-expressing progenitors.
Unlike the nTs and SpV nuclei, which develop in caudal hindbrain (Sieber et al., 2007; D'Autréaux et al., 2011) and in Ptf1a−/− mutants lack Pax2+ GABAergic neurons, Pax2+ GABAergic neurons in the Ptf1a−/− PrV nuclei were reduced in number but not lost. Interestingly, in Ptf1a−/− rh2–3, where PrV neurons arise (Oury et al., 2006), we observed a complete loss of dA4/dB1 neurons and misspecification of the Ptf1a lineage into Lmx1b+ neurons, which is comparable to that in more caudal rhombomeres. In contrast to control Ptf1aCre/+;nLacZ embryos, where Pax2+ PrV cells were β-gal+, in Ptf1a−/− (Ptf1aCre/YFP;nLacZ) mutants, Pax2+ PrV cells did not express β-gal. Therefore, it is likely that in Ptf1a mutants Pax2+ PrV cells belonging to the Ptf1a lineage become misspecified, and their loss is partially compensated by Pax2+ neurons originating from a Ptf1a− lineage, for example, dB4 cells, which express Pax2 but originate from Ptf1a−, more ventral progenitors (Fig. 4B–E).
In addition to viscerosensory, somatosensory, and ION defects, cochlear nuclei were also affected in Ptf1a−/− embryos (Fujiyama et al., 2009). Cochlear neurons are born in rh2–5 (Landsberg et al., 2005), and a subset of them arises from Ptf1a-expressing progenitors (Fujiyama et al., 2009). It is possible that the cell-fate misspecification that we describe in rh2–5 (Fig. 3Q, 4K) contributes to the development of Ptf1a−/− cochlear abnormalities. Currently, however, it is not clear how cochlear neurons correlate with dA/dB neurons in early rh2–5. Thus, additional studies are needed to determine whether Ptf1a regulates the development of cochlear neurons by specifying their fate.
Ptf1a as a regulator of neurogenesis in the developing CNS
Together with earlier Ptf1a studies, our data argue that suppressing the Lmx1b+ cell fate is a major function of Ptf1a conserved throughout the CNS, highlighting the similarities of fundamental cell-fate specification mechanisms in different CNS regions. Although in Ptf1a−/− hindbrain, a small fraction of Ptf1a lineage cells were misspecified into cerebellar granule cells and mossy fiber neurons (Pascual et al., 2007; Yamada et al., 2007), which are not known to express Lmx1b, the majority of Ptf1a lineage cells in Ptf1a−/− mutants, including those in the cerebellum (Millen et al., 2014), spinal cord (Glasgow et al., 2005), and brainstem (this study) transfated to Lmx1b+ neurons, which normally arise adjacent to the Ptf1a lineage in most CNS regions. Notably, even in rh2–6, where neurons originating from Ptf1a+ progenitors are normally flanked by Phox2b+/Lmx1b− dB2 cells, the loss of Ptf1a converts Ptf1a lineage cells into Lmx1b+ rather than dB2 cells (Fig. 3Q, 4K). Since the loss of a single gene, Ptf1a, converts large populations in the cerebellum, spinal cord, and rh2–7 into Lmx1b+ neurons, it is likely that Lmx1b+ cell fate is a default fate in dorsal CNS caudal to the mid-hindbrain boundary.
The development of mammalian CNS is controlled by a hierarchy of transcription factors that, first, subdivide the VZ along the dorsal–ventral axis into discrete progenitor domains, such as bHLH Atoh1, Ngn1/2, and Ascl1 in the spinal cord. Additional genes then establish specific programs in neurons arising from each domain (Gowan et al., 2001; Gross et al., 2002; Gifford et al., 2013; Gray, 2013). Since in rh2–7 Ptf1a is expressed in large progenitor domains producing multiple cell types, it is likely that Ptf1a is located relatively high in the hierarchy of transcription factors controlling cell-fate specification in the brainstem. In the CNS, Ptf1a regulates the fates of neurons generated in several distinct patterns of neurogenesis. In the cerebellum, Purkinje cells and molecular layer interneurons sequentially arise from the Ptf1a-expressing cerebellar VZ; in early rh2–7, dA4/dB1 cells arise from spatially distinct domains in the Ptf1+ VZ, while dBLa/dBLb neurons arise in a salt-and-pepper pattern in later rh7. Several examples illustrate that Ptf1a supports the production of distinct neuronal types by interacting with region-specific transcription factors. In the cerebellum, Gsx1 acts as a switch from Ptf1a-dependent production of Purkinje cells to the generation of molecular-layer interneurons (Seto et al., 2014). In caudal hindbrain, Olig3 interacts with Ptf1a during the differentiation of dA4 ION neurons (Storm et al., 2009). Additional studies are required to better understand temporal control of Ptf1a-related neurogenesis and to identify additional regulators subdividing the Ptf1a lineage into specific cell types.
In conclusion, we identify Ptf1a as a major regulator of cell-fate decisions in the mouse brainstem. Since homozygous PTF1A mutations were described in humans (Sellick et al., 2004), our study may help to delineate hindbrain pathology in these patients.
Footnotes
This work was supported by National Institutes of Health Grant R21-NS-077163 to VC and by the Neuroscience Institute of the University of Tennessee Health Science Center. We thank C.V. Wright (Vanderbilt University Medical Center, Nashville, TN), M. Magnuson (Vanderbilt University School of Medicine, Nashville, TN), J.F. Brunet (IBENS, Paris, France), M. Goulding (Salk Institute, La Jolla, CA), H. Edlund (University of Umea, Umea, Sweden), F.M. Zhou (University of Tennessee Health Science Center), E. Hedlund and T. Perlmann (Karolinska Institute, Stockholm, Sweden), and S. Arber (University of Basel, Basel, Switzerland) for mice and reagents; and K.J. Millen (Seattle Children's), and W.E. Armstrong, J.M. Ross, L. Fremuth, C.J. Bohl, K.M. Hamre, and M. Ennis (University of Tennessee Health Science Center) for helpful discussions and valuable comments on the manuscript.
References
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Blessing WW. The lower brainstem and bodily homeostasis. New York: Oxford UP; 1997. [Google Scholar]
- Borromeo MD, Meredith DM, Castro DS, Chang JC, Tung KC, Guillemot F, Johnson JE. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Development. 2014;141:2803–2812. doi: 10.1242/dev.105866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlison JS, Long Q, Fujitani Y, Wright CV, Magnuson MA. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev Biol. 2008;316:74–86. doi: 10.1016/j.ydbio.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Arata A, Mizuguchi R, Qian Y, Karunaratne A, Gray PA, Arata S, Shirasawa S, Bouchard M, Luo P, Chen CL, Busslinger M, Goulding M, Onimaru H, Ma Q. Tlx3 and Tlx1 are post-mitotic selector genes determining glutamatergic over GABAergic cell fates. Nat Neurosci. 2004;7:510–517. doi: 10.1038/nn1221. [DOI] [PubMed] [Google Scholar]
- Chizhikov VV, Lindgren AG, Currle DS, Rose MF, Monuki ES, Millen KJ. The roof plate regulates cerebellar cell-type specification and proliferation. Development. 2006;133:2793–2804. doi: 10.1242/dev.02441. [DOI] [PubMed] [Google Scholar]
- Cordes SP. Molecular genetics of cranial nerve development in mouse. Nat Rev Neurosci. 2001;2:611–623. doi: 10.1038/35090039. [DOI] [PubMed] [Google Scholar]
- Dai JX, Hu ZL, Shi M, Guo C, Ding YQ. Postnatal ontogeny of the transcription factor Lmx1b in the mouse central nervous system. J Comp Neurol. 2008;509:341–355. doi: 10.1002/cne.21759. [DOI] [PubMed] [Google Scholar]
- Dauger S, Pattyn A, Lofaso F, Gaultier C, Goridis C, Gallego J, Brunet JF. Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development. 2003;130:6635–6642. doi: 10.1242/dev.00866. [DOI] [PubMed] [Google Scholar]
- D'Autréaux F, Coppola E, Hirsch MR, Birchmeier C, Brunet JF. Homeoprotein Phox2b commands a somatic-to-visceral switch in cranial sensory pathways. Proc Natl Acad Sci U S A. 2011;108:20018–20023. doi: 10.1073/pnas.1110416108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farago AF, Awatramani RB, Dymecki SM. Assembly of the brainstem cochlear nuclear complex is revealed by intersectional and subtractive genetic fate maps. Neuron. 2006;50:205–218. doi: 10.1016/j.neuron.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Fujiyama T, Yamada M, Terao M, Terashima T, Hioki H, Inoue YU, Inoue T, Masuyama N, Obata K, Yanagawa Y, Kawaguchi Y, Nabeshima Y, Hoshino M. Inhibitory and excitatory subtypes of cochlear nucleus neurons are defined by distinct bHLH transcription factors, Ptf1a and Atoh1. Development. 2009;136:2049–2058. doi: 10.1242/dev.033480. [DOI] [PubMed] [Google Scholar]
- Gifford WD, Hayashi M, Sternfeld M, Tsai J, Alaynick WA, Pfaff SL. Spinal cord patterning. In: Rakic P, Rubenstein J, editors. Comprehensive developmental neuroscience: patterning and cell type specification in the developing CNS and PNS. New York: Elsevier; 2013. pp. 131–150. [Google Scholar]
- Glasgow SM, Henke RM, Macdonald RJ, Wright CV, Johnson JE. Ptf1a determines GABAergic over glutamatergic neuronal cell fate in the spinal cord dorsal horn. Development. 2005;132:5461–5469. doi: 10.1242/dev.02167. [DOI] [PubMed] [Google Scholar]
- Gowan K, Helms AW, Hunsaker TL, Collisson T, Ebert PJ, Odom R, Johnson JE. Crossinhibitory activities of Ngn1 and Math1 allow specification of distinct dorsal interneurons. Neuron. 2001;31:219–232. doi: 10.1016/S0896-6273(01)00367-1. [DOI] [PubMed] [Google Scholar]
- Gray PA. Transcription factors define the neuroanatomical organization of the medullary reticular formation. Front Neuroanat. 2013;7:7. doi: 10.3389/fnana.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross MK, Dottori M, Goulding M. Lbx1 specifies somatosensory association interneurons in the dorsal spinal cord. Neuron. 2002;34:535–549. doi: 10.1016/S0896-6273(02)00690-6. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K, Cholewa-Waclaw J, Nakada Y, Glasgow SM, Masui T, Henke RM, Wildner H, Martarelli B, Beres TM, Epstein JA, Magnuson MA, Macdonald RJ, Birchmeier C, Johnson JE. A nonclassical bHLH Rbpj transcription factor complex is required for specification of GABAergic neurons independent of Notch signaling. Genes Dev. 2008;22:166–178. doi: 10.1101/gad.1628008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino M, Nakamura S, Mori K, Kawauchi T, Terao M, Nishimura YV, Fukuda A, Fuse T, Matsuo N, Sone M, Watanabe M, Bito H, Terashima T, Wright CV, Kawaguchi Y, Nakao K, Nabeshima Y. Ptf1a, a bHLH transcriptional gene, defines GABAergic neuronal fates in cerebellum. Neuron. 2005;47:201–213. doi: 10.1016/j.neuron.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Huang X, Liu J, Ketova T, Fleming JT, Grover VK, Cooper MK, Litingtung Y, Chiang C. Transventricular delivery of Sonic hedgehog is essential to cerebellar ventricular zone development. Proc Natl Acad Sci U S A. 2010;107:8422–8427. doi: 10.1073/pnas.0911838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahagirdar V, Wagner CK. Ontogeny of progesterone receptor expression in the subplate of fetal and neonatal rat cortex. Cereb Cortex. 2010;20:1046–1052. doi: 10.1093/cercor/bhp165. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- Landsberg RL, Awatramani RB, Hunter NL, Farago AF, DiPietrantonio HJ, Rodriguez CI, Dymecki SM. Hindbrain rhombic lip is comprised of discrete progenitor cell populations allocated by Pax6. Neuron. 2005;48:933–947. doi: 10.1016/j.neuron.2005.11.031. [DOI] [PubMed] [Google Scholar]
- Maricich SM, Herrup K. Pax-2 expression defines a subset of GABAergic interneurons and their precursors in the developing murine cerebellum. J Neurobiol. 1999;41:281–294. doi: 10.1002/(SICI)1097-4695(19991105)41:2. 3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Millen KJ, Steshina EY, Iskusnykh IY, Chizhikov VV. Transformation of the cerebellum into more ventral brainstem fates causes cerebellar agenesis in the absence of Ptf1a function. Proc Natl Acad Sci U S A. 2014;111:E1777–E1786. doi: 10.1073/pnas.1315024111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomaksteinsky M, Kassabov S, Chettouh Z, Stoeklé HC, Bonnaud L, Fortin G, Kandel ER, Brunet JF. Ancient origin of somatic and visceral neurons. BMC Biol. 2013;11:53. doi: 10.1186/1741-7007-11-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oury F, Murakami Y, Renaud JS, Pasqualetti M, Charnay P, Ren SY, Rijli FM. Hoxa2- and rhombomere-dependent development of the mouse facial somatosensory map. Science. 2006;313:1408–1413. doi: 10.1126/science.1130042. [DOI] [PubMed] [Google Scholar]
- Pascual M, Abasolo I, Mingorance-Le Meur A, Martínez A, Del Rio JA, Wright CV, Real FX, Soriano E. Cerebellar GABAergic progenitors adopt an external granule cell-like phenotype in the absence of Ptf1a transcription factor expression. Proc Natl Acad Sci U S A. 2007;104:5193–5198. doi: 10.1073/pnas.0605699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development. 1997;124:4065–4075. doi: 10.1242/dev.124.20.4065. [DOI] [PubMed] [Google Scholar]
- Pickles JO. Auditory pathways: anatomy and physiology. Handb Clin Neurol. 2015;129:3–25. doi: 10.1016/B978-0-444-62630-1.00001-9. [DOI] [PubMed] [Google Scholar]
- Qian Y, Fritzsch B, Shirasawa S, Chen CL, Choi Y, Ma Q. Formation of brainstem (nor)adrenergic centers and first-order relay visceral sensory neurons is dependent on homeodomain protein Rnx/Tlx3. Genes Dev. 2001;15:2533–2545. doi: 10.1101/gad.921501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Shirasawa S, Chen CL, Cheng L, Ma Q. Proper development of relay somatic sensory neurons and D2/D4 interneurons requires homeobox genes Rnx/Tlx-3 and Tlx-1. Genes Dev. 2002;16:1220–1233. doi: 10.1101/gad.982802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash BG, Tomasi S, Lim HD, Suh CY, Vaccarino FM. Cortical gyrification induced by fibroblast growth factor 2 in the mouse brain. J Neurosci. 2013;33:10802–10814. doi: 10.1523/JNEUROSCI.3621-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Kong L, Zhang GR, Liu M, Wang X, Szabo G, Curthoys NP, Geller AI. Glutamatergic or GABAergic neuron-specific, long-term expression in neocortical neurons from helper virus-free HSV-1 vectors containing the phosphate-activated glutaminase, vesicular glutamate transporter-1, or glutamic acid decarboxylase promoter. Brain Res. 2007;1144:19–32. doi: 10.1016/j.brainres.2007.01.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok TJ, Cella F. The rat nervous system. Ed 2. New York: Academic; 1995. Precerebellar nuclei and red nucleus; pp. 277–308. [Google Scholar]
- Schambra U. Prenatal mouse brain atlas. New York: Springer; 2008. [Google Scholar]
- Schubert T, Huckfeldt RM, Parker E, Campbell JE, Wong RO. Assembly of the outer retina in the absence of GABA synthesis in horizontal cells. Neural Dev. 2010;5:15. doi: 10.1186/1749-8104-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, Gloyn AL, Edghill EL, Hattersley AT, Wellauer PK, Goodwin G, Houlston RS. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36:1301–1305. doi: 10.1038/ng1475. [DOI] [PubMed] [Google Scholar]
- Seto Y, Nakatani T, Masuyama N, Taya S, Kumai M, Minaki Y, Hamaguchi A, Inoue YU, Inoue T, Miyashita S, Fujiyama T, Yamada M, Chapman H, Campbell K, Magnuson MA, Wright CV, Kawaguchi Y, Ikenaka K, Takebayashi H, Ishiwata S, Ono Y, Hoshino M. Temporal identity transition from Purkinje cell progenitors to GABAergic interneuron progenitors in the cerebellum. Nat Commun. 2014;5:3337. doi: 10.1038/ncomms4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber MA, Storm R, Martinez-de-la-Torre M, Müller T, Wende H, Reuter K, Vasyutina E, Birchmeier C. Lbx1 acts as a selector gene in the fate determination of somatosensory and viscerosensory relay neurons in the hindbrain. J Neurosci. 2007;27:4902–4909. doi: 10.1523/JNEUROSCI.0717-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm R, Cholewa-Waclaw J, Reuter K, Bröhl D, Sieber M, Treier M, Müller T, Birchmeier C. The bHLH transcription factor Olig3 marks the dorsal neuroepithelium of the hindbrain and is essential for the development of brainstem nuclei. Development. 2009;136:295–305. doi: 10.1242/dev.027193. [DOI] [PubMed] [Google Scholar]
- Sudarov A, Turnbull RK, Kim EJ, Lebel-Potter M, Guillemot F, Joyner AL. Ascl1 genetics reveals insights into cerebellum local circuit assembly. J Neurosci. 2011;31:11055–11069. doi: 10.1523/JNEUROSCI.0479-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson DJ, Steshina EY, Wakenight P, Aldinger KA, Goldowitz D, Millen KJ, Chizhikov VV. Phenotypic and genetic analysis of the cerebellar mutant tmgc26, a new ENU-induced ROR-alpha allele. Eur J Neurosci. 2010;32:707–716. doi: 10.1111/j.1460-9568.2010.07330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite MR, Skaggs K, Kaviany P, Skidmore JM, Causeret F, Martin JF, Martin DM. Distinct populations of GABAergic neurons in mouse rhombomere 1 express but do not require the homeodomain transcription factor PITX2. Mol Cell Neurosci. 2012;49:32–43. doi: 10.1016/j.mcn.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolsey TA. Peritheral alteration and somatosensory development. In: Coleman JR, editor. Development of sensory systems in mammals. New York: Wiley; 1990. pp. 461–516. [Google Scholar]
- Xiang CX, Zhang KH, Johnson RL, Jacquin MF, Chen ZF. The transcription factor, Lmx1b, promotes a neuronal glutamate phenotype and suppresses a GABA one in the embryonic trigeminal brainstem complex. Somatosens Mot Res. 2012;29:1–12. doi: 10.3109/08990220.2011.650869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Terao M, Terashima T, Fujiyama T, Kawaguchi Y, Nabeshima Y, Hoshino M. Origin of climbing fiber neurons and their developmental dependence on Ptf1a. J Neurosci. 2007;27:10924–10934. doi: 10.1523/JNEUROSCI.1423-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Seto Y, Taya S, Owa T, Inoue YU, Inoue T, Kawaguchi Y, Nabeshima Y, Hoshino M. Specification of spatial identities of cerebellar neuron progenitors by ptf1a and atoh1 for proper production of GABAergic and glutamatergic neurons. J Neurosci. 2014;34:4786–4800. doi: 10.1523/JNEUROSCI.2722-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]