

Abstract
Impaired breathing, cardiac function, and arousal during and after seizures are important causes of morbidity and mortality. Previous work suggests that these changes are associated with depressed brainstem function in the ictal and post-ictal periods. Lower brainstem serotonergic systems are postulated to play an important role in cardiorespiratory changes during and after seizures, whereas upper brainstem serotonergic and other systems regulate arousal. However, direct demonstration of seizure-associated neuronal activity changes in brainstem serotonergic regions has been lacking. Here, we performed multiunit and single-unit recordings from medullary raphe and midbrain dorsal raphe nuclei in an established rat seizure model while measuring changes in breathing rate and depth as well as heart rate. Serotonergic neurons were identified by immunohistochemistry. Respiratory rate, tidal volume, and minute ventilation were all significantly decreased during and after seizures in this model. We found that population firing of neurons in the medullary and midbrain raphe on multiunit recordings was significantly decreased during the ictal and post-ictal periods. Single-unit recordings from identified serotonergic neurons in the medullary raphe revealed highly consistently decreased firing during and after seizures. In contrast, firing of midbrain raphe serotonergic neurons was more variable, with a mixture of increases and decreases. The markedly suppressed firing of medullary serotonergic neurons supports their possible role in simultaneously impaired cardiorespiratory function in seizures. Decreased arousal likely arises from depressed population activity of several neuronal pools in the upper brainstem and forebrain. These findings have important implications for preventing morbidity and mortality in people living with epilepsy.
SIGNIFICANCE STATEMENT Seizures often cause impaired breathing, cardiac dysfunction, and loss of consciousness. The brainstem and, specifically, brainstem serotonin neurons are thought to play an important role in controlling breathing, cardiac function, and arousal. We used an established rat seizure model to study the overall neuronal activity in the brainstem as well as firing of specific serotonin neurons while measuring cardiorespiratory function. Our results demonstrated overall decreases in brainstem neuronal activity and marked downregulation of lower brainstem serotonin neuronal firing in association with decreased breathing and heart rate during and after seizures. These findings point the way toward new treatments to augment brainstem function and serotonin, aiming to prevent seizure complications and reduce morbidity and mortality in people living with epilepsy.
Keywords: brainstem, consciousness, respiratory, serotonin, SUDEP, temporal lobe epilepsy
Introduction
Seizures can affect brain function both locally and remotely through different circuits and mechanisms causing impaired arousal, cardiorespiratory changes, and, in some cases, death. Repeated cardiorespiratory dysfunction during and after seizures may contribute to chronic hypoxic brain damage and to long-term deficits in epilepsy (Schridde et al., 2008; Choy et al., 2014). More acutely, sudden unexpected death in epilepsy (SUDEP) is a condition in which patients with epilepsy die suddenly, usually during or after a seizure and with no other identifiable cause of death. SUDEP is likely due to a combination of seizure-induced cardiovascular, breathing, and arousal impairment (Bateman et al., 2010; Richerson and Buchanan, 2011; Ryvlin et al., 2013; Sowers et al., 2013). Serotonin is known to play a key role in regulating all of these functions via arousal-modulating and cardiorespiratory control centers in the upper and lower brainstem, respectively (Bradley et al., 2002; Severson et al., 2003; Richerson, 2004; Buchanan and Richerson, 2010); however, the specific changes in electrical activity of brainstem serotonergic neurons during seizures have not previously been investigated directly.
Recent work from animal models has demonstrated that depression of upper brainstem ascending arousal systems during and after seizures may be necessary and sufficient for impaired conscious arousal (Blumenfeld et al., 2004b, 2012; Englot et al., 2008, 2009, 2010; Gummadavelli et al., 2015; Motelow et al., 2015; Furman et al., 2015). We propose that similar mechanisms may also depress lower brainstem modulatory function during seizures, including serotonergic neurons in the medullary raphe, contributing to cardiorespiratory impairment during and after seizures.
Previous work suggests that the pathophysiology of SUDEP overlaps with sudden infant death syndrome (SIDS), which has been linked to abnormalities of the brainstem serotonergic system leading to apnea and death (Buchanan and Richerson, 2009; Richerson and Buchanan, 2011; Sowers et al., 2013). Serotonergic neurons in the midbrain dorsal raphe project rostrally to modulate cognition, mood, and arousal, whereas serotonergic neurons in the medullary raphe project caudally to modulate breathing, cardiac, and other functions (Kinney et al., 2009; Buchanan and Richerson, 2010). Serotonergic 5-hydroxytryptamine (5-HT) neurons in both the medullary and midbrain raphe are central chemoreceptors and are highly sensitive to pH and CO2 (Richerson, 1995; Severson et al., 2003; Richerson, 2004). Lmx1bf/f/p mice lacking almost all 5-HT neurons showed decreased ventilation in response to PCO2 stimulation (Hodges et al., 2008), impaired arousal from sleep in response to inspired CO2 (Buchanan and Richerson, 2010), and increased mortality after electrically induced seizures (Buchanan et al., 2014).
This research aimed to determine changes in the firing of medullary and midbrain raphe neurons during and after seizures in an established rodent seizure model. We first investigated the population firing of neurons using multiunit recordings in the medullary and midbrain raphe and then identified single serotonergic neurons using juxtacellar recordings and immunohistochemistry. Parallel measurements of breathing plethysmography and heart rate by EKG enabled us to determine changes in brainstem serotonergic neuronal activity associated with impaired cardiorespiratory function during and after seizures.
Materials and Methods
Animals
All procedures were conducted under approved protocols of Yale University's Institutional Animal Care and Use Committee (IACUC). A total of 39 adult female Sprague Dawley rats (Charles River Laboratories) weighing 196–285 g were used in this research. Twenty-three animals were used for multiunit activity (MUA) recordings in the dorsal raphe and medullary raphe nuclei and 16 for juxtacellular single-unit activity recordings. Breathing data were obtained during and after seizures for 19 animals and EKG data for 12 animals.
Breathing measurements
Respiratory function was recorded via a modified head-out-plethysmography method, which is based on a previously described setup (Cummings and Frappell, 2009). We made minor changes that facilitated assessment of breathing parameters such as percentage change in minute ventilation, tidal volume, and breathing frequency while the rat remained in the stereotaxis in our acute seizure model. Briefly, we first fixed a mask made from a 1.5 inch piece of SILASTIC tubing (Tygon R-3603, outer diameter 5/8 inch, inner diameter 1/2 inch) over the snout of the animal with tissue glue (Loctite, D4236), leaving the front teeth accessible to the stereotaxis bite bar. Next, we sealed the opposite end of the tubing mask to a small-volume chamber, which was constructed by juxtaposing the concave faces of the rubber gaskets from one 50 ml and one 20 ml syringe as described previously (Cummings and Frappell, 2009). Two small connectors (tube-to-tube, 1/8 inch to1/16 inch reducing straight connectors, McMaster Carr, 53055K514) were inserted into the other side of the gasket chamber for two tubes, one for gas inflow and one for outflow. Inflow of room air was provided by an air pump (Whisper 10 Aquarium; Tetra), and outflow by the wall vacuum controlled by a subminiature vacuum regulator (Airtrol, V-800-30). Before all experiments, the steady-state flow rate of both the inflow and outflow was adjusted to 160 ml/min using a direct reading air flowmeter (Cole Parmer, EW-32003-10). To measure breathing, a respiratory flow head (ADInstruments, MLT10L) was inserted 10 cm from the inflow side of the chamber, and a differential pressure transducer (ultra-low pressure/high sensitivity pressure transducer DC002NDR5; Honeywell) was connected to both sides of the flow head to measure the pressure drop across the resistance of the flow head wire mesh. Breathing was monitored throughout the experiments and signals from the pressure transducer were digitized at 1000 Hz and recorded using a CED Power 1401 and Spike2 software (CED).
EKG measurements
Two copper electrodes (30-40-2, male amphenol pins; FHC) were attached to the two sides of the rat chest wall to obtain EKG data. The chest skin was shaved, Quik-Gel (#92000015; Compumedics Neuromedical Supplies) applied around the electrodes to increase conductance, and sterile adhesive bandages were used to fix the electrodes in place. EKG data were amplified (1000×) with a differential amplifier (A-M Systems, 1800) and then digitized at 1000 Hz using a CED Power 1401 and Spike2 software (CED).
MUA recordings from medullary raphe and dorsal raphe
As described previously (Englot et al., 2008), for surgical procedures, animals were first deeply anesthetized with ketamine/xylazine (90/15 mg/kg, i.m.). Responsiveness to pain was checked every 15 min by toe pinch. A heating pad was used to keep the body temperature constant at ∼37°C. A bipolar stainless steel electrode (50–100 kΩ resistance; Plastics One, E363/2-2TW) was placed stereotactically in the dorsal hippocampus (HC) on the right side with the following coordinates, all in mm relative to bregma: anteroposterior (AP) −3.8; mediolateral (ML) +2.5; superior–inferior (SI) −3.2 for stimulation and for recording local field potential (LFP) signals. A steel screw (0–80 × 3/32; Plastics One) was implanted into the skull just caudal to the hippocampal burr hole to help fix the electrode by applying acrylic dental cement (Lang Dental Manufacturing, powder: REF 1220, jet liquid: REF 1403). The two tips of the electrodes were shaved off 0.5 mm of insulation, separated by ∼0.5 mm, and put into the brain in the coronal plane. For recording LFP and MUA in the medullary raphe and dorsal raphe, we placed a high-impedance monopolar electrode (2–4 MΩ resistance; FHC, UEWMGGSEDNNM) with the following coordinates: AP −11.0 to −14.0, ML −0.3 to +0.3, SI −9.0 to −11.0 for medullary raphe; and AP −7.3 to −8.8, ML −0.3 to +0.3, SI −6.2 to −7.0 for midbrain dorsal raphe (approached at 30° to the midline). All stereotactic coordinates (Paxinos and Watson, 1998) are final electrode tip locations in millimeters relative to bregma. Seizure induction and electrophysiological recordings took place between 1 and 4 h after electrode implantation.
The methods for induction of partial and secondarily generalized limbic seizures were described in detail previously (Englot et al., 2008). In brief, seizures were induced from a lightly anesthetized state attained by waiting at least 1 h after the most recent dose of ketamine/xylazine, but ensuring continued unresponsiveness to toe pinch (checked every 15 min) and immediately administering supplemental anesthetic if any movement or other response such as increased heart rate or respirations occurred. The lightly anesthetized state thus established reduced confounding effects of anesthetics while maintaining a humane experimental paradigm approved by our IACUC. We delivered a 2 s, 60 Hz, biphasic square-wave 1 ms per phase stimulus train to the bipolar hippocampal electrode using an isolated pulse stimulator (A-M Systems, 2100) with stimuli titrated between 150 and 1500 μA to induce seizures lasting at least 30 s after the stimulus. HC LFP signals were amplified (×1000) and filtered (1–500 Hz) using a Microelectrode AC Amplifier (A-M Systems, 1800). Medullary and midbrain raphe signals were broad-band filtered from 0.1 Hz to 10 kHz (×1000 gain) using the same amplifier and then filtered with an analog filter (unity gain; Krohn-Hite, model 3364) into LFP (0.1–100 Hz) and MUA signals (400 Hz to 10 kHz). All electrophysiology signals were digitized and recorded (sampling rate 1 kHz for LFP, 20 kHz for MUA) using a Power 1401 (CED) and Spike2 software (CED). At the conclusion of experiments, the location of all electrodes were verified by histology (described below).
Juxtacellular recordings from medullary raphe and dorsal raphe
Animal preparation, electrodes in HC, and seizure induction were conducted in the same way as described above. Extracellular single-unit activity recordings were acquired using the juxtacellular method (Pinault, 1996; Motelow et al., 2015). Briefly, glass electrodes were used to collect single-unit activity data of both medullary raphe and dorsal raphe regions targeting the same coordinates as above with a micromanipulator (Sutter Instruments, MPC-325). 1.5 mm × 100 mm borosilicate glass capillaries (#1B150F-4, World Precision Instruments) were pulled on a Flaming/Brown micropipette puller (Sutter Instruments, P-1000 horizontal puller), which were then bumped under a microscope to produce a flat electrode tip and filled with 3% Neurobiotin (Vector Laboratories, SP-1120) in saline (0.9% NaCl). Only those electrodes with a resistance of 15–30 MΩ were used during experiments. An Axoclamp-2B amplifier (Molecular Devices, ×10 gain, current-clamp mode) was used to acquire single-unit activity signals digitized at 20,000 Hz using a Power 1401 and Spike2 software (CED). Once a neuron was stably recorded from during the baseline, seizure, and recovery periods, it was labeled by passing current pulses (0.7–50 nA, pulse duration 150 milliseconds, 3 Hz) through the electrode tip to drive firing as described previously (Ros et al., 2009; Motelow et al., 2015). Locations of recorded cells were obtained by histology as described in the next section.
Immunohistochemistry and microscopy
At the end of all experiments, animals were euthanized via intraperitoneal injection of Euthasol (Schein) to verify histologically the location of electrodes and neurons. Animals were perfused through the cardiac ventricle with heparinized saline, followed by 500 ml of cold fresh 4% paraformaldehyde (PFA) solution, and then brains were collected and postfixed for 24–72 h in PFA.
Brains were sliced at 100 μm thickness for MUA recordings and 60 μm thickness for juxtacellular cellular recordings using a vibratome (Leica, VT1000S) at the region surrounding the level of recordings. For MUA recordings in the midbrain or medullary raphe, slices were incubated in a series of solutions at room temperature listed below to obtain staining of serotonergic neurons: 30 min in blocking buffer containing 5% donkey serum (Sigma-Aldrich, D9663) in PBS with 0.3% Triton-X (PBS-T); overnight in primary antibody of monoclonal mouse anti-tryptophan hydroxylase (Sigma-Aldrich, T0678) in blocking buffer (1:500); 3 10 min washes of PBS-T; 2 h in secondary antibody of Alexa Fluor 647 donkey anti-mouse IgG (H+L) (Life Technologies, A-31571) in blocking buffer (1:500); and then 2 10 min PBS-T washes and 1 10 min PBS wash. Slices were mounted for microscopy using Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, H-1200) to label cell nuclei and sealed with nail polish. The location of multiunit recording electrode tracts and serotonergic neurons were then identified using a Leica DM5500 Microsystems microscope and the Leica LAS Microimage system.
For juxtacellular recordings, 30–40 slices were collected and incubated in a diluted cyanine 3-conjugated to streptavidin (Jackson ImmunoResearch) in blocking buffer 1:1000) for 3 h. After three 10 min washes in PBS-T, slices were “wet mounted” and inspected under the Leica DM5500 Microsystems to identify Neurobiotin-filled cells. The slices containing the cell bodies were then stained for serotonergic neurons and mounted for microscopy as described above. Confocal images were obtained on a Zeiss LSM 710 Duo NLO/multiphoton microscope using a C-Apochromat 63×/1.2 W Corr objective to determine whether Neurobiotin-filled cells were also stained for tryptophan hydroxylase.
Data analysis
Breathing data analysis.
Digitized breathing data from the pressure transducer were analyzed initially in Spike2 and then using in-house software written in MATLAB (The MathWorks). The peak pressure of each respiration was determined using a built-in function of Spike2. The times of these events were exported to MATLAB, which was used to calculate the respiratory rate. Because the breathing trace was proportional to flow, we obtained a measure proportional to tidal volume by calculating the integral of the breathing trace amplitude (absolute value) and dividing by the number of breaths per unit time (Cummings and Frappell, 2009). Minute ventilation was defined as the average tidal volume during an epoch multiplied by the number of breaths and divided by the number of minutes in that epoch. Breathing data were analyzed during epochs of baseline (30–0 s before stimulus), seizure period (the whole period of hippocampal seizure activity based on polyspike activity in the LFP), and the post-ictal period (0–30 s after seizure). All values were expressed as percentage change relative to preseizure baseline, or 100 × (value − baseline)/baseline. When more than one seizure was triggered in an animal, we consistently used the first seizure's breathing data from each animal for analysis. Statistical significance was assessed at p of 0.05 with Holm-Bonferroni correction for multiple comparisons in the ictal and post-ictal periods.
MUA analysis.
Recordings of MUA were analyzed using in-house software written on MATLAB (R2009a, Mathworks). Baseline, seizure and post-ictal epoch were defined as for the breathing data analysis. Root mean squared (RMS) voltage was calculated by squaring the signal, taking the mean in one-second nonoverlapping bins and then taking the square root. For group analysis we calculated percentage change from the 30 s of baseline before seizure initiation, using a statistical threshold p of 0.05 with Holm-Bonferroni correction as above. When more than one seizure was obtained per animal we used a conservative approach by first taking averages within animals and then performed statistics across animals (sample size = number of animals), although very similar results were obtained if instead statistics were performed across seizures. We used RMS as an estimate of action potential firing because this approach has been validated previously under normal conditions and during seizures (Logothetis et al., 2001; Shmuel et al., 2006; Englot et al., 2009; Mishra et al., 2011) and template matching may be unreliable for analysis of MUA during seizures (Schridde et al., 2008).
Juxtacellular analysis.
Spike sorting on the juxtacellular recordings was performed using Spike2 (CED, v5.20a) and recordings were then analyzed using in-house software written on MATLAB (R2009a, Mathworks). Analyses were performed only on recorded serotonergic neurons identified by histology. Group firing rate analyses of single-unit activity (SUA) for each group (serotonergic neurons for midbrain and medullary raphe) were performed using paired two-tailed t tests comparing the firing rate over the course of the seizure (seizure) and the first 30 s after seizure (post-ictal) with the last 30 s before seizure initiation (baseline). In animals with multiple seizures or multiple neurons recorded we again took averages first within animals and then performed statistics across animals. Significance threshold was p of 0.05, Holm-Bonferroni corrected.
EKG analysis.
Digitized EKG data were analyzed initially in Spike2 and then using in-house software written in MATLAB. The peak of the EKG trace for each systolic-diastolic cycle was determined using a built-in function of Spike2. The times of these events were exported to MATLAB which was used to calculate the heart rate. Baseline, seizure and post-ictal epochs were defined as for the breathing and unit activity data analyses, and similarly percentage change in heart rate was calculated for the ictal and post-ictal periods with Holm-Bonferroni corrected significance threshold p of 0.05.
Results
Impaired cardiorespiratory function in the ictal and post-ictal periods
We collected breathing data during acute seizures in 19 animals with simultaneous MUA or juxtacellular recordings, and EKG data under the same conditions from 12 of these animals. Breathing was markedly impaired during and after seizures, with average minute ventilation decreased by 38% during seizures and by 49% in the post-ictal period (both p < 0.001) due to simultaneous decreases in both respiratory rate and tidal volume (Fig. 1A). Representative examples of impaired breathing during and after seizures are also shown in the lower traces of Figures 2B, 3B, 5C, and 7C.
Figure 1.

Effects of seizures on cardiorespiratory function and brainstem neuronal activity. A, Cardiorespiratory effects of seizures induced by 2 s of hippocampal stimulation. Mean minute ventilation, respiratory rate, tidal volume, and heart rate all decrease in the ictal and post-ictal periods compared with 30 s of preseizure baseline. B, Effects of seizures on neuronal activity in the medullary raphe and midbrain dorsal raphe nuclei. Mean MUA from the medullary and midbrain raphe and SUA from medullary 5-HT neurons decreased in the ictal and post-ictal periods. Mean SUA of 5-HT neurons in the midbrain dorsal raphe was not significantly changed by seizures. Locations of recording electrodes and neurons are shown in Figures 4 and 6. Sample sizes (n, animals) are indicated. One seizure per animal was analyzed for A. For B, the number of seizures analyzed were 63 for MUA in medulla, 58 for MUA in midbrain, 8 for SUA in medulla, and 10 for SUA in midbrain. Error bars indicate SEM. *p ≤ 0.05; **p < 0.005, Holm–Bonferroni corrected.
Figure 2.

Decreased MUA in medullary raphe and reduced breathing during seizure. A, Seizure induced by 2 s stimulation of HC. After the stimulus, fast polyspike activity is seen in the HC LFP. MUA from the medullary raphe shows marked suppression of neuronal firing during the seizure with gradual recovery in the post-ictal period. Respiratory trace shows breathing flow amplitude (proportional to volume) and rate both decreased markedly during the ictal and post-ictal periods. B, Expanded segments of data from baseline, seizure, post-ictal, and recovery periods from the boxed regions in A.
Figure 3.

Decreased MUA in midbrain dorsal raphe and reduced breathing during seizure. A, Seizure induced by 2 s of stimulation of HC. After the stimulus, fast polyspike activity is seen in the HC LFP. MUA from the midbrain dorsal raphe shows marked suppression of neuronal firing during the seizure with gradual recovery in the post-ictal period. Respiratory trace shows breathing flow amplitude (proportional to volume) and rate both decreased markedly during the ictal and post-ictal periods. B, Expanded segments of data from baseline, seizure, post-ictal, and recovery periods from the boxed regions in A.
Figure 5.

Medullary raphe 5-HT neuron decreases firing during and after seizure. A, Seizure induced by 2 s stimulation of HC. After the stimulus, fast polyspike activity is seen in the HC LFP. Single unit activity (SUA) from the medullary raphe serotonergic neuron shows marked suppression of neuronal firing during the seizure with gradual recovery in the post-ictal period. Respiratory trace shows breathing flow amplitude and rate both decreased markedly during the ictal and post-ictal periods. B, Expanded segments of data from baseline, seizure, post-ictal, and recovery periods from the boxed regions in A.
Figure 7.

Midbrain raphe 5-HT neuron does not decrease firing during seizure. A, Seizure induced by 2 s stimulation of HC. After the stimulus, fast polyspike activity is seen in the HC LFP. Single-unit activity (SUA) from the midbrain dorsal raphe serotonergic neuron continues firing without major overall changes during or after the seizure. Respiratory trace again shows breathing flow amplitude and rate both decreased markedly during the ictal and post-ictal periods. B, Expanded segments of data from baseline, seizure, post-ictal, and recovery periods from the boxed regions in A.
Average heart rate showed a more modest decrease of 6% during seizures, with a slightly more prominent 9% decrease in the post-ictal period (p < 0.05; Fig. 1A).
MUA decreases in medullary and midbrain raphe during and after seizures
MUA recordings provide a window into the overall population firing of neurons in local brain regions. We targeted our MUA recordings to the medullary and midbrain raphe nuclei because these regions are known to contain serotonergic and other neurons important for control of cardiorespiratory function and arousal (Bradley et al., 2002; Severson et al., 2003; Jordan, 2005; Buchanan and Richerson, 2010). In both the medullary and midbrain raphe, MUA was markedly reduced during seizures, as well as in the post-ictal period (Figs. 2, 3). On average, MUA in the medullary raphe was reduced by 29 ± 8% (mean ± SEM) during seizures and by 34 ± 6% in the post-ictal period; MUA in the midbrain raphe was reduced by 25 ± 7% during seizures and by 34 ± 6% in the post-ictal period (p < 0.001 for all; Fig. 1B). These findings demonstrate robust decreases in firing of neurons in the midbrain and medullary raphe regions. Locations of electrodes were determined histologically and confirmed that many of the neurons surrounding the electrode tips were serotonergic (Fig. 4). However, with MUA recordings, it was not possible to distinguish firing of serotonergic from other neurons contributing to the changes in population activity observed during seizures.
Figure 4.

Serotonergic neurons in regions of medullary and midbrain MUA recordings. A, Histology example from medullary raphe MUA recording: a coronal section at approximately AP −10.3 mm. B, Example from midbrain dorsal raphe MUA recording: coronal section at approximately −7.64 mm. Red boxes in A1 and B1 represent the general regions where serotonergic nuclei are located in the medulla and midbrain, respectively. A2 and B2 show an overview of tryptophan hydroxylase (TPOH) and nuclear DAPI staining of the target slices. A3 and B3 are expanded views of white boxes in A2 and B2, showing serotonergic and other neurons in the vicinity of the MUA electrode tips. Scale bars: A2, 1 mm; B2, 500 μm; A3, 50 μm; B3, 100 μm. A1 and A2 are reproduced with permission from Paxinos and Watson (1998).
Average firing of serotonergic neurons decreases in medulla but not in midbrain raphe
To identify the firing rate changes of identified 5-HT neurons during and after seizures, we next performed extracellular single-unit recordings and labeled individual 5-HT neurons using the juxtacellular method in both the medulla and midbrain. With this approach, we found that firing of medullary raphe serotonergic neurons was markedly reduced during both the ictal and post-ictal periods at the same time that breathing was severely depressed (Fig. 5A). On average, the firing rate of identified serotonergic neurons was reduced by 40 ± 16% during seizures and by 38 ± 18% in the post-ictal period (p < 0.05 for both) (Fig. 1B). Recorded serotonergic neurons were identified by combined staining for neurobiotin from the recording electrode and tryptophan hydroxylase (Fig. 6A). The locations of all recovered medullary raphe serotonergic neurons recorded during seizures are shown in Figure 6C.
Figure 6.
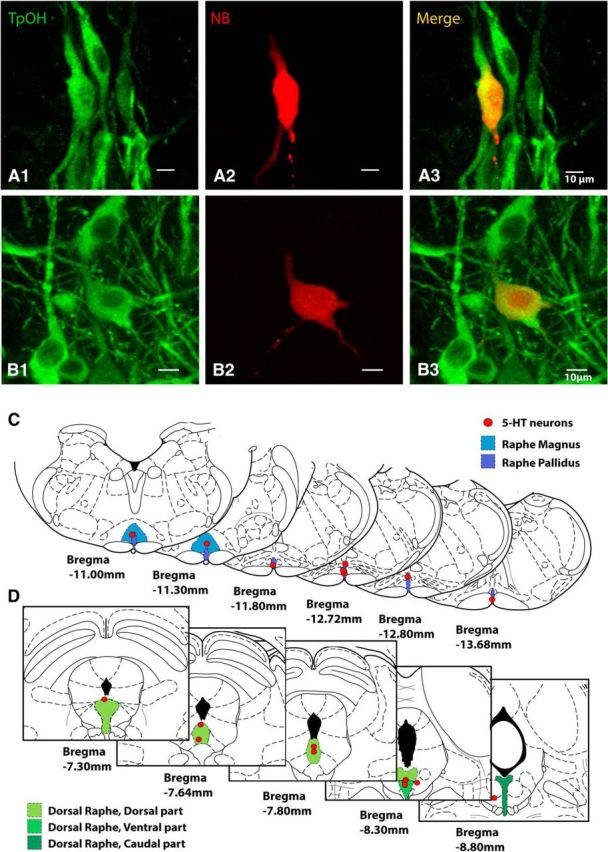
Histology of 5-HT neurons studied by juxtacellular recordings in medulla and midbrain. A, Labeled medullary raphe 5-HT cell recorded juxtacellularly, stained for tryptophan hydroxylase (TPOH) in A1, Neurobiotin (NBiotin) in A2, and merge in A3. B, Labeled midbrain dorsal raphe 5-HT cell recorded juxtacellularly, stained for TPOH in B1, Neurobiotin in B2, and merge in B3. Scale bars, 10 μm. C, Locations of all recorded 5-HT neurons in the medulla (n = 8). Identified 5-HT neurons were located in regions of raphe magnus and raphe pallidus. D, Locations of all recorded 5-HT neurons in the midbrain (n = 10). Identified 5-HT neurons were located in the following regions of the dorsal raphe dorsal, ventral, and caudal subnuclei. Coronal section schematics reproduced with permission from Paxinos and Watson (1998).
Interestingly, although the midbrain dorsal raphe MUA population firing was markedly reduced during and after seizures, as already described (Fig. 3), serotonergic neurons in this region did not show consistently reduced firing (Fig. 7). On average, the firing rate of midbrain raphe serotonergic neurons showed nonsignificant increases of 22 ± 25% during seizures and 16 ± 38% in the post-ictal period (both NS; Fig. 1B). Locations of all recorded midbrain raphe serotonergic neurons are shown in Figure 6D.
The firing of medullary raphe serotonergic neurons decreased consistently (in 8 of 8 neurons) during seizures, as shown in raster plots for individual cells (Fig. 8A,B). Midbrain raphe serotonergic neurons in contrast showed more variable changes in firing, with 7 of 10 neurons showing increases and 3 of 10 showing decreases during seizures (Fig. 8C,D; χ2 = 9.16, p < 0.003; for changes in medullary vs midbrain 5-HT neurons).
Figure 8.
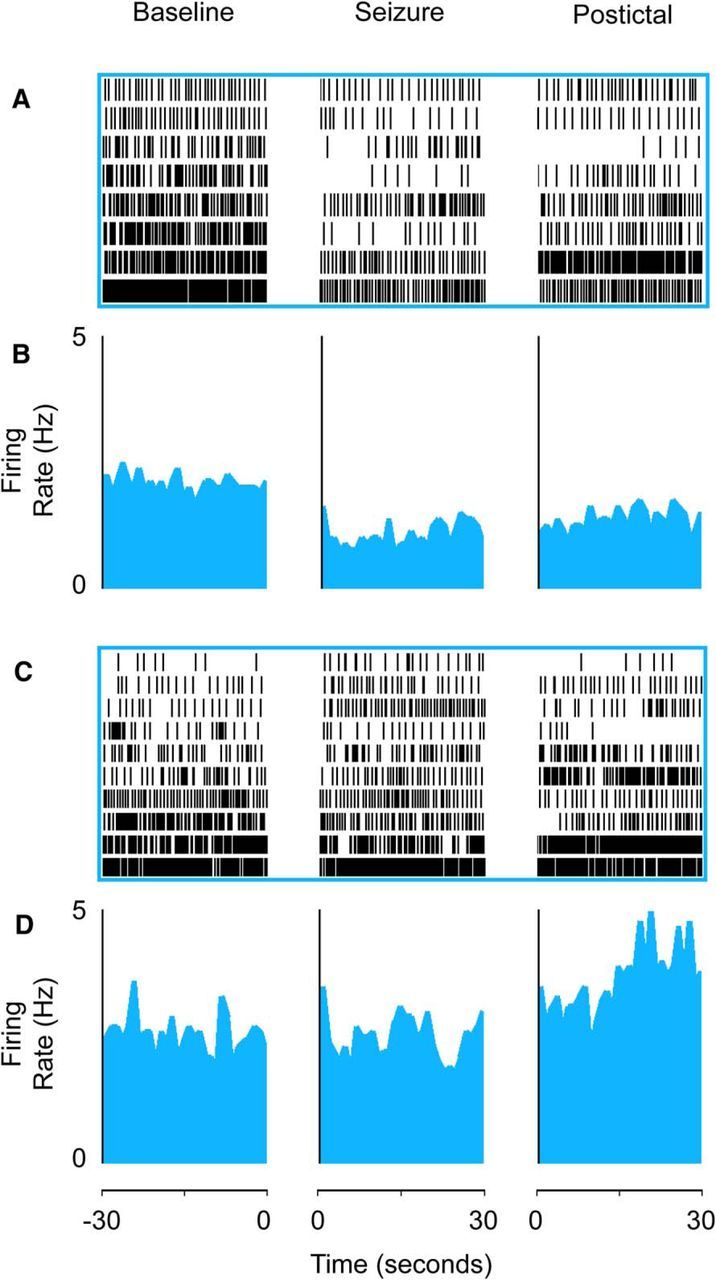
Serotonergic neurons in medulla decrease firing during seizures, whereas firing of midbrain serotonin neurons is mixed. A, B, Medullary raphe serotonergic neurons showed decreased firing during seizures and in the post-ictal period. A, Raster plot of recovered 5-HT medullary raphe cells (n = 8 cells from 8 animals). B, Mean firing rate histogram data. C, D, Midbrain raphe serotonergic neurons had variable changes in firing rate during seizures. C, Raster plot of recovered 5-HT midbrain raphe neurons in dorsal raphe (n = 10 cells from 8 animals). D, Mean firing rate histogram data. Neurons in raster plots (A, C) are ordered based on baseline firing rate for visualization. Histogram data (B, D) are presented as curvilinear plots with firing rates in 1 s nonoverlapping bins. Data are from the 30 s baseline immediately before seizures, the first 30 s after seizure onset, and the first 30 s after seizure end in the post-ictal period. Anatomical locations of all recorded neurons are shown in Figure 7.
Discussion
We investigated neuronal activity in the medulla and midbrain raphe during ictal and post-ictal cardiorespiratory impairment in an established rodent seizure model. Using recordings of MUA, we found markedly reduced population firing of neurons in the medullary and midbrain raphe nuclei during and after seizures. Juxtacellular recordings from single histologically verified serotonergic neurons revealed highly consistent reduced firing of 5-HT neurons in the medulla during and after seizures; however, the firing of midbrain 5-HT neurons was more variable. These finding suggest a model in which depressed firing of specific serotonergic neurons in the medulla may be most strongly associated with reduced output of nearby medullary cardiorespiratory control centers, whereas more general depressed activity in midbrain raphe and other nearby upper brainstem modulatory regions leads to depressed arousal (Motelow et al., 2015; Furman et al., 2015).
Although cardiac, breathing, and arousal effects of seizures have long been recognized, the specific mechanisms by which seizures cause these changes have been relatively neglected (Surges and Sander, 2012; Sowers et al., 2013; Massey et al., 2014; Sedigh-Sarvestani et al., 2015). Changes in cardiorespiratory and arousal functions with seizures have consequences for both chronic and acute morbidity and mortality in epilepsy. Acute cardiovascular changes, hypoventilation, and apnea can significantly increase morbidity and mortality in seizures (Goldman et al., 2009; Moseley et al., 2013). In addition, over time, repeated seizures with intense neuronal activity in vulnerable regions such as the hippocampus depletes local oxygen levels (Schridde et al., 2008; Choy et al., 2014). This may be exacerbated by systemic hypoxia and hypoperfusion during seizures leading to chronic hypoxic brain damage, ultimately contributing to long-term epilepsy comorbidities including cognitive impairment and psychosocial dysfunction (Selassie et al., 2014; Shorvon, 2014; Mula and Cock, 2015).
Furthermore, acute cardiorespiratory changes in the ictal and post-ictal periods may create a state of increased vulnerability to catastrophic cardiorespiratory arrest and SUDEP (Bateman et al., 2010; Richerson and Buchanan, 2011; Ryvlin et al., 2013; Sowers et al., 2013; Massey et al., 2014). SUDEP is the most common cause of premature death in people with epilepsy (Ficker, 2000; Pedley and Hauser, 2002; Shorvon and Tomson, 2011). Recent work suggests that cardiac dysfunction (Surges et al., 2010; Surges and Sander, 2012), impaired respiratory drive, and decreased arousal in the ictal and post-ictal periods are all important factors in triggering SUDEP (Richerson and Buchanan, 2011; Ryvlin et al., 2013; Sowers et al., 2013; Massey et al., 2014). Although the current model does not investigate SUDEP per se, it does provide novel insights into the fundamental mechanisms for brainstem impairment induced by seizures, particularly in key serotonergic modulatory regions that may contribute to a vulnerable state of cardiorespiratory and arousal impairment increasing SUDEP risk. A recent landmark study of SUDEP cases during video/EEG monitoring revealed marked post-ictal cardiac and respiratory abnormalities, followed by apnea and then finally asystole (Ryvlin et al., 2013). Control of both breathing and cardiovascular function depends on the medulla. It has been proposed that, as in SIDS, ictal and post-ictal breathing is depressed because of a reduced brainstem serotonergic response to CO2 (Buchanan and Richerson, 2009; Richerson and Buchanan, 2011; Sowers et al., 2013; Massey et al., 2014). Normally, PCO2-sensitive 5-HT neurons in the medullary raphe stimulate breathing through projections to other medullary breathing control centers such as the pre-Bötzinger complex (Bradley et al., 2002; Richerson, 2004). Reduced 5-HT can cause post-ictal respiratory arrest and death in animal models (Brennan et al., 1997; Buchanan et al., 2014) that are preventable by a selective-serotonin reuptake inhibitor (Faingold et al., 2010; Faingold et al., 2011). The present findings showing consistently reduced firing of serotonergic neurons in the medullary raphe during and after seizures provide key direct mechanistic evidence for suppression of medullary 5-HT neuronal activity in the ictal and post-ictal periods. The results further support a possible role for serotonin-based treatments to prevent these potentially deadly changes.
Altered cardiac function is also common during and after seizures; these include arrhythmias, tachycardia, and bradycardia, as we observed in our model (Goldman et al., 2009; Moseley et al., 2013; Sedigh-Sarvestani et al., 2015). Cardiac control depends on lower brainstem circuits, including parasympathetic outputs from the dorsal vagal nucleus and sympathetic outputs from the rostral ventrolateral medulla, regulated by nucleus tractus solitarius and caudal ventrolateral medulla (Blessing, 1997). The cardiac control regions are under major neuromodulatory influence from medullary serotonergic as well as noradrenergic and cholinergic inputs (Blessing, 1997; Jordan, 2005; Dehkordi et al., 2007). Our finding of markedly decreased firing in medullary raphe serotonergic neurons during and after seizures contributes to this picture of impaired lower brainstem neuromodulatory control of cardiac function associated with seizures.
The mechanisms for depressed lower brainstem function with seizures are presently unknown, but may resemble depressed upper brainstem control of arousal, in which descending inhibition has been postulated to play an important role (Englot and Blumenfeld, 2009; Li et al., 2015; Motelow et al., 2015). Converging evidence supports the “network inhibition hypothesis” in which seizures propagate to GABAergic inhibitory neurons, which in turn depress subcortical arousal in the upper brainstem, leading to loss of consciousness (Norden and Blumenfeld, 2002; Blumenfeld, 2012). Prior work from both human patients (Blumenfeld et al., 2004a, 2004b, 2009; Englot et al., 2010; Cunningham et al., 2014) and rodent seizure models (Englot et al., 2008, 2009; Gummadavelli et al., 2015; Li et al., 2015; Motelow et al., 2015; Furman et al., 2015) suggest that depressed cortical function and impaired consciousness during seizures can be produced by seizure propagation to subcortical structures, leading to inhibition of upper brainstem arousal circuits. Impaired consciousness during and after seizures significantly affects quality of life and may also reduce patients' ability to wake up when lying prone with an obstructed airway, thereby contributing to SUDEP (Buchanan and Richerson, 2010; Massey et al., 2014; Liebenthal et al., 2015). As an extension of the network inhibition hypothesis, seizure propagation to GABAergic regions may also lead to depressed lower brainstem function, including critical medullary regions causing impaired cardiorespiratory function. Descending inhibitory inputs to the medulla could potentially arise from a number of areas. Recent work has shown that seizure propagation to the amygdala may play an important role in suppressing breathing, especially given known inhibitory projections from the amygdala to the brainstem (Dlouhy et al., 2015).
Further work is needed to more fully understand the mechanisms of the decreased neuronal activity that we observed during and after seizures in the midbrain and medullary raphe nuclei and in identified serotonergic neurons in the medulla. In particular, it will be important to determine whether propagation of seizure activity to specific regions outside of the seizure onset zone are necessary and sufficient to produce the observed depressed neuronal activity in the brainstem. The present study did not differentiate between partial seizures and partial seizures with secondary generalization to cortex and other brain regions that may influence the degree of cardiorespiratory impairment and depressed brainstem function. Both partial and secondary generalized seizures are known to affect cardiorespiratory function and the present findings likely represent a mixture of these seizure types. Additional studies are needed with simultaneous recordings in potential sites of seizure propagation, including the cortex, insula, amygdala, and other subcortical regions, to evaluate their role in depressed brainstem function and cardiorespiratory impairment. Future mechanistic work, including optogenetic activation and inhibition of these propagation sites (Furman et al., 2015), will also help to identify the network nodes responsible for these changes. Another important direction for future studies includes the role of additional neurotransmitters aside from serotonin, including acetylcholine, norepinephrine, glutamate, and others in the cardiorespiratory system and arousal changes occurring during seizures. Prior work has already demonstrated a potentially important role for depressed subcortical cholinergic systems in impaired cortical arousal and loss of consciousness (Li et al., 2015; Motelow et al., 2015; Furman et al., 2015). It is interesting, for example, that we observed decreased overall neuronal population activity in the upper brainstem medullary raphe nuclei with multiunit recordings during seizures, whereas single-unit activity in identified serotonergic neurons in the midbrain dorsal raphe did not consistently show decreased firing. It is likely that other nonserotonergic neurons contribute to the overall reduced activity in the upper brainstem during seizures (Motelow et al., 2015), leading to depressed arousal, although consciousness was not monitored in the present study. Nonserotonergic neurons in fact make up a substantial proportion of raphe neurons (Jacobs and Azmitia, 1992; Hendricks et al., 1999; Cerpa et al., 2014), so caution is necessary in comparing single-unit recordings from identified serotonergic neurons with multiunit recordings, which, due to electrode geometry, sample from a much wider population of neurons. Other important factors that could have contributed to the more variable serotonergic firing in the midbrain dorsal raphe include effects of partial versus secondarily generalized seizures and variable levels of anesthestics, including ketamine, known to modulate 5-HT neuronal activity (McCardle and Gartside, 2012; Massey et al., 2015), with possible differential effects in the midbrain versus medulla, all of which should be investigated further. Moreover, specific subpopulations of serotonergic neurons in the dorsal raphe may be responsible for depressed arousal in seizures, whereas other serotonergic neurons in the same region are less involved; this is another important direction for future study. Unlike serotonin neurons in the midbrain, identified serotoninergic neurons in the medulla showed highly consistent reduced firing during and after seizures.
In conclusion, the present study demonstrates overall decreased neuronal firing in the brainstem raphe nuclei during and after seizures, as well as specific consistent decreased firing of serotonergic neurons in the medulla during times when ictal and post-ictal cardiorespiratory function are depressed. This is an important step in directly demonstrating marked changes in lower brainstem serotonin function during seizures for the first time. In addition, the availability of a controlled animal model of brainstem and cardiorespiratory impairment in acute seizures could greatly facilitate future studies of the networks controlling these changes. These investigations will hopefully lead to improved treatments to prevent the morbidity and mortality associated with cardiorespiratory impairment among those living with epilepsy.
Footnotes
This work was supported by the National Institutes of Health (Grants R01 NS066974, R21 NS083783, and P30 NS052519 to H.B.; Grants P20 NS076916 and U54NS041069 to G.B.R.; Grant MSTP TG T32GM07205 to J.E.M.); the VA Medical Center (G.B.R.); the Epilepsy Foundation (Postdoctoral Research and Training Award to M.F.); the China Scholarship Council (Postgraduate Scholarship Program Award CSC 201206370075 to Q.Z.); the China Scholarship Council (Postgraduate Scholarship Program award CSC to W.L.); the Howard Hughes Medical Institute (Citizens United for Research against Epilepsy Medical Student Fellowship to A.G. and A.K.); the Loughridge Williams Foundation; and the Betsy and Jonathan Blattmachr family. We thank Ruonan Jia for help with Figure 1 and members of the William B.J. Cafferty Laboratory for providing the Leica fluorescence microscope.
The authors declare no competing financial interests.
References
- Bateman LM, Spitz M, Seyal M. Ictal hypoventilation contributes to cardiac arrhythmia and SUDEP: report on two deaths in video-EEG-monitored patients. Epilepsia. 2010;51:916–920. doi: 10.1111/j.1528-1167.2009.02513.x. [DOI] [PubMed] [Google Scholar]
- Blessing WW. The lower brainstem and bodily homeostasis. Oxford: OUP; 1997. [Google Scholar]
- Blumenfeld H. Impaired consciousness in epilepsy. Lancet Neurol. 2012;11:814–826. doi: 10.1016/S1474-4422(12)70188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H, Rivera M, McNally KA, Davis K, Spencer DD, Spencer SS. Ictal neocortical slowing in temporal lobe epilepsy. Neurology. 2004a;63:1015–1021. doi: 10.1212/01.WNL.0000141086.91077.CD. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, McNally KA, Vanderhill SD, Paige AL, Chung R, Davis K, Norden AD, Stokking R, Studholme C, Novotny EJ, Jr, Zubal IG, Spencer SS. Positive and negative network correlations in temporal lobe epilepsy. Cereb Cortex. 2004b;14:892–902. doi: 10.1093/cercor/bhh048. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, Varghese GI, Purcaro MJ, Motelow JE, Enev M, McNally KA, Levin AR, Hirsch LJ, Tikofsky R, Zubal IG, Spencer SS, Paige AL. Cortical and subcortical networks in human secondarily generalized tonic-clonic seizures. Brain. 2009;132:999–1012. doi: 10.1093/brain/awp028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SR, Pieribone VA, Wang W, Severson CA, Jacobs RA, Richerson GB. Chemosensitive serotonergic neurons are closely associated with large medullary arteries. Nat Neurosci. 2002;5:401–402. doi: 10.1038/nn848. [DOI] [PubMed] [Google Scholar]
- Brennan TJ, Seeley WW, Kilgard M, Schreiner CE, Tecott LH. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16:387–390. doi: 10.1038/ng0897-387. [DOI] [PubMed] [Google Scholar]
- Buchanan GF, Richerson GB. Role of chemoreceptors in mediating dyspnea. Respir Physiol Neurobiol. 2009;167:9–19. doi: 10.1016/j.resp.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF, Richerson GB. Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci U S A. 2010;107:16354–16359. doi: 10.1073/pnas.1004587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan GF, Murray NM, Hajek MA, Richerson GB. Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol. 2014;592:4395–4410. doi: 10.1113/jphysiol.2014.277574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa V, Gonzalez A, Richerson GB. Diphtheria toxin treatment of Pet-1-Cre floxed diphtheria toxin receptor mice disrupts thermoregulation without affecting respiratory chemoreception. Neuroscience. 2014;279:65–76. doi: 10.1016/j.neuroscience.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy M, Dubé CM, Patterson K, Barnes SR, Maras P, Blood AB, Hasso AN, Obenaus A, Baram TZ. A novel, noninvasive, predictive epilepsy biomarker with clinical potential. J Neurosci. 2014;34:8672–8684. doi: 10.1523/JNEUROSCI.4806-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings KJ, Frappell PB. Breath-to-breath hypercapnic response in neonatal rats: temperature dependency of the chemoreflexes and potential implications for breathing stability. Am J Physiol Regul Integr Comp Physiol. 2009;297:R124–R134. doi: 10.1152/ajpregu.91011.2008. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Chen WC, Shorten A, McClurkin M, Choezom T, Schmidt CP, Chu V, Bozik A, Best C, Chapman M, Furman M, Detyniecki K, Giacino JT, Blumenfeld H. Impaired consciousness in partial seizures is bimodally distributed. Neurology. 2014;82:1736–1744. doi: 10.1212/WNL.0000000000000404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehkordi O, Millis RM, Dennis GC, Jazini E, Williams C, Hussain D, Jayam-Trouth A. Expression of alpha-7 and alpha-4 nicotinic acetylcholine receptors by GABAergic neurons of rostral ventral medulla and caudal pons. Brain Res. 2007;1185:95–102. doi: 10.1016/j.brainres.2007.09.027. [DOI] [PubMed] [Google Scholar]
- Dlouhy BJ, Gehlbach BK, Kreple CJ, Kawasaki H, Oya H, Buzza C, Granner MA, Welsh MJ, Howard MA, Wemmie JA, Richerson GB. Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J Neurosci. 2015;35:10281–10289. doi: 10.1523/JNEUROSCI.0888-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englot DJ, Blumenfeld H. Consciousness and epilepsy: why are complex-partial seizures complex? Prog Brain Res. 2009;177:147–170. doi: 10.1016/S0079-6123(09)17711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englot DJ, Yang L, Hamid H, Danielson N, Bai X, Marfeo A, Yu L, Gordon A, Purcaro MJ, Motelow JE, Agarwal R, Ellens DJ, Golomb JD, Shamy MC, Zhang H, Carlson C, Doyle W, Devinsky O, Vives K, Spencer DD, et al. Impaired consciousness in temporal lobe seizures: role of cortical slow activity. Brain. 2010;133:3764–3777. doi: 10.1093/brain/awq316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englot DJ, Mishra AM, Mansuripur PK, Herman P, Hyder F, Blumenfeld H. Remote effects of focal hippocampal seizures on the rat neocortex. J Neurosci. 2008;28:9066–9081. doi: 10.1523/JNEUROSCI.2014-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englot DJ, Modi B, Mishra AM, DeSalvo M, Hyder F, Blumenfeld H. Cortical deactivation induced by subcortical network dysfunction in limbic seizures. J Neurosci. 2009;29:13006–13018. doi: 10.1523/JNEUROSCI.3846-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faingold CL, Randall M, Tupal S. DBA/1 mice exhibit chronic susceptibility to audiogenic seizures followed by sudden death associated with respiratory arrest. Epilepsy Behav. 2010;17:436–440. doi: 10.1016/j.yebeh.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Faingold CL, Tupal S, Randall M. Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav. 2011;22:186–190. doi: 10.1016/j.yebeh.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Ficker DM. Sudden unexplained death and injury in epilepsy. Epilepsia. 2000;4:S7–S12. doi: 10.1111/j.1528-1157.2000.tb01519.x. [DOI] [PubMed] [Google Scholar]
- Furman M, Zhan Q, Lerner BA, McCafferty C, Motelow JE, Meng J, Ma C, Buchanan GF, Witten IB, Deisseroth K, Cardin JA, Blumenfeld H. Optogenetic stimulation of cholinergic brainstem neurons during focal limbic seizures: effects on cortical physiology. Epilepsia. 2015;56:e198–e202. doi: 10.1111/epi.13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gummadavelli A, Motelow JE, Smith N, Zhan Q, Schiff ND, Blumenfeld H. Thalamic stimulation to improve level of consciousness after seizures: evaluation of electrophysiology and behavior. Epilepsia. 2015;56:114–124. doi: 10.1111/epi.12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks T, Francis N, Fyodorov D, Deneris ES. The ETS domain factor Pet-1 is an early and precise marker of central serotonin neurons and interacts with a conserved element in serotonergic genes. J Neurosci. 1999;19:10348–10356. doi: 10.1523/JNEUROSCI.19-23-10348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF, Richerson GB. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008;28:2495–2505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol Rev. 1992;72:165–229. doi: 10.1152/physrev.1992.72.1.165. [DOI] [PubMed] [Google Scholar]
- Jordan D. Vagal control of the heart: central serotonergic (5-HT) mechanisms. Exp Physiol. 2005;90:175–181. doi: 10.1113/expphysiol.2004.029058. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol. 2009;4:517–550. doi: 10.1146/annurev.pathol.4.110807.092322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Motelow JE, Zhan Q, Hu YC, Kim R, Chen WC, Blumenfeld H. Cortical network switching: possible role of the lateral septum and cholinergic arousal. Brain Stimul. 2015;8:36–41. doi: 10.1016/j.brs.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebenthal JA, Wu S, Rose S, Ebersole JS, Tao JX. Association of prone position with sudden unexpected death in epilepsy. Neurology. 2015;84:703–709. doi: 10.1212/WNL.0000000000001260. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A. Neurophysiological investigation of the basis of the fMRI signal. Nature. 2001;412:150–157. doi: 10.1038/35084005. [DOI] [PubMed] [Google Scholar]
- Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol. 2014;10:271–282. doi: 10.1038/nrneurol.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey CA, Iceman KE, Johansen SL, Wu Y, Harris MB, Richerson GB. Isoflurane abolishes spontaneous firing of serotonin neurons and masks their pH/CO(2) chemosensitivity. J Neurophysiol. 2015;113:2879–2888. doi: 10.1152/jn.01073.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCardle CE, Gartside SE. Effects of general anaesthetics on 5-HT neuronal activity in the dorsal raphe nucleus. Neuropharmacology. 2012;62:1787–1796. doi: 10.1016/j.neuropharm.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Mishra AM, Ellens DJ, Schridde U, Motelow JE, Purcaro MJ, DeSalvo MN, Enev M, Sanganahalli BG, Hyder F, Blumenfeld H. Where fMRI and electrophysiology agree to disagree: corticothalamic and striatal activity patterns in the WAG/Rij rat. J Neurosci. 2011;31:15053–15064. doi: 10.1523/JNEUROSCI.0101-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley B, Bateman L, Millichap JJ, Wirrell E, Panayiotopoulos CP. Autonomic epileptic seizures, autonomic effects of seizures, and SUDEP. Epilepsy Behav. 2013;26:375–385. doi: 10.1016/j.yebeh.2012.08.020. [DOI] [PubMed] [Google Scholar]
- Motelow JE, Li W, Zhan Q, Mishra AM, Sachdev RN, Liu G, Gummadavelli A, Zayyad Z, Lee HS, Chu V, Andrews JP, Englot DJ, Herman P, Sanganahalli BG, Hyder F, Blumenfeld H. Decreased subcortical cholinergic arousal in focal seizures. Neuron. 2015;85:561–572. doi: 10.1016/j.neuron.2014.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mula M, Cock HR. More than seizures: improving the lives of people with refractory epilepsy. Eur J Neurol. 2015;22:24–30. doi: 10.1111/ene.12603. [DOI] [PubMed] [Google Scholar]
- Norden AD, Blumenfeld H. The role of subcortical structures in human epilepsy. Epilepsy Behav. 2002;3:219–231. doi: 10.1016/S1525-5050(02)00029-X. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pedley TA, Hauser WA. Sudden death in epilepsy: a wake-up call for management. Lancet. 2002;359:1790–1791. doi: 10.1016/S0140-6736(02)08726-3. [DOI] [PubMed] [Google Scholar]
- Pinault D. A novel single-cell staining procedure performed in vivo under electrophysiological control: morpho-functional features of juxtacellularly labeled thalamic cells and other central neurons with biocytin or Neurobiotin. J Neurosci Methods. 1996;65:113–136. doi: 10.1016/0165-0270(95)00144-1. [DOI] [PubMed] [Google Scholar]
- Richerson GB. Response to CO2 of neurons in the rostral ventral medulla in vitro. J Neurophysiol. 1995;73:933–944. doi: 10.1152/jn.1995.73.3.933. [DOI] [PubMed] [Google Scholar]
- Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci. 2004;5:449–461. doi: 10.1038/nrn1409. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Buchanan GF. The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia. 2011;52:28–38. doi: 10.1111/j.1528-1167.2010.02908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros H, Sachdev RN, Yu Y, Sestan N, McCormick DA. Neocortical networks entrain neuronal circuits in cerebellar cortex. J Neurosci. 2009;29:10309–10320. doi: 10.1523/JNEUROSCI.2327-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, Boon P, Crespel A, Dworetzky BA, Høgenhaven H, Lerche H, Maillard L, Malter MP, Marchal C, Murthy JM, Nitsche M, Pataraia E, Rabben T, Rheims S, Sadzot B, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12:966–977. doi: 10.1016/S1474-4422(13)70214-X. [DOI] [PubMed] [Google Scholar]
- Schridde U, Khubchandani M, Motelow JE, Sanganahalli BG, Hyder F, Blumenfeld H. Negative BOLD with large increases in neuronal activity. Cereb Cortex. 2008;18:1814–1827. doi: 10.1093/cercor/bhm208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedigh-Sarvestani M, Blumenfeld H, Loddenkemper T, Bateman LM. Seizures and brain regulatory systems: Consciousness, sleep, and autonomic systems. J Clin Neurophysiol. 2015;32:188–193. doi: 10.1097/WNP.0000000000000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selassie AW, Wilson DA, Martz GU, Smith GG, Wagner JL, Wannamaker BB. Epilepsy beyond seizure: a population-based study of comorbidities. Epilepsy Res. 2014;108:305–315. doi: 10.1016/j.eplepsyres.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Severson CA, Wang W, Pieribone VA, Dohle CI, Richerson GB. Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci. 2003;6:1139–1140. doi: 10.1038/nn1130. [DOI] [PubMed] [Google Scholar]
- Shmuel A, Augath M, Oeltermann A, Logothetis NK. Negative functional MRI response correlates with decreases in neuronal activity in monkey visual area V1. Nat Neurosci. 2006;9:569–577. doi: 10.1038/nn1675. [DOI] [PubMed] [Google Scholar]
- Shorvon S. What epilepsy comorbidities are important to model in the laboratory? Clinical perspectives. Adv Exp Med Biol. 2014;813:265–271. doi: 10.1007/978-94-017-8914-1_21. [DOI] [PubMed] [Google Scholar]
- Shorvon S, Tomson T. Sudden unexpected death in epilepsy. Lancet. 2011;378:2028–2038. doi: 10.1016/S0140-6736(11)60176-1. [DOI] [PubMed] [Google Scholar]
- Sowers LP, Massey CA, Gehlbach BK, Granner MA, Richerson GB. Sudden unexpected death in epilepsy: fatal post-ictal respiratory and arousal mechanisms. Respir Physiol Neurobiol. 2013;189:315–323. doi: 10.1016/j.resp.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surges R, Sander JW. Sudden unexpected death in epilepsy: mechanisms, prevalence, and prevention. Curr Opin Neurol. 2012;25:201–207. doi: 10.1097/WCO.0b013e3283506714. [DOI] [PubMed] [Google Scholar]
- Surges R, Adjei P, Kallis C, Erhuero J, Scott CA, Bell GS, Sander JW, Walker MC. Pathologic cardiac repolarization in pharmacoresistant epilepsy and its potential role in sudden unexpected death in epilepsy: a case-control study. Epilepsia. 2010;51:233–242. doi: 10.1111/j.1528-1167.2009.02330.x. [DOI] [PubMed] [Google Scholar]