

Abstract
Current regimens used to treat pulmonary Mycobacterium abscessus disease have limited efficacy. There is an urgent need for new drugs and optimized combinations and doses. We performed hollow-fiber-system studies in which M. abscessus was exposed to moxifloxacin lung concentration-time profiles similar to human doses of between 0 and 800 mg/day. The minimum bactericidal concentration and MIC were 8 and 2 mg/liter, respectively, in our M. abscessus strain, suggesting bactericidal activity. Measurement of the moxifloxacin concentrations in each hollow-fiber system revealed an elimination rate constant (kel) of 0.11 ± 0.05 h−1 (mean ± standard deviation) (half-life of 9.8 h). Inhibitory sigmoid maximal effect (Emax) modeling revealed that the highest Emax was 3.15 ± 1.84 log10 CFU/ml on day 3, and the exposure mediating 50% of Emax (EC50) was a 0- to 24-h area under the concentration time curve (AUC0–24)-to-MIC ratio of 41.99 ± 31.78 (r2 = 0.99). The EC80 was an AUC0–24/MIC ratio of 102.11. However, no moxifloxacin concentration killed the bacteria to burdens below the starting inoculum. There was regrowth beyond day 3 in all doses, with replacement by a resistant subpopulation that had an MIC of >32 mg/liter by the end of the experiment. A quadratic function best described the relationship between the AUC0–24/MIC ratio and the moxifloxacin-resistant subpopulation. Monte Carlo simulations of 10,000 patients revealed that the 400- to 800-mg/day doses would achieve or exceed the EC80 in ≤12.5% of patients. The moxifloxacin susceptibility breakpoint was 0.25 mg/liter, which means that almost all M. abscessus clinical strains are moxifloxacin resistant by these criteria. While moxifloxacin's efficacy against M. abscessus was poor, formal combination therapy studies with moxifloxacin are still recommended.
INTRODUCTION
Mycobacterium abscessus is a rapidly growing mycobacterium that is notorious because of resistance to most antibiotics (1). Pulmonary disease due to M. abscessus infection is chronic and relentless. Current regimens used to treat M. abscessus consist of a combination of amikacin, a macrolide, and either cefoxitin or imipenem; however, the regimens fail in most patients (2). Based on static in vitro models, amikacin is considered the key antibiotic in the treatment regimens (3–6). We recently demonstrated that the efficacy of amikacin based on concentration-time profiles achievable in human lungs as recapitulated in the hollow-fiber-system model of M. abscessus (HFS-M. abscessus) was poor, with failure to kill the bacteria below stasis (7). This, as well as clinical experience of high failure rates of amikacin-based regimens, means that there is an urgent need to find new antibiotics and optimize their doses. Here, we approached this by conducting a recommended formal pharmacokinetic/pharmacodynamic (PK/PD) evaluation of alternative antibiotics with potential bactericidal activity (8).
The 8-methoxy fluoroquinolone, moxifloxacin, has been shown to have excellent efficacy against Mycobacterium tuberculosis, Mycobacterium avium, and Mycobacterium kansasii (9–12). In addition, continuation regimens currently used to treat pulmonary M. abscessus include moxifloxacin in the combination, starting from the second month of therapy. Given these prior successes of moxifloxacin, as well as its current role in the treatment of M. abscessus, we tested it against this species. We performed formal moxifloxacin PK/PD work in the hollow-fiber-system model of pulmonary M. abscessus. We exposed M. abscessus to moxifloxacin concentration-time profiles as encountered in lungs of patients with pneumonia (13). Repetitive day-to-day sampling of hollow-fiber systems is a major advantage in identifying the evolution of bacteria in response to the periodic fluctuation of antibiotic concentrations, vital given M. abscessus' propensity to develop acquired drug resistance (8, 14–17).
MATERIALS AND METHODS
Bacteria, antibiotic, and growth conditions.
Stock cultures of M. abscessus ATCC 19977 (American Type Culture Collection, Manassas, VA), stored at −80°C in Middlebrook 7H9 broth supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC; Remel, Lenexa, KS) and 15% glycerol, were used for all the experiments. One vial was thawed before each assay and incubated for 24 to 48 h at 30°C to achieve logarithmic growth phase. Moxifloxacin hydrochloride was purchased from the Baylor University Medical Center pharmacy. On the day of use, the moxifloxacin was diluted in sterile water to desired concentrations for the assays.
Antimicrobial susceptibility testing and mutation frequency.
The MIC was identified using broth macrodilution in Middlebrook 7H9 broth (here termed “broth”), as well as by use of the Etest (bioMérieux, Durham, NC). In addition to the turbidity test, the CFU per ml were enumerated for each concentration evaluated in the broth macrodilution test. In this test, the MIC was defined as the lowest concentration associated with ≥99% decrease in CFU/ml compared to the growth of the untreated control and the minimum bactericidal concentration (MBC) as the concentration corresponding to >99.9% kill. Mutation frequency was determined for the inoculum by culturing 0.2 ml on Middlebrook 7H10 agar plates (here termed “agar”) supplemented with 3 times the moxifloxacin MIC. Cultures were incubated for 5 days.
Exposure-response studies in the hollow-fiber system.
The hollow-fiber-system model of pulmonary M. abscessus (HFS-M. abscessus) has been used previously to perform PK/PD evaluation of amikacin (7). Twenty milliliters of 6 log10 CFU/ml M. abscessus in log phase was inoculated into the peripheral compartment of each of seven hollow-fiber cartridges (FiberCell Systems, Frederick, MD). Doses that mimicked the non-protein-bound plasma 0- to 24-h area under the concentration time curve (AUC0–24), peak concentrations, and time to maximum concentration achieved in humans treated with moxifloxacin doses of 0, 25, 50, 100, 200, 400, and 800 mg were administered to the central compartment once daily via computerized syringe pumps (18). These exposures were chosen because they represent the range of clinically tolerated doses of moxifloxacin. Treatment was for 21 days of daily therapy. A plasma-to-lung epithelial lining fluid penetration ratio of 1 was assumed, based on the literature (13, 19, 20). As an example, the standard 400-mg-a-day dose was expected to achieve a peak concentration of 4.2 mg/liter and a half-life of 10 h, translating to an AUC0–24/MIC ratio of 28.3. The actual moxifloxacin concentrations achieved in all the systems were validated by repetitive sampling of 1 ml from the central compartment of each HFS-M. abscessus during the first 3 days at 0, 1, 6, 9, 12, 18, 23.5, 25, 30, 33, 36, 42, and 47.5 h postdose. In order to quantify the M. abscessus burden, 1 ml of the peripheral compartment culture contents was removed from each system on days 0, 1, 2, 3, 5, 7, 10, 14, and 21. The samples were washed with saline to avoid antibiotic carryover, after which samples were serially diluted and cultured on agar. To quantify the moxifloxacin-resistant M. abscessus CFU/ml, the same samples were also inoculated onto agar supplemented with 3 times the moxifloxacin MIC.
Drug assay.
The moxifloxacin concentrations in the samples collected from the central compartment of the HFS-M. abscessus were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Moxifloxacin and moxifloxacin-13CD3 (internal standard) were purchased from Sigma (St. Louis, MO) and Santa Cruz Biotech (Santa Cruz, CA), respectively. Calibrator, controls, and internal standard were included in each analytical run for quantitation. Stock solutions of moxifloxacin and the internal standard were prepared in 80:20 methanol-water at a concentration of 1 mg/ml and stored at −20°C. A 7-point calibration curve was prepared by diluting moxifloxacin stock solution in drug-free medium (0.1, 0.2, 1, 2, 5, 10, and 20 mg/liter). Quality control samples were prepared by spiking medium with stock standards for two levels of controls. Samples were prepared in 96-well microtiter plates by the addition of 10 μl of calibrator, quality controls, or sample to 190 μl of 0.1% formic acid in water containing 10 mg/liter internal standard, followed by vortexing. Chromatographic separation was achieved on an Acquity UPLC high-strength silica (HSS) T3 analytical column (1.8 μm particle size, 50 by 2.1 mm; Waters) maintained at 30°C at a flow of 0.2 ml/min with a binary gradient and a total run time of 6 min. The observed ion values (m/z) of the fragment ions were as follows: for moxifloxacin, m/z 402.2→384.2, and for the internal standard, moxifloxacin-13CD3, m/z 406.2→388.3. Sample injection and separation were performed by using an Acquity UPLC column interfaced with a Xevo TQ mass spectrometer (Waters). All data were collected using MassLynx version 4.1 with software change note 810 (SCN810). The limit of quantitation for this assay was 0.1 mg/liter. The inter- and intraday variations were 1.5% and 9.4%, respectively.
Pharmacokinetics and pharmacodynamics modeling.
All drug concentrations from each of the HFS-M. abscessus units at all time points were comodeled using ADAPT 5 software (Biomedical Simulations Resource, University of Southern California). The steps used in the pharmacokinetic parameter analysis were as described in detail in prior studies (10, 21). The pharmacokinetic parameter estimates identified were used to calculate the observed AUC0–24 values and AUC0–24/MIC ratios. Exposure-response was modeled using the inhibitory sigmoid maximal effect (Emax) model, a standard model recommended for examining the effects of antibiotics whether they are static or cidal, for both clinical and laboratory data (22–24). Total bacterial burden was used as the response parameter, while drug exposure was expressed as the AUC0–24/MIC ratio. For moxifloxacin resistance emergence, we used the quadratic model that we identified previously, with drug exposure examined versus the size of the moxifloxacin-resistant subpopulation on each sampling day (8, 25).
Monte Carlo simulations.
In order to put our findings into clinical context, we performed a 10,000-patient Monte Carlo simulation to identify two important clinical aspects. First, we wanted to identify the clinical dose best able to achieve or exceed the EC80, which is the exposure mediating 80% of the maximal kill (Emax). This exposure is considered optimal since Emax is on an asymptote and it is independent of whether a drug is cidal or static. Second, we wanted to identify the susceptibility breakpoint, which we considered the MIC below which >10% of patients achieve the EC80 with standard dosing or with the highest dose that can be administered. The steps in the Monte Carlo simulations were as detailed in recommendations elsewhere and in our prior work (8, 26). We examined moxifloxacin doses of 400 mg a day, 600 mg a day, and 800 mg a day. We entered the two-compartment model pharmacokinetic parameter estimates and covariance identified by Kees et al. into subroutine PRIOR of ADAPT 5, as shown in Table 1 (27). We assumed a 1:1 AUC0–24 ratio in the lung versus plasma (13). An MIC distribution from South Korea (28) was used, and data on MIC distribution from Nijmegen, the Netherlands, and the Dallas-Fort Worth metroplex, United States, were included for comparison of the impact of the new susceptibility breakpoint.
TABLE 1.
Moxifloxacin pharmacokinetic parameter estimates utilized in Monte Carlo simulations
| Pharmacokinetic parameter | Observed in patients (entered into subroutine PRIOR) |
Simulated for 10,000 patients |
||
|---|---|---|---|---|
| Parameter estimate | IIVa as %CV | Parameter estimate | IIV as %CV | |
| Total clearance (liters/h) | 11.3 | 23.7 | 11.3 | 14.58 |
| Intercompartmental clearance (liters/h) | 47.7 | —b | 47.7 | 19.19 |
| Absorption rate constant (h−1) | 1.09 | 135 | 1.09 | 143 |
| Central vol (liters) | 55.6 | — | 55.6 | 18.40 |
| Peripheral vol (liters) | 59.6 | 15.3 | 59.6 | 14.99 |
IIV, interindividual variability.
—, fixed in the pharmacokinetic model by Kees et al. (27).
RESULTS
In this study, the moxifloxacin MIC for the M. abscessus laboratory strain was 2 mg/liter. In addition, the concentration associated with >99.9% kill, or MBC, was 8 mg/liter. Thus, the MBC/MIC ratio was 4, which means that moxifloxacin would be considered bactericidal according to the standard convention (29). The frequency of mutation to 3 times the moxifloxacin MIC was (2.11 ± 0.16) × 10−5 in repeat experiments.
In the HFS-M. abscessus dose-effect studies, the moxifloxacin concentrations measured in each HFS-M. abscessus unit revealed an elimination rate constant (kel) of 0.11 ± 0.05 h−1, which translates to a half-life of 9.8 h, consistent with the moxifloxacin pharmacokinetics in a recent 241-patient clinical study (18). As an example, the r2 for the intended drug concentrations, exposures, and half-life of the 400-mg standard dose versus the observed concentrations was 0.98, which means the intended pharmacokinetics were recapitulated well. Figure 1 shows the time-kill curves for each of the moxifloxacin exposures in the HFS-M. abscessus. None of the exposures evaluated achieved a considerable killing effect of the M. abscessus.
FIG 1.
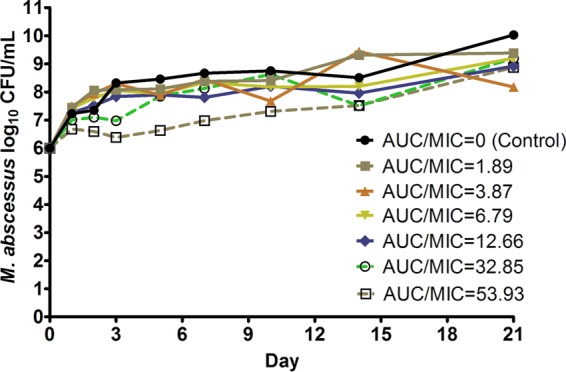
Moxifloxacin exposure-effect against M. abscessus in the HFS. None of the moxifloxacin exposures evaluated attained killing below the starting inoculum of 6.0 log10 CFU/ml at any point during the study. Regrowth was observed after day 3 in all systems.
The relationship between the AUC0–24/MIC ratio and the M. abscessus burden in the HFS-M. abscessus is shown in Fig. 2; the highest Emax was encountered on day 3. The day 3 parameters were an Emax of 3.15 ± 1.84 log10 CFU/ml, a Hill slope of 1.56 ± 0.63, and an EC50 that was an AUC0–24/MIC ratio of 41.99 ± 31.78 (r2 = 0.987). Based on the inhibitory sigmoid Emax relationship for day 3, the EC80 was calculated as a non-protein-bound AUC0–24/MIC ratio of 102.11. The Emax appears high mainly because in nontreated controls, M. abscessus grew to large bacterial burdens of about 10 log10 CFU/ml, starting from 6.0 log10 CFU/ml at the start of the experiment. Indeed, the data in Fig. 2 show that even on day 3, moxifloxacin never reduced the bacterial burden below the starting inoculum (stasis).
FIG 2.
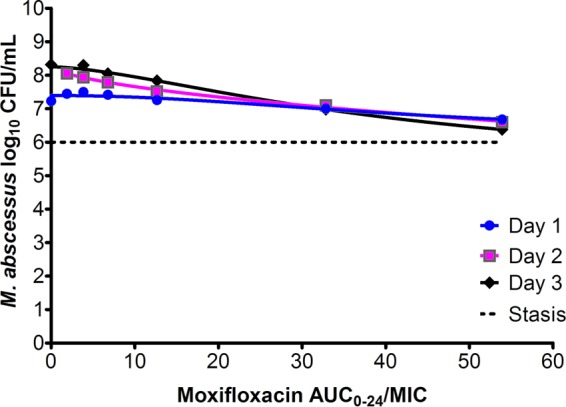
Moxifloxacin exposure versus M. abscessus burden. Inhibitory sigmoid Emax curves are shown for the first 3 days, prior to replacement of microbial population by drug-resistant subpopulation. The highest Emax was encountered on day 3, but not even that produced kill below the stasis level.
The data in Fig. 3 show that acquired drug resistance emerged after 10 days of exposure. These data further show that the moxifloxacin-resistant subpopulation had replaced the total population in the systems exposed to the 3 highest doses by day 21.The relationship between the size of the resistant subpopulation and the moxifloxacin exposure was described by a system of changing U-shaped curves, in effect a quadratic function. However, even at the highest AUC0–24/MIC ratios tested, moxifloxacin was not able to suppress acquired drug resistance. Indeed, the MICs of cultures from the systems exposed to the 3 highest doses had changed from 2 mg/liter to >32 mg/liter by day 21.
FIG 3.
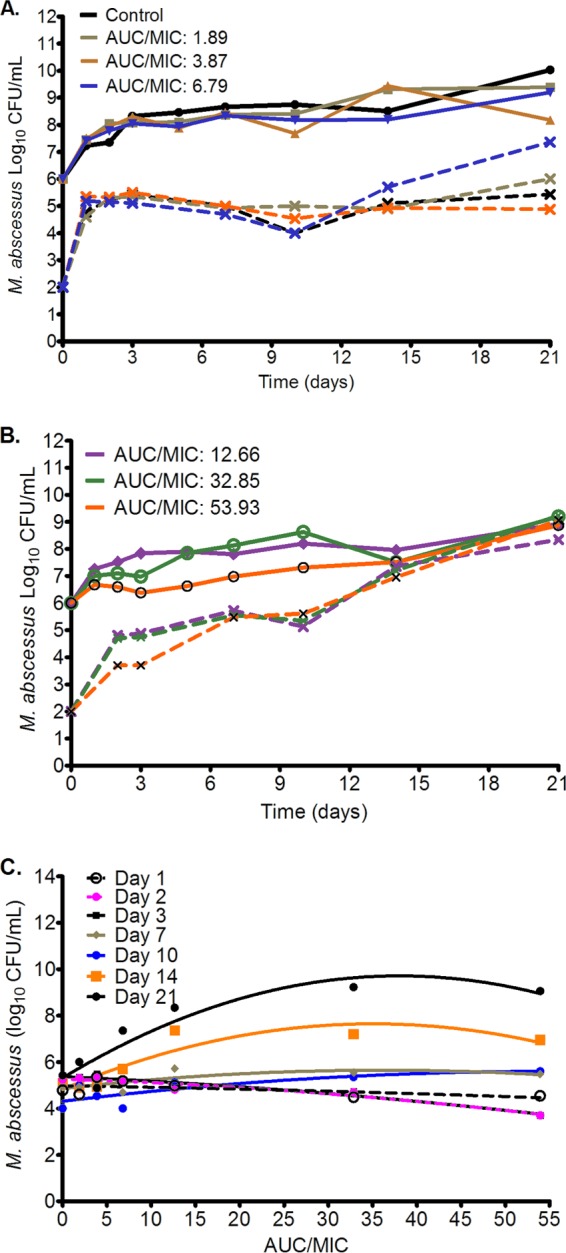
Changes in total and moxifloxacin-resistant subpopulations with time. Total M. abscessus population (solid lines) and moxifloxacin-resistant subpopulation (hatched lines) over the course of 21 days of treatment with different moxifloxacin AUC0–24/MIC exposures. (A) Control and three lower exposures. While the drug-resistant subpopulation increased with time, especially beyond day 10, it had not completely replaced the total population in these lower doses, with the highest dose among these lower doses associated with the largest moxifloxacin-resistant subpopulation. (B) At the three highest exposures, the total population had been completely replaced by the moxifloxacin-resistant subpopulation by day 14. (C) The relationship between moxifloxacin AUC/MIC and size of subresistant subpopulation is described by systems of evolving U-shaped curves. At day 1 it was a straight line, then it switched to an inverted U-shaped curve that was more pronounced by day 21.
Figure 4 shows the result of Monte Carlo simulations of 10,000 simulated subjects, based on the largest published moxifloxacin MIC distribution of M. abscessus, which is from South Korea (28). Overall, the cumulative fraction of response, or proportion of patients that achieved or exceeded EC80, given the MIC distribution, was poor for all doses. For the standard dose of 400 mg daily, only 4.7% of patients would achieve the EC80, for 600 mg a day, 9.7% would, and for 800 mg a day, 12.5% would. If the penetration ratio of moxifloxacin AUC achieved in the lung versus plasma were 2, as suggested by two outlier pharmacokinetic studies (19, 20), the cumulative fraction of response would become 12.5% for 400 mg a day, 21.5% for 600 mg a day, and 26.6% for 800 mg a day. Either way, standard and high doses of moxifloxacin performed poorly.
FIG 4.
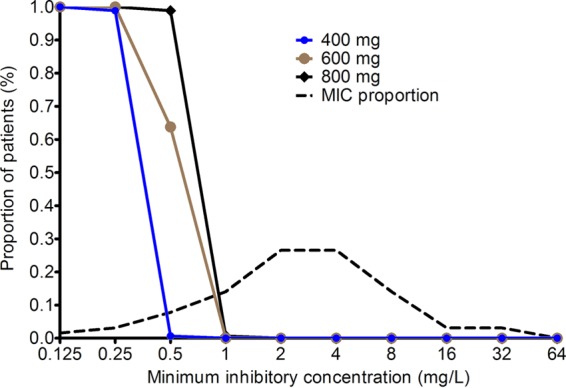
Probability of target attainment of three different moxifloxacin doses in 10,000 patients. Target attainment probability at each MIC fell to 0% after the 0.25-mg/liter MIC for a dose of 400 mg and to just above 60% for 600 mg. The target attainment probability for a dose of 800 mg a day plummeted after the 0.5-mg/liter MIC. Since the proportion of clinical isolates with an MIC of >0.25 mg/liter is large (and 0% of these achieve the EC80), the fraction of all patients who will achieve the EC80 is very small.
The data in Fig. 4 also show the target attainment at different moxifloxacin MICs. These data show that at the standard dose and at 600 mg a day, the target attainment to achieve EC80 fell to below 0.9 (i.e., 90%) after the MIC of 0.25 mg/liter. This is the PK/PD-derived moxifloxacin susceptibility breakpoint for M. abscessus, since above this MIC, most patients will not achieve optimal microbial effect. It should be borne in mind that even this optimal kill would still not kill below the bacterial burden at the start of treatment. If the dose was increased to 800 mg a day, the susceptibility breakpoint only changed by one tube dilution. In contrast, the epidemiologic cutoff point for the South Korean MIC distribution would be 16 mg/liter. In Fig. 5, we show the MIC distributions of M. abscessus from our two clinical practice locations in the Netherlands and the Dallas-Fort Worth metroplex in Texas. Given the MIC distributions shown in Fig. 5 and the proposed susceptibility breakpoint of 0.25 mg/liter, then almost all M. abscessus isolates in North Texas and the Netherlands as of 2015 were intrinsically resistant to moxifloxacin. Accordingly, the cumulative fraction of responses in these two locales would fall to 0% even for a dose of 800 mg a day.
FIG 5.
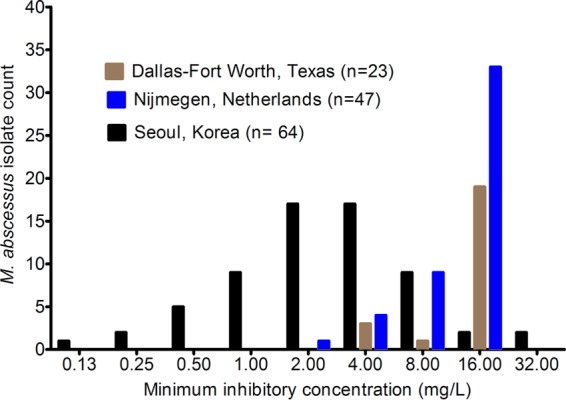
Moxifloxacin MIC distribution in three different practice locations. The MIC distribution from South Korea was used for the Monte Carlo simulations; the other two settings are used as comparators. Notably, the MIC distributions from the Netherlands and from Texas, United States, show most isolates to be resistant to moxifloxacin.
DISCUSSION
Moxifloxacin and quinolones in general are considered ideal antibiotics given that they are bactericidal, are active against a broad spectrum of bacteria, are available both in oral and parenteral formulations, have a broad distribution in organs and tissues, and can achieve therapeutic concentrations both in the intra- and extracellular environment (21, 30). Indeed, these agents have now found broad use in the treatment of many slow-growing and rapidly growing mycobacteria. The MBC/MIC ratio calculated in this study would seem to indicate that the drug would work well against M. abscessus. However, in our formal PK/PD study, we found that moxifloxacin does not reduce the bacterial burden below the starting inoculum even at high exposures and that resistance arises fairly quickly during monotherapy. Therefore, the meaning of standard MBC/MIC assays used to determine bactericidal effect is questionable.
Moxifloxacin has been used for M. abscessus pulmonary disease at 400 mg/daily, after the discontinuation of parenteral antibiotics that are administered during the first 1 to 2 months. In that role, it is used as part of combination therapy. However, we show that its performance in the HFS-M. abscessus does not inspire continued confidence even for that role. Inhibitory sigmoid Emax-based target exposures such as EC80 and EC90 have been used to identify the susceptibility breakpoints of several bacteria, including mycobacteria (8, 10–12, 31–36). We chose the EC80 exposure as the optimal kill exposure that we have found to best translate to the clinic for other mycobacteria, such as M. tuberculosis and M. avium, and indeed, to other Gram-negative and Gram-positive organisms, even with drugs that do not kill below stasis (31–35). Indeed, indices of microbial kill, such as 3.0 log10 CFU/ml, that are commonly used in work with Gram-positive cocci and Gram-negative bacilli are rarely achieved in work with mycobacteria with a single agent. Nevertheless, an approach similar to ours has been used to identify susceptibility breakpoints, even at exposures that are merely associated with stasis, for several drugs and bacteria (31–36). Work done with M. tuberculosis in the hollow-fiber system to identify the EC80 in the inhibitory sigmoid Emax model for drugs that kill below stasis, as well as those that do not, in tandem with Monte Carlo simulations, has been shown to predict optimal clinical exposures, doses, and susceptibility breakpoints to within 94% of the value later identified in the clinic, based on FDA and European Medicines Agency (EMA) submissions (8, 37–41). The hollow-fiber work that identified these exposures and breakpoints later confirmed in the clinic was done with single strains of M. tuberculosis, with the MIC distribution taken into account only at the stage of Monte Carlo simulations. We report here on moxifloxacin dosing and optimal exposures using the same hollow-fiber system and the same inhibitory sigmoid Emax models and EC80 exposures in tandem with Monte Carlo simulations, demonstrating limited activity at doses up to 800 mg a day, and we expect the same degree of accuracy as in tuberculosis.
In the treatment of M. abscessus, moxifloxacin is used as part of combination therapy. Thus, it could be that it would show efficacy in combination therapy. Formal combination therapy studies with moxifloxacin in the treatment of M. abscessus will need to be performed in the HFS-M. abscessus in order to explore potential synergy with other agents before definitive recommendations can be made to remove it from current regimens. So far, we have examined high-dose moxifloxacin in combination with tigecycline and ceftaroline and found that it did not add to the effect of these drugs (unpublished data). Thus, the findings for moxifloxacin monotherapy are likely to be borne out for its use in combination therapy.
Acquired moxifloxacin resistance has been described in mycobacteria, particularly for M. tuberculosis (11). In the absence of other companion antibiotics, moxifloxacin monotherapy rapidly leads to resistance, as was the case here for M. abscessus. Unfortunately, in this study, no moxifloxacin exposure was associated with suppression of resistance. The relationship between acquired moxifloxacin resistance and the moxifloxacin AUC/MIC was best described by a quadratic function, similar to our findings with M. tuberculosis (25). However, the underlying mechanism for moxifloxacin resistance in M. abscessus deserves further exploration; DNA gyrase is still an interesting bacterial target against which new inhibitors are being developed, with evaluation of their potential for use against M. abscessus and other nontuberculous mycobacteria (42).
Finally, according to guidelines, the current breakpoint to determine moxifloxacin resistance to rapidly growing mycobacteria, such as M. abscessus, is ≥4 mg/liter in cation-adjusted Mueller-Hinton broth (43), which in a wide MIC distribution (28) will barely allow the use of moxifloxacin in 50% of cases. A 1.0-mg/liter susceptibility breakpoint has also been used by the CLSI (43). In this study, although using a different type of broth (Middlebrook 7H9), we found a PK/PD-derived breakpoint of 0.25 mg/liter. Examination of the MIC distributions in our clinical practices demonstrated that at this proposed breakpoint, most isolates would be a priori resistant to moxifloxacin. This comports well with failure of the moxifloxacin-containing regimens in clinical practice. Our findings suggest that the poor outcome observed when treating M. abscessus is no more than a reflection of the amount of intrinsically resistant isolates that are circulating and infecting patients. The approach that utilizes the hollow-fiber-system model and the EC80 or EC90 followed by Monte Carlo simulations has been found to be highly accurate in predicting MICs above which combination therapy fails in clinical studies, even for drugs that do not kill below the stasis line, in the case of tuberculosis and disease caused by other more mundane bacteria, such as Gram-negative rods (25, 44–52). Thus, our findings are likely accurate, but this will require clinical validation in the future. Such confirmation will require multivariable pharmacometric analyses of clinical data, which do not currently exist for moxifloxacin and M. abscessus pulmonary disease (53). Indeed, no such data exist, as far as we know, for any of the drugs used in treatment of pulmonary M. abscessus, since there is a lack of clinical trials and large prospective clinical cohort studies for this disease. Thus, the PK/PD-derived breakpoint is a proposed breakpoint to be used for clinical decision making until a large-enough data set can better identify further breakpoints.
There are some limitations in this study. We performed our evaluation with one laboratory strain. Although the strain used was the type strain, ideally this work should be extended to clinical isolates with different susceptibility profiles. Indeed, the type strain was not from a patient with pneumonia but from an abscess after trauma. It is not clear whether strains that cause lung disease are intrinsically different from those that cause extrapulmonary disease. Nevertheless, even when single strains have been used in PK/PD studies in the hollow-fiber system with other bacteria, the results have been found to be fairly accurate and to allow generalizations (11, 25, 37–41, 44–52). Moreover, here we only report the effect of moxifloxacin alone, and there is still a need to evaluate its performance as part of combination therapy. However, studies evaluating different moxifloxacin-based combinations to explore the potential of synergistic combinations, as well to explore the intracellular model for pulmonary disease, have been performed and are being prepared for publication.
In summary, we used a recently developed preclinical model for M. abscessus disease to evaluate and then identify the efficacy of moxifloxacin, which demonstrated rapid appearance of resistance. Efficacy was poor, and moxifloxacin even at high doses is not expected to work in the clinic. We used the PK/PD studies to identify and propose a new moxifloxacin susceptibility breakpoint of 0.25 mg/liter.
ACKNOWLEDGMENTS
We thank Robert Cavagnolo from med fusion clin-labs, Lewisville, TX, USA, for kindly allowing us to have access to moxifloxacin MIC data.
Conflict of interest declaration: T.G. is a consultant for Astellas Pharma USA, as well as LuminaCare Solutions; he founded Jacaranda Biomed, Inc.
Funding Statement
The funding agencies played no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother 67:810–818. doi: 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- 2.Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. 2011. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis 52:565–571. doi: 10.1093/cid/ciq237. [DOI] [PubMed] [Google Scholar]
- 3.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K. 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 175:367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 4.Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. 2015. Time-kill kinetics of antibiotics active against rapidly growing mycobacteria. J Antimicrob Chemother 70:811–817. doi: 10.1093/jac/dku431. [DOI] [PubMed] [Google Scholar]
- 5.van Ingen J, Ferro BE, Hoefsloot W, Boeree MJ, van Soolingen D. 2013. Drug treatment of pulmonary nontuberculous mycobacterial disease in HIV-negative patients: the evidence. Expert Rev Anti Infect Ther 11:1065–1077. doi: 10.1586/14787210.2013.830413. [DOI] [PubMed] [Google Scholar]
- 6.Lee MR, Sheng WH, Hung CC, Yu CJ, Lee LN, Hsueh PR. 2015. Mycobacterium abscessus complex infections in humans. Emerg Infect Dis 21:1638–1646. doi: 10.3201/2109.141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferro BE, Srivastava S, van Ingen J, Deshpande D, Sherman C, Pasipanodya JG, van Soolingen D, Mouton JW, Gumbo T. 2016. Amikacin pharmacokinetics/pharmacodynamics in a novel hollow-fiber Mycobacterium abscessus disease model. Antimicrob Agents Chemother 60:1242–1248. doi: 10.1128/AAC.02282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gumbo T, Angulo-Barturen I, Ferrer-Bazaga S. 2015. Pharmacokinetic-pharmacodynamic and dose-response relationships of antituberculosis drugs: recommendations and standards for industry and academia. J Infect Dis 211(Suppl 3):S96–S106. doi: 10.1093/infdis/jiu610. [DOI] [PubMed] [Google Scholar]
- 9.Sindelar G, Zhao X, Liew A, Dong Y, Lu T, Zhou J, Domagala J, Drlica K. 2000. Mutant prevention concentration as a measure of fluoroquinolone potency against mycobacteria. Antimicrob Agents Chemother 44:3337–3343. doi: 10.1128/AAC.44.12.3337-3343.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deshpande D, Srivastava S, Meek C, Leff R, Hall GS, Gumbo T. 2010. Moxifloxacin pharmacokinetics/pharmacodynamics and optimal dose and susceptibility breakpoint identification for treatment of disseminated Mycobacterium avium infection. Antimicrob Agents Chemother 54:2534–2539. doi: 10.1128/AAC.01761-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gumbo T, Louie A, Deziel MR, Parsons LM, Salfinger M, Drusano GL. 2004. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis 190:1642–1651. doi: 10.1086/424849. [DOI] [PubMed] [Google Scholar]
- 12.Srivastava S, Pasipanodya J, Sherman CM, Meek C, Leff R, Gumbo T. 2015. Rapid drug tolerance and dramatic sterilizing effect of moxifloxacin monotherapy in a novel hollow-fiber model of intracellular Mycobacterium kansasii disease. Antimicrob Agents Chemother 59:2273–2279. doi: 10.1128/AAC.04441-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon N, Sampol E, Albanese J, Martin C, Arvis P, Urien S, Lacarelle B, Bruguerolle B. 2003. Population pharmacokinetics of moxifloxacin in plasma and bronchial secretions in patients with severe bronchopneumonia. Clin Pharmacol Ther 74:353–363. doi: 10.1016/S0009-9236(03)00201-7. [DOI] [PubMed] [Google Scholar]
- 14.Pasipanodya JG, Nuermberger E, Romero K, Hanna D, Gumbo T. 2015. Systematic analysis of hollow fiber model of tuberculosis experiments. Clin Infect Dis 61(Suppl 1):S10–S17. doi: 10.1093/cid/civ425. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava S, Gumbo T. 2011. In vitro and in vivo modeling of tuberculosis drugs and its impact on optimization of doses and regimens. Curr Pharm Des 17:2881–2888. doi: 10.2174/138161211797470192. [DOI] [PubMed] [Google Scholar]
- 16.Gumbo T, Lenaerts AJ, Hanna D, Romero K, Nuermberger E. 2015. Nonclinical models for antituberculosis drug development: a landscape analysis. J Infect Dis 211(Suppl 3):S83–S95. doi: 10.1093/infdis/jiv183. [DOI] [PubMed] [Google Scholar]
- 17.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, van Oers NS, Gumbo T. 2012. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother 56:4806–4815. doi: 10.1128/AAC.05546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zvada SP, Denti P, Sirgel FA, Chigutsa E, Hatherill M, Charalambous S, Mungofa S, Wiesner L, Simonsson US, Jindani A, Harrison T, McIlleron HM. 2014. Moxifloxacin population pharmacokinetics and model-based comparison of efficacy between moxifloxacin and ofloxacin in African patients. Antimicrob Agents Chemother 58:503–510. doi: 10.1128/AAC.01478-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Capitano B, Mattoes HM, Shore E, O'Brien A, Braman S, Sutherland C, Nicolau DP. 2004. Steady-state intrapulmonary concentrations of moxifloxacin, levofloxacin, and azithromycin in older adults. Chest 125:965–973. doi: 10.1378/chest.125.3.965. [DOI] [PubMed] [Google Scholar]
- 20.Soman A, Honeybourne D, Andrews J, Jevons G, Wise R. 1999. Concentrations of moxifloxacin in serum and pulmonary compartments following a single 400 mg oral dose in patients undergoing fibre-optic bronchoscopy. J Antimicrob Chemother 44:835–838. doi: 10.1093/jac/44.6.835. [DOI] [PubMed] [Google Scholar]
- 21.Musuka S, Srivastava S, Siyambalapitiyage Dona CW, Meek C, Leff R, Pasipanodya J, Gumbo T. 2013. Thioridazine pharmacokinetic-pharmacodynamic parameters “wobble” during treatment of tuberculosis: a theoretical basis for shorter-duration curative monotherapy with congeners. Antimicrob Agents Chemother 57:5870–5877. doi: 10.1128/AAC.00829-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gumbo T. 2011. General principles of antimicrobial therapy, p 1365–1382. In Brunton LL, Chabner BA, Knollmann BC (ed), Goodman & Gilman's the pharmacological basis of therapeutics, 12th ed McGraw Hill Medical, New York, NY. [Google Scholar]
- 23.Mouton JW, Punt N, Vinks AA. 2007. Concentration-effect relationship of ceftazidime explains why the time above the MIC is 40 percent for a static effect in vivo. Antimicrob Agents Chemother 51:3449–3451. doi: 10.1128/AAC.01586-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller AE, Punt N, Mouton JW. 2013. Optimal exposures of ceftazidime predict the probability of microbiological and clinical outcome in the treatment of nosocomial pneumonia. J Antimicrob Chemother 68:900–906. doi: 10.1093/jac/dks468. [DOI] [PubMed] [Google Scholar]
- 25.Gumbo T, Dona CS, Meek C, Leff R. 2009. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother 53:3197–3204. doi: 10.1128/AAC.01681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pasipanodya J, Gumbo T. 2011. An oracle: antituberculosis pharmacokinetics-pharmacodynamics, clinical correlation, and clinical trial simulations to predict the future. Antimicrob Agents Chemother 55:24–34. doi: 10.1128/AAC.00749-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kees MG, Schaeftlein A, Haeberle HA, Kees F, Kloft C, Heininger A. 2013. Population pharmacokinetics and pharmacodynamic evaluation of intravenous and enteral moxifloxacin in surgical intensive care unit patients. J Antimicrob Chemother 68:1331–1337. doi: 10.1093/jac/dkt040. [DOI] [PubMed] [Google Scholar]
- 28.Koh WJ, Jeon K, Lee NY, Kim BJ, Kook YH, Lee SH, Park YK, Kim CK, Shin SJ, Huitt GA, Daley CL, Kwon OJ. 2011. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am J Respir Crit Care Med 183:405–410. doi: 10.1164/rccm.201003-0395OC. [DOI] [PubMed] [Google Scholar]
- 29.Pankey GA, Sabath LD. 2004. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin Infect Dis 38:864–870. doi: 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- 30.Owens RC Jr, Ambrose PG. 2001. Pharmacodynamics of quinolones, 155–176. In Nightingale CH, Murakawa T, Ambrose PG (ed), Antimicrobial pharmacodynamics in theory and clinical practice, 1st ed Marcel Dekker, New York, NY. [Google Scholar]
- 31.Drusano GL, Preston SL, Hardalo C, Hare R, Banfield C, Andes D, Vesga O, Craig WA. 2001. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob Agents Chemother 45:13–22. doi: 10.1128/AAC.45.1.13-22.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deshpande D, Srivastava S, Meek C, Leff R, Gumbo T. 2010. Ethambutol optimal clinical dose and susceptibility breakpoint identification by use of a novel pharmacokinetic-pharmacodynamic model of disseminated intracellular Mycobacterium avium. Antimicrob Agents Chemother 54:1728–1733. doi: 10.1128/AAC.01355-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srivastava S, Musuka S, Sherman C, Meek C, Leff R, Gumbo T. 2010. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis 201:1225–1231. doi: 10.1086/651377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deshpande D, Pasipanodya JG, Gumbo T. 2016. Azithromycin dose to maximize efficacy and suppress acquired drug resistance in pulmonary Mycobacterium avium disease. Antimicrob Agents Chemother 60:2157–2163. doi: 10.1128/AAC.02854-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Labreche MJ, Graber CJ, Nguyen HM. 2015. Recent updates on the role of pharmacokinetics-pharmacodynamics in antimicrobial susceptibility testing as applied to clinical practice. Clin Infect Dis 61:1446–1452. doi: 10.1093/cid/civ498. [DOI] [PubMed] [Google Scholar]
- 36.Ambrose PG, Meagher AK, Passarell JA, Van Wart SA, Cirincione BB, Rubino CM, Korth-Bradley JM, Babinchak T, Ellis-Grosse E. 2009. Use of a clinically derived exposure-response relationship to evaluate potential tigecycline-Enterobacteriaceae susceptibility breakpoints. Diagn Microbiol Infect Dis 63:38–42. doi: 10.1016/j.diagmicrobio.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 37.Gumbo T, Pasipanodya JG, Romero K, Hanna D, Nuermberger E. 2015. Forecasting accuracy of the hollow fiber model of tuberculosis for clinical therapeutic outcomes. Clin Infect Dis 61(Suppl 1):S25–S31. doi: 10.1093/cid/civ427. [DOI] [PubMed] [Google Scholar]
- 38.Gumbo T, Pasipanodya JG, Nuermberger E, Romero K, Hanna D. 2015. Correlations between the hollow fiber model of tuberculosis and therapeutic events in tuberculosis patients: learn and confirm. Clin Infect Dis 61(Suppl 1):S18–S24. doi: 10.1093/cid/civ426. [DOI] [PubMed] [Google Scholar]
- 39.Cavaleri M, Manolis E. 2015. Hollow fiber system model for tuberculosis: the European Medicines Agency experience. Clin Infect Dis 61(Suppl 1): S1–S4. doi: 10.1093/cid/civ484. [DOI] [PubMed] [Google Scholar]
- 40.Romero K, Clay R, Hanna D. 2015. Strategic regulatory evaluation and endorsement of the hollow fiber tuberculosis system as a novel drug development tool. Clin Infect Dis 61(Suppl 1):S5–S9. doi: 10.1093/cid/civ424. [DOI] [PubMed] [Google Scholar]
- 41.Chilukuri D, McMaster O, Bergman K, Colangelo P, Snow K, Toerner JG. 2015. The hollow fiber system model in the nonclinical evaluation of antituberculosis drug regimens. Clin Infect Dis 61(Suppl 1):S32–S33. doi: 10.1093/cid/civ460. [DOI] [PubMed] [Google Scholar]
- 42.Locher CP, Jones SM, Hanzelka BL, Perola E, Shoen CM, Cynamon MH, Ngwane AH, Wiid IJ, van Helden PD, Betoudji F, Nuermberger EL, Thomson JA. 2015. A novel inhibitor of gyrase B is a potent drug candidate for treatment of tuberculosis and nontuberculosis mycobacterial infections. Antimicrob Agents Chemother 59:1455–1465. doi: 10.1128/AAC.04347-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clinical Laboratory Standards Institute. 2011. Susceptibility testing of Mycobacteria, Nocardiae and other aerobic Actinomycetes—approved standard M24-A2, 2nd ed CLSI, Wayne, PA. [PubMed] [Google Scholar]
- 44.Booker BM, Smith PF, Forrest A, Bullock J, Kelchlin P, Bhavnani SM, Jones RN, Ambrose PG. 2005. Application of an in vitro infection model and simulation for reevaluation of fluoroquinolone breakpoints for Salmonella enterica serotype Typhi. Antimicrob Agents Chemother 49:1775–1781. doi: 10.1128/AAC.49.5.1775-1781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Defife R, Scheetz MH, Feinglass JM, Postelnick MJ, Scarsi KK. 2009. Effect of differences in MIC values on clinical outcomes in patients with bloodstream infections caused by gram-negative organisms treated with levofloxacin. Antimicrob Agents Chemother 53:1074–1079. doi: 10.1128/AAC.00580-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gumbo T. 2010. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother 54:1484–1491. doi: 10.1128/AAC.01474-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chigutsa E, Pasipanodya JG, Visser ME, van Helden PD, Smith PJ, Sirgel FA, Gumbo T, McIlleron H. 2015. Impact of nonlinear interactions of pharmacokinetics and MICs on sputum bacillary kill rates as a marker of sterilizing effect in tuberculosis. Antimicrob Agents Chemother 59:38–45. doi: 10.1128/AAC.03931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gumbo T, Pasipanodya JG, Wash P, Burger A, McIlleron H. 2014. Redefining multidrug-resistant tuberculosis based on clinical response to combination therapy. Antimicrob Agents Chemother 58:6111–6115. doi: 10.1128/AAC.03549-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gumbo T, Chigutsa E, Pasipanodya J, Visser M, van Helden PD, Sirgel FA, McIlleron H. 2014. The pyrazinamide susceptibility breakpoint above which combination therapy fails. J Antimicrob Chemother 69:2420–2425. doi: 10.1093/jac/dku136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williamson DA, Roberts SA, Bower JE, Vaughan R, Newton S, Lowe O, Lewis CA, Freeman JT. 2012. Clinical failures associated with rpoB mutations in phenotypically occult multidrug-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis 16:216–220. doi: 10.5588/ijtld.11.0178. [DOI] [PubMed] [Google Scholar]
- 51.van Ingen J, Aarnoutse R, de Vries G, Boeree MJ, van Soolingen D. 2011. Low-level rifampicin-resistant Mycobacterium tuberculosis strains raise a new therapeutic challenge. Int J Tuberc Lung Dis 15:990–992. doi: 10.5588/ijtld.10.0127. [DOI] [PubMed] [Google Scholar]
- 52.Christianson S, Voth D, Wolfe J, Sharma MK. 2014. Re-evaluation of the critical concentration for ethambutol antimicrobial sensitivity testing on the MGIT 960. PLoS One 9(9):e108911. doi: 10.1371/journal.pone.0108911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ambrose PM. 2016. Rational susceptibility test interpretive criteria. Perspectives from the USCAST. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM374226.pdf.