

Abstract
Sofosbuvir displays a high phenotypic barrier to resistance, and it is a component of several combination therapies for hepatitis C virus (HCV) infections. HCV fitness can be a determinant of decreased sensitivity to direct-acting antiviral agents such as telaprevir or daclatasvir, but fitness-dependent decreased drug sensitivity has not been established for drugs with a high phenotypic barrier to resistance. Low- and high-fitness HCV populations and biological clones derived from them were used to infect Huh-7.5 hepatoma cells. Sofosbuvir efficacy was analyzed by measuring virus progeny production during several passages and by selection of possible sofosbuvir resistance mutations determined by sequencing the NS5B-coding region of the resulting populations. Sofosbuvir exhibited reduced efficacy against high-fitness HCV populations, without the acquisition of sofosbuvir-specific resistance mutations. A reduced sofosbuvir efficacy, similar to that observed with the parental populations, was seen for high-fitness individual biological clones. In independently derived high-fitness HCV populations or clones passaged in the presence of sofosbuvir, M289L was selected as the only substitution in the viral polymerase NS5B. In no case was the sofosbuvir-specific resistance substitution S282T observed. High HCV fitness can lead to decreased sensitivity to sofosbuvir, without the acquisition of specific sofosbuvir resistance mutations. Thus, fitness-dependent drug sensitivity can operate with HCV inhibitors that display a high barrier to resistance. This mechanism may underlie treatment failures not associated with selection of sofosbuvir-specific resistance mutations, linked to in vivo fitness of pretreatment viral populations.
INTRODUCTION
Hepatitis C virus (HCV) infection affects about 2.3% of the world population, with treatment and patient management costs being an important burden for health systems (1). Treatment efficacy, quantified as the rate of sustained viral response, has improved markedly with the introduction of direct-acting antiviral agents (DAAs) (2). DAAs include inhibitors of the viral protease NS3-4A (telaprevir [TPV], boceprevir, simeprevir, paritaprevir/ritonavir, asunaprevir, etc.), of nonstructural protein NS5A (daclatasvir [DCV], ledipasvir, ombitasvir, etc.), and the polymerase NS5B (nonnucleoside analogues, such as dasabuvir, and one nucleoside analogue, such as sofosbuvir [SOF]). Many DAAs have been licensed for human use, and others are still in preclinical and clinical assessment.
A major issue in antiviral treatments is the selection of inhibitor-resistant mutants leading to treatment failure. Selection is influenced by genetic and phenotypic barriers to resistance. The genetic barrier depends on the number and type of mutations needed for the RNA to encode amino acid substitutions needed to confer resistance. The phenotypic barrier is determined by the fitness cost inflicted upon the virus by the mutations associated with resistance. Barriers vary depending on the nature of the antiviral agent, the viral genomic nucleotide sequence, and population parameters. For statistical reasons a large, replicating viral population increases the likelihood that required mutations will occur with genomes harboring fitness-restoring compensatory mutations (reviewed in reference 3). In the case of HCV, inhibitors of NS3-4A and NS5A and nonnucleoside inhibitors of NS5B generally exhibit a low phenotypic barrier to resistance while nucleoside analogues display a high barrier (4–8).
Recent results suggest that HCV fitness may be a determinant of decreased sensitivity to antiviral agents (9). Passage of the parental population HCV (passage 0, p0) [based on the JFH-1-based chimera Jc1FLAG2(p7-nsGluc2A)] in Huh-7.5 reporter cells resulted in increased fitness, as expected from continued virus replication in the same environment (3). Fitness levels of the populations at passage 45 (HCV p45) and at passage 100 (HCV p100) were 1.9 ± 0.3 and 2.2 ± 0.4, respectively, relative to the fitness of HCV p0 (fitness 1). These fitness increases were accompanied by an expansion of the mutant spectrum, with a minimum mutation frequency increase of 1.7-fold and 2.6-fold for HCV p45 and HCV p100, respectively, relative to HCV p0 (9). The passaged populations exhibited decreased sensitivity to the anti-HCV inhibitors TPV, DCV, cyclosporine (CsA), ribavirin (RBV), and alpha interferon (IFN-α) (9, 10). In no case was resistance associated with specific mutations in the viral genome. Further proof was provided by parallel kinetics of progeny production over a 1,000-fold range of multiplicities of infection (MOIs) in the presence of the drugs and by maintenance of the resistance phenotypes in biological clones isolated from the passaged HCV populations (9). It was concluded that fitness or a fitness-associated trait was a drug resistance determinant for HCV (9, 11, 12).
This prior study did not examine a DAA with a high phenotypic resistance barrier such as SOF, the prodrug of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine, which, in its triphosphate form, acts as chain terminator during HCV RNA elongation (13, 14). SOF has been used in monotherapy and in various combinations with ribavirin and other DAAs (ledipasvir, simeprevir, etc.) with average sustained virological response (SVR) rates exceeding 90% (see, for example, references 15 to 20, among others). Mutations associated with SOF resistance have been described previously. Substitution S282T in NS5B, which confers resistance to 2′-C-methylated nucleotide analogues, was first described in replicon assays (21) and subsequently selected in vitro using HCV replicons of genotypes 1 to 6 (22). For genotype 1a, S282T was found in combination with I434M. For genotype 2a, substitution T179A, M289L, or I293L was required for resistance together with S282T, while substitutions M343T and H479P increased the replication capacity of replicons carrying S282T (23). Regarding SOF resistance in vivo, NS5B S282T has been found in a patient infected with HCV genotype 2b who relapsed after SOF monotherapy in a phase 2 trial (17). In another study, 99.8% of the viral population harbored the S282T substitution after 4 weeks of SOF monotherapy, despite the low frequency of the mutant at baseline (24). S282T was also found in one subject who relapsed 4 weeks after SOF monotherapy (25) and in two patients infected with genotype 1 who did not achieve a sustained virological response after treatment with SOF/RBV (26) or SOF/ledipasvir (19). Recently, substitutions S282G/C/R were detected in nine patients, and in silico models suggest that these substitutions may be associated with SOF resistance (27). A mutant with a L159F/L320F double substitution emerged in one genotype 1b-infected patient, and the virus showed reduced susceptibility to SOF and mericitabine in cell culture (28). HCV genotype 3 with NS5B substitutions L159F and V321A were isolated from patients in phase 3 clinical trials with SOF-based regimens, but these substitutions did not affect overall susceptibility to SOF; in other patients that presented partial responses or relapse to SOF treatment, NS5B substitutions found included L159F, C316N, S282R, and/or L320F, but their clinical significance has not been established (reviewed in reference 29). Structural bioinformatics analyses from next-generation sequencing (NGS) data of virus from patients participating in clinical trials with SOF confirmed that substitutions L159F, V321A, C316N, and S282R were associated with treatment failure (7). Thus, despite a generally accepted high phenotypic barrier to achieve SOF resistance, both in vitro assays and clinical results suggest that a number of substitutions in NS5B—which differ depending on the viral genotype—can confer diminished susceptibility to this inhibitor, with S282T playing a key role (30).
Given the remarkable clinical impact of SOF-based HCV treatments, it was of interest to investigate if passaged, high-fitness HCV populations displayed decreased sensitivity to SOF in the absence of SOF-specific resistance mutations. In the present report we show that this is indeed the case, suggesting that inherent pretreatment fitness of HCV can alter sensitivity to sofosbuvir-based regimens, without the need for selection of specific resistance mutants.
MATERIALS AND METHODS
Cells, viruses, infections, and titration of infectivity.
The origin of the Huh-7 Lunet, Huh-7.5, and Huh-7.5 reporter cell lines, conditions for their growth, preparation of HCV, and titration of infectivity have been described previously (9, 10, 31–33). Huh-7 Lunet cells were used for electroporation of RNA transcribed from plasmid Jc1FLAG2(p7-nsGluc2A) (genotype 2a) or its GNN replication-defective mutant used as a negative control (10). Supernatants of the passaged electroporated cells were pooled, the virus was concentrated 20-fold, and the concentrate was used to infect Huh-7.5 reporter cells to prepare the parental virus HCV p0; further details are described in Perales et al. (10). HCV p0 was subjected to 100 serial passages in Huh-7.5 reporter cells. For the first passage, 4 × 105 Huh-7.5 reporter cells were infected with HCV p0 at a multiplicity of infection (MOI) of 0.5 50% tissue culture infective doses (TCID50)/cell. For subsequent passages, the same numbers of cells were infected with the virus contained in 0.5 ml of cell culture supernatant from the previous infection; at passage 60 the volume of supernatant used for infection was diluted gradually up to 20-fold to limit cytopathology. The MOIs at different passages ranged from 0.2 to 5 TCID50/cell. Passage (p) numbers are identified as follow: HCV p45 and HCV p100 are the HCV p0 population subjected to 45 and 100 serial passages, respectively, in Huh-7.5 reporter cells.
To titrate HCV infectivity, viral samples were serially diluted and added to Huh-7.5 cells seeded 15 h earlier in 96-well plates (6,400 cells/well). At 3 days postinfection, the cells were washed with phosphate-buffered saline (PBS), fixed with cold methanol, and stained with anti-NS5A monoclonal antibody 9E10 (10, 33).
Sofosbuvir treatment.
A stock solution of 10 mM SOF (Selleck Chemicals) was prepared in dimethyl sulfoxide (DMSO) and stored at −80°C. Prior to use, the solution was diluted in Dulbecco's modified Eagle's medium (DMEM) to reach the desired concentration. SOF toxicity (the SOF concentration needed to kill 50% of the cells, or cytotoxic concentration 50 [CC50]), and inhibitory activity (the SOF concentration needed to inhibit the infectious progeny production by 50% [IC50]) were measured as previously described (9).
Preparation of biological clones from hepatitis C virus populations.
HCV p0, HCV p45, and HCV p100 were treated with mild detergent (sodium deoxycholate at 0.01%) for 10 min at room temperature, diluted, and applied to Huh-7.5 cell monolayers in M96 wells. The cell culture supernatant of a well with a single cluster of Huh-7.5-infected cells was used to infect 1 × 105 Huh-7.5 cells in an M24 well, followed by an infection of 4 × 105 Huh-7.5 cells in an M6 dish. Finally, 1 × 106 Huh-7.5 cells were infected in a p60 dish under standard infection conditions (9).
RNA extraction, cDNA synthesis, PCR amplification, and nucleotide sequencing.
Intracellular viral RNA was extracted from infected cells at each passage using a Qiagen RNeasy kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. HCV RNA was reverse transcribed using avian myeloblastosis (AMV) reverse transcriptase (Promega), and the PCR amplification of the NS5B-coding region was carried out using AccuScript (Agilent Technologies) with oligonucleotide primers Jc1-NS5B-F1, 5′-TGGTCTACTTGCTCCGAGGAGG (sense orientation; the 5′ nucleotide corresponds to genomic residue 7625), and Jc1-NS5B-R4, 5′-AGTTAGCTATGGAGTGTACCTAG (antisense orientation; the 5′ nucleotide corresponds to genomic residue 9476; residue numbering is that of HCV strain JFH-1). Amplification products were analyzed by agarose gel electrophoresis, using HindIII-digested ϕ29 DNA as a molecular mass standard. Amplification controls in the absence of RNA were run in parallel to ascertain the absence of contamination by undesired templates.
Nucleotide sequences were determined on the two strands of the amplified DNA according to standard procedures (10, 34).
Statistical analyses.
To determine the statistical significance of differences in serial-passage experiments, two-way analysis of variance (ANOVA) was performed with the SPSS, version 13.0, statistical package (SPSS, Inc.).
RESULTS
Dose-dependent inhibition of HCV by sofosbuvir in cell culture.
To quantify the toxicity of SOF for Huh-7.5 reporter cells and its inhibitory activity on progeny production by the parental HCV p0 population, CC50 and IC50s were obtained (Fig. 1A and B). The IC50 (20 ± 3 nM) was at least 2,000-fold higher than the CC50 (>50 μM), yielding a therapeutic index (TI), defined as CC50/IC50, of >2,000. The TI value allowed exploration of the inhibitory effect over a 12-fold range of SOF concentrations in the course of three serial passages of HCV p0 (Fig. 1C). The results indicate a dose-dependent inhibition of HCV p0 progeny production under conditions of undetectable toxicity for the host Huh-7.5 reporter cells.
FIG 1.
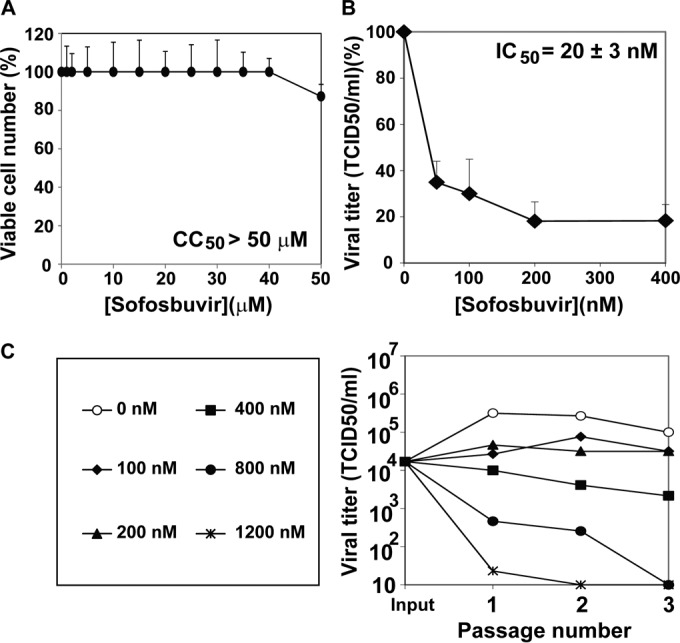
Inhibition of hepatitis C virus by sofosbuvir. (A) Determination of 50% cytotoxic concentration (CC50). Huh-7.5 reporter cells (1.3 × 104) were incubated with the indicated concentration of sofosbuvir (SOF) during 72 h at 37°C, and the numbers of viable cells were counted using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide], as described previously (9). (B) Determination of the drug concentration required for 50% inhibition (IC50) of infectious HCV yield. Huh-7.5 reporter cells (1.0 × 105) were infected with 3 × 103 TCID50 of HCV p0 in the presence of the indicated concentrations of SOF. Virus titers were determined in the cell culture supernatants at 72 h postinfection. Viral titers are given as the percentages of the titers obtained in the infection in the absence of SOF. (C) Progeny production in the course of three serial passages of HCV p0 in the presence of increasing concentrations of SOF, as indicated. Infection conditions are those explained in the description of panel B. Procedures are detailed in Materials and Methods.
Resistance to sofosbuvir of multiply passaged HCV p0.
To test if passaged HCV displayed resistance to SOF, HCV p0, HCV p45, and HCV p100 were subjected to 10 serial infections in the absence or presence of SOF (800 nM and 1,200 nM) (Fig. 2). In the absence of SOF, HCV p100 showed increased progeny production during the first five passages compared with result with HCV p0 or HCV p45, indicative of its high fitness (9). Passage in the presence of 800 nM SOF resulted in loss of infectivity of HCV p0 by passage 4 but survival of HCV p45 and HCV p100, with at least a 103-fold higher progeny production than that of HCV p0 (the difference of progeny production between HCV p45 or HCV p100 and HCV p0 was statistically significant [P < 0.001; ANOVA test], but the difference between results with HCV p45 and HCV p100 was not statistically significant [P > 0.05; ANOVA test]). In the presence of 1,200 nM SOF, the three populations were inhibited; but while the infectivity of HCV p0 was not detectable by passage 2, HCV p100 survived at least during five passages, and HCV p45 increased its viral production from passages 8 to 10 (Fig. 2) (the difference in progeny production between HCV p45 and HCV p0 or HCV p100 was statistically significant [P < 0.001; ANOVA test], but the difference between HCV p0 and HCV p100 was not significant [P > 0.05; ANOVA test]). Thus, HCV passaged in cell culture in the absence of SOF exhibited significant resistance to SOF.
FIG 2.
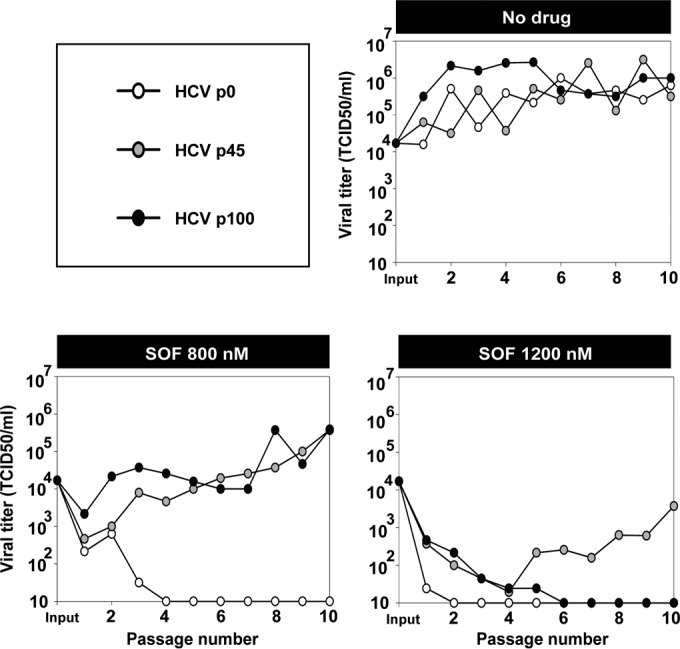
Infectious progeny production of hepatitis C virus populations passaged in the absence or presence of sofosbuvir. HCV p0, HCV p45, and HCV p100 were subjected to 10 serial passages in the absence (no drug) or the presence of 800 nM or 1,200 nM sofosbuvir (SOF). For the first passage, 4 × 105 Huh-7.5 reporter cells were infected with 1.2 × 104 TCID50 of the indicated virus, to give a multiplicity of infection (MOI) of 0.03 TCID50/cell. For successive infections, 4 × 105 Huh-7.5 reporter cells were infected with the virus contained in 500 μl of the cell culture supernatant from the previous infection, yielding a range of MOIs of 1.25 × 10−5 to 3.95 TCID50/cell. Infections were allowed to proceed for 72 h. The abscissa axes indicate the limits of detection of virus titers. Infection conditions are further detailed in Materials and Methods.
Mutations in the NS5B-coding regions of high-fitness HCV populations passaged in the absence or presence of sofosbuvir.
The consensus sequence of the NS5B-coding region of HCV p0, HCV p45, and HCV p100 did not reveal detectable substitutions previously associated with SOF resistance, except for T179A present in ∼50% of genomes in HCV p100 (Fig. 3). T179A has not been considered a SOF resistance substitution per se in the absence of S282T. When tested as a single replacement, T179A remained at low frequency levels at passage 5 in the presence of 800 nM SOF and was undetectable by passage 10. Instead, M289L was dominant at passage 10 (Fig. 3). When HCV p100 was subjected to five passages in the presence of 1,200 nM SOF, T179A was dominant, and M289L was present in about 25% of the genomes; this viral population did not survive beyond passage 5 (Fig. 2). M289L also became dominant upon passage of HCV p45 in the presence of 800 nM or 1,200 nM SOF (Fig. 3). Selection of this substitution raised the possibility that the SOF resistance of HCV p45 and HCV p100 was due to the presence of genomes with this mutation at low frequency even though it was not detected in the population consensus sequences. To address this, SOF resistance was examined using biological viral clones isolated from the HCV p0, HCV p45, and HCV p100 populations.
FIG 3.
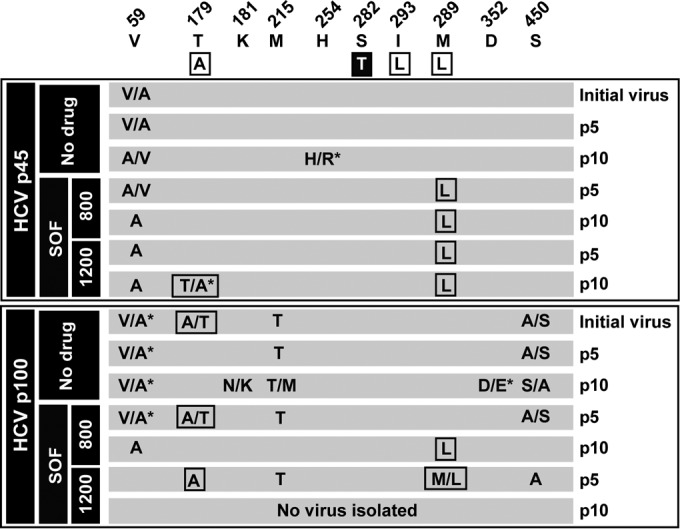
Amino acid substitutions in the consensus sequence of the NS5B (polymerase)-coding region of the hepatitis C virus passaged in the absence or presence of sofosbuvir. Viral populations and sofosbuvir (SOF) concentrations (nanomolar) are indicated in the filled boxes on the left; the virus passage number (p) is given in the last column. The upper row indicates the amino acids where substitutions have been found and their positions in the NS5B protein. The boxed amino acids in the second row are those that have been related to SOF resistance in HCV genotype 2a, with the major resistance substitution S282T highlighted. The two panels below the sequence describe the amino acid substitutions in each population, with those that have been related to SOF resistance boxed. A slash between two unmarked amino acids indicates that both were present at about 50% frequency in the consensus sequence, according to the peaks in the sequencing data; a slash with an asterisk indicates dominance (around 70% frequency) of the first amino acid in the pair. The complete repertoire of synonymous and nonsynonymous mutations found in the viral populations is listed in Table S1 in the supplemental material. Procedures for nucleotide sequencing are described in Materials and Methods.
Passage of biological clones of hepatitis C virus in the absence or presence of sofosbuvir.
Three biological clones from each of the HCV p0, HCV p45, and HCV p100 populations were isolated by endpoint virus dilution and successive infections, as detailed in Materials and Methods. Critical to interpreting subsequent results is that the number of rounds of amplification undergone by the individual clones to obtain the working stocks was at least 3-fold lower than the number of rounds involved in the preparation of HCV p0 from the initial plasmid transcript (9). A low number of replication rounds ensures a mutant spectrum diversification lower than that of the reference HCV p0 population.
Each of the nine biological clones was subjected to three passages in the absence or presence of 800 nM SOF. Despite variations among individual clones from the same population, the average progeny production at passage 3 by the HCV p45 and HCV p100 clones was 8.5-fold and 100.9-fold, respectively, higher than the average production by the HCV p0 clones (P = 0.7 and P = 0.02, respectively; ANOVA test) (Fig. 4). Since minority HCV genomes harboring a SOF resistance mutation should have been excluded by the cloning process, this result suggests that viral fitness or a fitness-related trait can confer SOF resistance to HCV without the presence of specific SOF resistance mutations. This differential sensitivity to SOF did not vary over the 630,000-fold range of MOI values involved in the passages (Fig. 4).
FIG 4.

Infectious progeny production of biological clones of hepatitis C virus passaged in the absence or presence of sofosbuvir. Biological clones and their parental populations are indicated in the upper boxes, and the absence or presence of sofosbuvir (SOF) is indicated at left. For the first passage, 4 × 105 Huh-7.5 reporter cells were infected with 1.2 × 104 TCID50 of the corresponding clone, to give a multiplicity of infection (MOI) of 0.03 TCID50/cell. For successive infections, 4 × 105 Huh-7.5 reporter cells were infected with the virus contained in 500 μl of the cell culture supernatant from the previous infection, yielding a range of MOIs of 1.25 × 10−5 to 7.9 TCID50/cell. Infections were allowed to proceed for 72 h. The abscissa axes indicate the limits of detection of virus titers. Procedures to obtain the biological clones and infection conditions are described in Materials and Methods.
Evidence of a dual fitness-dependent mechanism of drug resistance.
To investigate the possible selection of SOF resistance mutations during passage of biological clones in the presence of SOF, the NS5B-coding regions of clones passaged in the absence or presence of SOF that yielded sufficient viral RNA were sequenced. The results indicate that in some (but not all) populations, mutations leading to an amino acid substitution previously related to SOF resistance were selected, reaching partial or complete dominance (Fig. 5). The most significant substitutions were T179A and M289L, for opposite reasons. In our experimental system T179A can be excluded as a bona fide SOF resistance mutation because it occurred only in a biological clone from HCV p0 in the absence and presence of SOF. M289L became partially or totally dominant in two out of the six clones tested and always in the presence of SOF. In no case was the SOF resistance substitution S282T selected (Fig. 5). In conclusion, high HCV basal fitness is a factor of diminished sensitivity to SOF and a parameter that can promote selection of specific mutations related to SOF resistance.
FIG 5.

Amino acid substitutions in the consensus sequence of the NS5B (polymerase)-coding region of biological clones of hepatitis C virus passaged in the absence or presence of sofosbuvir. The parental viral populations from which the corresponding biological clones were retrieved (identified with the same code used in Fig. 4) and the sofosbuvir (SOF) concentration (nanomolar) are indicated in the filled boxes on the left; the virus passage number (p) is given in the last column. Biological clones not included in the nucleotide sequence analysis are those for which no sufficient viral RNA was obtained (compare with Fig. 4). The upper row includes the amino acids where substitutions have been found and their positions in the NS5B protein. The boxed amino acids in the second row are those that have been related to SOF resistance in HCV genotype 2a, with the major resistance substitution S282T highlighted. The six panels below the sequence describe the amino acid substitutions in each population, with those that have been related to SOF resistance boxed. A slash between two unmarked amino acids means that both were present at about 50% frequency in the consensus sequence according to the peaks in the sequencing data; a slash with an asterisk indicates dominance (around 70% frequency) of the first amino acid in the pair. The complete repertoire of synonymous and nonsynonymous mutations found in the viral populations is listed in Table S2 in the supplemental material. Procedures for nucleotide sequencing are described in Materials and Methods.
DISCUSSION
In this report we have documented a fitness-dependent resistance to the high-barrier anti-HCV DAA SOF. This drug was chosen because of its importance in current HCV therapies (e.g., references 15 to 20 and 35 to 40). The results show that HCV p45 and HCV p100 are significantly more resistant than HCV p0 to SOF. A comparison with the previous results with other inhibitors shows quantitative differences. While the inhibition by IFN-α, ribavirin, TPV, DCV, and CsA was 513-, 412-, 453-, 13,658-, and 309-fold, respectively, greater for HCV p0 than HCV p100, for SOF this difference was only 34-fold (compare Fig. 2 with Fig. 3 in reference 9). For this comparison, the drug concentrations used were 6.0- to 50-fold higher than the corresponding IC50s, and titers were measured at passage 3 in infections carried out at an MOI of 0.03 TCID50/cell. Additional work is needed to determine if the lower difference observed with SOF than with the other DAAs may relate to its higher resistance barrier.
In a study with HCV genotype 2a replicons, Lam and colleagues observed that the major SOF resistance substitution S282T was selected only when replicon carrier cells were treated with SOF concentrations that were 70-fold higher than the IC50 (23). Since the SOF concentrations used in our HCV inhibition experiments were 32- to 48-fold higher than the IC50, it is not unexpected that S282T was not selected in any of the populations or biological clones passaged in the presence of SOF.
The molecular basis and the possible clinical impact of the basal HCV fitness level on treatment response are unknown. Both in the cell culture system and among clinical samples of HCV, mutations that alter HCV fitness have been described (10, 41). As a possible mechanism, we proposed a competition between the number of replicating genomes at each replicative unit in an infected cell and the concentration of inhibitor that reaches the replicative units (9). This model is plausible if viral fitness is essentially due to increased intracellular viral replication. The other model is an increased stability of the replication complex, with longer time for the functional HCV RNA to decay away in the absence of replication. Experiments to test these possibilities are in progress. Regarding the possible clinical significance of our findings, the main difficulty is to estimate the basal HCV fitness at the onset of the treatment. Theoretical studies have inferred fitness landscapes from sequence data, but the majority of them have ignored intrahost heterogeneity and have regarded viruses as “strains” rather than mutant clouds. An interesting approach has been taken by N. Beerenwinkel and colleagues to infer fitness landscapes from deep-sequencing data (42, 43). They likened HIV-1 fitness parameters to the mutant spectrum composition using quasispecies theory with a number of assumptions to permit the computation (mutation-selection equilibrium, use of a limited haplotype subset, uniform mutation rate, etc.). Use of these computational tools together with clinical data that may be associated with high HCV fitness (duration of the infection, viral load, and mutant spectrum complexity) (44, 45) may provide a predictive tool for inhibitor resistance independent of specific mutations, and such efforts are in progress.
It has been observed that treatment failures are more frequent in patients that have remained infected for a long time, and it has been well established that prolonged infections in the same environment result in a fitness gain (reviewed in reference 3). There is recent evidence of patients that do not respond to treatment in whom resistance mutations are not detected after HCV rebound. In a cohort of 1,797 patients subjected to TPV monotherapy, 28% were nonresponders, and the virus in 23% did not include detectable TPV resistance mutations (46). In a large cohort of 1,662 patients treated with SOF either alone or in combination with ribavirin or ribavirin and pegylated IFN-α, 18% of patients did not attain an SVR; in many cases treatment failure could not be attributed to specific resistance mutations (25). The authors proposed that residual HCV replication might occur in the liver during drug treatment, with undetectable RNA in the blood. Other modeling studies have shown that low-level virus replication may occur in the presence of inhibitor, without the selection of drug-resistant variants (47). In some patients that did not respond to a combination of TPV, pegylated IFN-α, and ribavirin, no resistance mutations were detected (48). In another study, no SOF resistance mutations were observed after virological relapse of HCV genotype 2 in patients subjected to SOF-based combination therapies (49).
If a fitness-related mechanism of decreased HCV sensitivity operated in vivo, it would mean that some treatment failures are expected, independent of specific resistance mutations. The fitness cost of resistance mutations and fitness rescue due to compensatory mutations are two well-established mechanisms by which fitness can impact the development of antiviral resistance. The results presented here introduce new roles of fitness in connection with the management of HCV infections: a direct influence on multidrug resistance and the enhanced probability that high fitness, by virtue of its associated capacity to explore sequence space, can promote selection of drug resistance mutations or mutations that facilitate the selection of bona fide resistance mutations. It should be noted that current standard methodologies to assess the influence of specific mutations identified in breakthrough viruses during treatment in HCV drug resistance consist of introducing the mutations in either infectious HCV clones or subgenomic replicons and measuring their resistance levels in cell culture. Such methods will not uncover possible fitness effects on resistance since fitness is dependent on the sequence and population context of the infectious virus as it replicates in the natural liver environment (reviewed in reference 3). A major issue raised by our study is that bona fide SOF-resistant variants may not be selected or even selectable under current anti-HCV regimens. However, differences in the treatment regimen and the duration necessary to achieve an SVR may be due to fitness differences in the starting viral population. This may underpin the decreased SVR efficacy associated with SOF for HCV genotype 3, with SOF requiring longer treatment durations or combination with additional DAAs.
Repeating an issue already raised during early days of the AIDS epidemic, the value of resistance testing is being debated for guiding HCV treatment decisions (50). As a general note, molecular information has always been beneficial to improving personalized antiviral treatments (reviewed in reference 3) unless pan-genomic, fully effective new combination treatments become available. The present study on fitness-dependent SOF resistance provides an additional incentive for resistance testing in patients: the possibility that treatment failure may occur in the absence of selection of mutants with specific resistance mutations. This information should contribute to a better understanding of the influence exerted by quasispecies dynamics in vivo and of treatment outcomes. Finally, the observations with HCV beg the question of possible fitness-dependent effects of inhibitor sensitivity in other viruses.
Supplementary Material
ACKNOWLEDGMENTS
Centro de Investigación en Red de Enfermedades Hepáticas y Digestivas (CIBERehd) is funded by Instituto de Salud Carlos III. C.P. is supported by the Miguel Servet program of the Instituto de Salud Carlos III (CP14/00121).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00581-16.
REFERENCES
- 1.Horner SM, Naggie S. 2015. Successes and challenges on the road to cure hepatitis C. PLoS Pathog 11:e1004854. doi: 10.1371/journal.ppat.1004854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawlotsky JM. 2015. Hepatitis C treatment: the data flood goes on-an update from the liver meeting 2014. Gastroenterology 148:468–479. doi: 10.1053/j.gastro.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Domingo E. 2016. Virus as populations. Academic Press, Elsevier, Amsterdam. [Google Scholar]
- 4.Gotte M. 2014. Resistance to nucleotide analogue inhibitors of hepatitis C virus NS5B: mechanisms and clinical relevance. Curr Opin Virol 8:104–108. doi: 10.1016/j.coviro.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Pawlotsky JM. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 6.De Luca A, Bianco C, Rossetti B. 2014. Treatment of HCV infection with the novel NS3/4A protease inhibitors. Curr Opin Pharmacol 18:9–17. doi: 10.1016/j.coph.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Donaldson EF, Harrington PR, O'Rear JJ, Naeger LK. 2015. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology 61:56–65. doi: 10.1002/hep.27375. [DOI] [PubMed] [Google Scholar]
- 8.McCown MF, Rajyaguru S, Le Pogam S, Ali S, Jiang WR, Kang H, Symons J, Cammack N, Najera I. 2008. The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob Agents Chemother 52:1604–1612. doi: 10.1128/AAC.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheldon J, Beach NM, Moreno E, Gallego I, Pineiro D, Martinez-Salas E, Gregori J, Quer J, Esteban JI, Rice CM, Domingo E, Perales C. 2014. Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus. J Virol 88:12098–12111. doi: 10.1128/JVI.01860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perales C, Beach NM, Gallego I, Soria ME, Quer J, Esteban JI, Rice C, Domingo E, Sheldon J. 2013. Response of hepatitis C virus to long-term passage in the presence of alpha interferon: multiple mutations and a common phenotype. J Virol 87:7593–7607. doi: 10.1128/JVI.02824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perales C, Domingo E. 2016. Antiviral strategies based on lethal mutagenesis and error threshold. Curr Top Microbiol Immunol 392:323–339. doi: 10.1007/82_2015_459. [DOI] [PubMed] [Google Scholar]
- 12.Perales C, Quer J, Gregori J, Esteban JI, Domingo E. 2015. Resistance of hepatitis C virus to inhibitors: complexity and clinical implications. Viruses 7:5746–5766. doi: 10.3390/v7112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fung A, Jin Z, Dyatkina N, Wang G, Beigelman L, Deval J. 2014. Efficiency of incorporation and chain termination determines the inhibition potency of 2′-modified nucleotide analogs against hepatitis C virus polymerase. Antimicrob Agents Chemother 58:3636–3645. doi: 10.1128/AAC.02666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. 2010. Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 53:7202–7218. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 15.Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, Nahass R, Ghalib R, Gitlin N, Herring R, Lalezari J, Younes ZH, Pockros PJ, Di Bisceglie AM, Arora S, Subramanian GM, Zhu Y, Dvory-Sobol H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Sulkowski M, Kwo P. 2014. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 370:1483–1493. doi: 10.1056/NEJMoa1316366. [DOI] [PubMed] [Google Scholar]
- 16.Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, Romero-Gomez M, Zarski JP, Agarwal K, Buggisch P, Foster GR, Brau N, Buti M, Jacobson IM, Subramanian GM, Ding X, Mo H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Mangia A, Marcellin P. 2014. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 370:1889–1898. doi: 10.1056/NEJMoa1402454. [DOI] [PubMed] [Google Scholar]
- 17.Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM. 2013. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 368:34–44. doi: 10.1056/NEJMoa1208953. [DOI] [PubMed] [Google Scholar]
- 18.Asselah T. 2014. Daclatasvir plus sofosbuvir for HCV infection: an oral combination therapy with high antiviral efficacy. J Hepatol 61:435–438. doi: 10.1016/j.jhep.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 19.Lawitz E, Poordad FF, Pang PS, Hyland RH, Ding X, Mo H, Symonds WT, McHutchison JG, Membreno FE. 2014. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 383:515–523. doi: 10.1016/S0140-6736(13)62121-2. [DOI] [PubMed] [Google Scholar]
- 20.Lawitz E, Sulkowski MS, Ghalib R, Rodriguez-Torres M, Younossi ZM, Corregidor A, DeJesus E, Pearlman B, Rabinovitz M, Gitlin N, Lim JK, Pockros PJ, Scott JD, Fevery B, Lambrecht T, Ouwerkerk-Mahadevan S, Callewaert K, Symonds WT, Picchio G, Lindsay KL, Beumont M, Jacobson IM. 2014. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet 384:1756–1765. doi: 10.1016/S0140-6736(14)61036-9. [DOI] [PubMed] [Google Scholar]
- 21.Migliaccio G, Tomassini JE, Carroll SS, Tomei L, Altamura S, Bhat B, Bartholomew L, Bosserman MR, Ceccacci A, Colwell LF, Cortese R, De Francesco R, Eldrup AB, Getty KL, Hou XS, LaFemina RL, Ludmerer SW, MacCoss M, McMasters DR, Stahlhut MW, Olsen DB, Hazuda DJ, Flores OA. 2003. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J Biol Chem 278:49164–49170. doi: 10.1074/jbc.M305041200. [DOI] [PubMed] [Google Scholar]
- 22.Rajyaguru S, Xu S, Hebner C, Svarovskaia E, Gontcharova V, Doehle B, Miller M, Mo H. 2013. Sofosbuvir selects the NS5B S282T mutation in vitro in genotype 1-6 replicons and is not cross-resistant to resistance-associated variants selected by other classes of antiviral inhibitors. Hepatology 58(Suppl S1):739A.23483581 [Google Scholar]
- 23.Lam AM, Espiritu C, Bansal S, Micolochick Steuer HM, Niu C, Zennou V, Keilman M, Zhu Y, Lan S, Otto MJ, Furman PA. 2012. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob Agents Chemother 56:3359–3368. doi: 10.1128/AAC.00054-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedskog C, Dvory-Sobol H, Gontcharova V, Martin R, Ouyang W, Han B, Gane EJ, Brainard D, Hyland RH, Miller MD, Mo H, Svarovskaia E. 2015. Evolution of the HCV viral population from a patient with S282T detected at relapse after sofosbuvir monotherapy. J Viral Hepat 22:871–881. doi: 10.1111/jvh.12405. [DOI] [PubMed] [Google Scholar]
- 25.Svarovskaia ES, Dvory-Sobol H, Parkin N, Hebner C, Gontcharova V, Martin R, Ouyang W, Han B, Xu S, Ku K, Chiu S, Gane E, Jacobson IM, Nelson DR, Lawitz E, Wyles DL, Bekele N, Brainard D, Symonds WT, McHutchison JG, Miller MD, Mo H. 2014. Infrequent development of resistance in genotype 1-6 hepatitis C virus-infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin Infect Dis 59:1666–1674. doi: 10.1093/cid/ciu697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osinusi A, Meissner EG, Lee YJ, Bon D, Heytens L, Nelson A, Sneller M, Kohli A, Barrett L, Proschan M, Herrmann E, Shivakumar B, Gu W, Kwan R, Teferi G, Talwani R, Silk R, Kotb C, Wroblewski S, Fishbein D, Dewar R, Highbarger H, Zhang X, Kleiner D, Wood BJ, Chavez J, Symonds WT, Subramanian M, McHutchison J, Polis MA, Fauci AS, Masur H, Kottilil S. 2013. Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics: a randomized clinical trial. JAMA 310:804–811. doi: 10.1001/jama.2013.109309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji H, Kozak RA, Biondi MJ, Pilon R, Vallee D, Liang BB, La D, Kim J, Van Domselaar G, Leonard L, Sandstrom P, Brooks J. 2015. Next generation sequencing of the hepatitis C virus NS5B gene reveals potential novel S282 drug resistance mutations. Virology 477:1–9. doi: 10.1016/j.virol.2014.12.037. [DOI] [PubMed] [Google Scholar]
- 28.Tong X, Le Pogam S, Li L, Haines K, Piso K, Baronas V, Yan JM, So SS, Klumpp K, Najera I. 2014. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low-level resistance to the HCV polymerase inhibitors mericitabine and sofosbuvir. J Infect Dis 209:668–675. doi: 10.1093/infdis/jit562. [DOI] [PubMed] [Google Scholar]
- 29.Childs-Kean LM, Hand EO. 2015. Simeprevir and sofosbuvir for treatment of chronic hepatitis C infection. Clin Ther 37:243–267. doi: 10.1016/j.clinthera.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 30.Lontok E, Harrington P, Howe A, Kieffer T, Lennerstrand J, Lenz O, McPhee F, Mo H, Parkin N, Pilot-Matias T, Miller V. 2015. Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology 62:1623–1632. doi: 10.1002/hep.27934. [DOI] [PubMed] [Google Scholar]
- 31.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, MacDonald MR, Bhatia SN, Rice CM. 2010. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat Biotechnol 28:167–171. doi: 10.1038/nbt.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 34.Agudo R, Ferrer-Orta C, Arias A, de la Higuera I, Perales C, Perez-Luque R, Verdaguer N, Domingo E. 2010. A multi-step process of viral adaptation to a mutagenic nucleoside analogue by modulation of transition types leads to extinction-escape. PLoS Pathog 6:e1001072. doi: 10.1371/journal.ppat.1001072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohli A, Kapoor R, Sims Z, Nelson A, Sidharthan S, Lam B, Silk R, Kotb C, Gross C, Teferi G, Sugarman K, Pang PS, Osinusi A, Polis MA, Rustgi V, Masur H, Kottilil S. 2015. Ledipasvir and sofosbuvir for hepatitis C genotype 4: a proof-of-concept, single-centre, open-label phase 2a cohort study. Lancet Infect Dis 15:1049–1054. doi: 10.1016/S1473-3099(15)00157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobson IM, Gordon SC, Kowdley KV, Yoshida EM, Rodriguez-Torres M, Sulkowski MS, Shiffman ML, Lawitz E, Everson G, Bennett M, Schiff E, Al-Assi MT, Subramanian GM, An D, Lin M, McNally J, Brainard D, Symonds WT, McHutchison JG, Patel K, Feld J, Pianko S, Nelson DR. 2013. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 368:1867–1877. doi: 10.1056/NEJMoa1214854. [DOI] [PubMed] [Google Scholar]
- 37.Kowdley KV, Lawitz E, Crespo I, Hassanein T, Davis MN, DeMicco M, Bernstein DE, Afdhal N, Vierling JM, Gordon SC, Anderson JK, Hyland RH, Dvory-Sobol H, An D, Hindes RG, Albanis E, Symonds WT, Berrey MM, Nelson DR, Jacobson IM. 2013. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 381:2100–2107. doi: 10.1016/S0140-6736(13)60247-0. [DOI] [PubMed] [Google Scholar]
- 38.Kowdley KV, Gordon SC, Reddy KR, Rossaro L, Bernstein DE, Lawitz E, Shiffman ML, Schiff E, Ghalib R, Ryan M, Rustgi V, Chojkier M, Herring R, Di Bisceglie AM, Pockros PJ, Subramanian GM, An D, Svarovskaia E, Hyland RH, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Pound D, Fried MW. 2014. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med 370:1879–1888. doi: 10.1056/NEJMoa1402355. [DOI] [PubMed] [Google Scholar]
- 39.Zeuzem S, Dusheiko GM, Salupere R, Mangia A, Flisiak R, Hyland RH, Illeperuma A, Svarovskaia E, Brainard DM, Symonds WT, Subramanian GM, McHutchison JG, Weiland O, Reesink HW, Ferenci P, Hezode C, Esteban R. 2014. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med 370:1993–2001. doi: 10.1056/NEJMoa1316145. [DOI] [PubMed] [Google Scholar]
- 40.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM, Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S, Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG, Sheikh AM, Younossi Z, Gane EJ. 2013. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 368:1878–1887. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 41.Stross C, Shimakami T, Haselow K, Ahmad MQ, Zeuzem S, Lange CM, Welsch C. 2016. Natural HCV variants with increased replicative fitness due to NS3 helicase mutations in the C-terminal helix α18. Sci Rep 6:19526. doi: 10.1038/srep19526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seifert D, Di Giallonardo F, Metzner KJ, Gunthard HF, Beerenwinkel N. 2015. A framework for inferring fitness landscapes of patient-derived viruses using quasispecies theory. Genetics 199:191–203. doi: 10.1534/genetics.114.172312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seifert D, Beerenwinkel N. 2016. Estimating fitness of viral quasispecies from next-generation sequencing data. Curr Top Microbiol Immunol 392:181–200. doi: 10.1007/82_2015_462. [DOI] [PubMed] [Google Scholar]
- 44.Farci P, Purcell RH. 2000. Clinical significance of hepatitis C virus genotypes and quasispecies. Semin Liver Dis 20:103–126. [PubMed] [Google Scholar]
- 45.Farci P. 2011. New insights into the HCV quasispecies and compartmentalization. Semin Liver Dis 31:356–374. doi: 10.1055/s-0031-1297925. [DOI] [PubMed] [Google Scholar]
- 46.Sullivan JC, De Meyer S, Bartels DJ, Dierynck I, Zhang EZ, Spanks J, Tigges AM, Ghys A, Dorrian J, Adda N, Martin EC, Beumont M, Jacobson IM, Sherman KE, Zeuzem S, Picchio G, Kieffer TL. 2013. Evolution of treatment-emergent resistant variants in telaprevir phase 3 clinical trials. Clin Infect Dis 57:221–229. doi: 10.1093/cid/cit226. [DOI] [PubMed] [Google Scholar]
- 47.Perales C, Agudo R, Manrubia SC, Domingo E. 2011. Influence of mutagenesis and viral load on the sustained low-level replication of an RNA virus. J Mol Biol 407:60–78. doi: 10.1016/j.jmb.2011.01.026. [DOI] [PubMed] [Google Scholar]
- 48.Sato M, Maekawa S, Komatsu N, Tatsumi A, Miura M, Muraoka M, Suzuki Y, Amemiya F, Takano S, Fukasawa M, Nakayama Y, Yamaguchi T, Uetake T, Inoue T, Sato T, Sakamoto M, Yamashita A, Moriishi K, Enomoto N. 2015. Deep sequencing and phylogenetic analysis of variants resistant to interferon-based protease inhibitor therapy in chronic hepatitis induced by genotype 1b hepatitis C virus. J Virol 89:6105–6116. doi: 10.1128/JVI.03127-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foster GR, Pianko S, Brown A, Forton D, Nahass RG, George J, Barnes E, Brainard DM, Massetto B, Lin M, Han B, McHutchison JG, Subramanian GM, Cooper C, Agarwal K. 2015. Efficacy of sofosbuvir plus ribavirin with or without peginterferon-alfa in patients with HCV genotype 3 infection and treatment-experienced patients with cirrhosis and HCV genotype 2 Infection. Gastroenterology 149:1462–1470. doi: 10.1053/j.gastro.2015.07.043. [DOI] [PubMed] [Google Scholar]
- 50.Schneider MD, Sarrazin C. 2014. Antiviral therapy of hepatitis C in 2014: do we need resistance testing? Antiviral Res 105:64–71. doi: 10.1016/j.antiviral.2014.02.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.