

Abstract
A screen for agents that potentiated the activity of paromomycin (PAR), a 4,5-linked aminoglycoside (AG), against wild-type Pseudomonas aeruginosa identified the RNA polymerase inhibitor rifampin (RIF). RIF potentiated additional 4,5-linked AGs, such as neomycin and ribostamycin, but not the clinically important 4,6-linked AGs amikacin and gentamicin. Potentiation was absent in a mutant lacking the AmgRS envelope stress response two-component system (TCS), which protects the organism from AG-generated membrane-damaging aberrant polypeptides and, thus, promotes AG resistance, an indication that RIF was acting via this TCS in potentiating 4,5-linked AG activity. Potentiation was also absent in a RIF-resistant RNA polymerase mutant, consistent with its potentiation of AG activity being dependent on RNA polymerase perturbation. PAR-inducible expression of the AmgRS-dependent genes htpX and yccA was reduced by RIF, suggesting that AG activation of this TCS was compromised by this agent. Still, RIF did not compromise the membrane-protective activity of AmgRS, an indication that it impacted some other function of this TCS. RIF potentiated the activities of 4,5-linked AGs against several AG-resistant clinical isolates, in two cases also potentiating the activity of the 4,6-linked AGs. These cases were, in one instance, explained by an observed AmgRS-dependent expression of the MexXY multidrug efflux system, which accommodates a range of AGs, with RIF targeting of AmgRS undermining mexXY expression and its promotion of resistance to 4,5- and 4,6-linked AGs. Given this link between AmgRS, MexXY expression, and pan-AG resistance in P. aeruginosa, RIF might be a useful adjuvant in the AG treatment of P. aeruginosa infections.
INTRODUCTION
Pseudomonas aeruginosa is a common nosocomial pathogen (1) and a major cause of morbidity and mortality in patients with cystic fibrosis (CF) (2, 3). Treatment of P. aeruginosa infections is complicated by the organism's innate resistance to many antimicrobials, a product of its impressive intrinsic resistome (4) and its access to an array of acquired resistance mechanisms (5, 6), with difficult-to-treat multidrug-resistant (MDR) (7) and extremely drug-resistant (8, 9) P. aeruginosa organisms becoming increasingly common. In the face of this intrinsic and acquired multidrug resistance, the use of agents historically used less commonly owing to issues of toxicity (e.g., the polymyxins) (10, 11) and the use of drug combination therapy (12, 13) are increasingly promoted. Still, despite much in vitro evidence for synergistic drug combinations being effective against MDR P. aeruginosa (14–20), the clinical benefits of drug combinations are less obvious (13, 21, 22).
Aminoglycosides (AGs) have a long history in the management of P. aeruginosa infections, particularly in the case of lung infections in patients with cystic fibrosis (23, 24), and are often used in combination with β-lactams (25–27) owing to a well-established synergy between these two antimicrobial classes (18, 20, 26–29). β-Lactam synergy with AGs has been suggested to result from β-lactam-promoted AG uptake owing to cell wall damage or lessening of this barrier. Still, although a β-lactam-promoted increase in the uptake of the AG streptomycin has been seen in various bacteria (30–32), including P. aeruginosa (33), synergy between these agents in the absence of β-lactam-enhanced AG uptake has also been noted (32). Fosfomycin, another cell wall synthesis inhibitor, has also been shown to potentiate AG activity against Gram-negative bacteria, including P. aeruginosa (34, 35). Still, AGs are ototoxic (36) and nephrotoxic (37), which has hitherto limited their use in treating P. aeruginosa infections more generally.
Given the increasing prevalence of multidrug-resistant P. aeruginosa and the paucity of useful antipseudomonal agents, AGs may become increasingly important in managing P. aeruginosa infections (38). To possibly limit issues with toxicity, these agents can, perhaps, more routinely be partnered with compounds that potentiate their activity and thus enable the use of lower doses of AGs, which should be less toxic (39, 40). In addition to the aforementioned antimicrobials, a number of AG potentiators have been described in the literature. AG potentiation as a result of metabolite-promoted (41) and alkaline pH-promoted (42) generation of a proton motive force that drives AG uptake has, for example, been reported for Escherichia coli and P. aeruginosa, respectively. Methylxanthines (a class of bronchodilators that includes caffeine) (43, 44), inhibitors of quorum sensing (45–47), a quorum-sensing signal (diffusible signal factor) from Xanthomonas campestris (48), and green light (49) have all been shown to potentiate AG activity against P. aeruginosa, although the mechanistic details of potentiation are unknown.
In attempting to potentiate AG activity, however, AG resistance mechanisms will be a confounding problem, and therefore, potentiators that target these mechanisms would be of particular interest. AG-modifying enzymes (AMEs) are major determinants of AG resistance in a variety of bacteria, including P. aeruginosa (6, 50), and there are a number of reports describing AME inhibitors (51, 52). Still, there is little, if any, indication that these are effective in enhancing AG susceptibility in intact organisms, particularly AG-resistant strains. AMEs occur infrequently in CF lung isolates (53–55), however, where the AG-exporting (56) MexXY-OprM multidrug efflux system (57, 58) and the AmgRS two-component system (TCS) (59, 60) that responds to and protects P. aeruginosa from the adverse effects of AG-generated membrane-damaging aberrant polypeptides (60, 61) are major determinants of AG resistance (6, 50, 62; K. Poole, H. Fetar, and M. G. Surette, unpublished data). Thus, AG potentiators that target these might be useful adjuvants for antipseudomonal therapy of CF lung infections. In the current study, we identified a compound, the antimicrobial rifampin (RIF), which potentiates AG activity against laboratory and clinical isolates of P. aeruginosa. Moreover, this potentiation is absent in a mutant lacking AmgRS, an indication that RIF somehow targets this TCS and compromises its contribution to AG resistance.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Plasmid pEX18Tc and its derivatives were maintained in E. coli with 5 (in L broth) or 10 (on L agar) μg/ml of tetracycline and selected in P. aeruginosa with 50 μg/ml of tetracycline. A ΔamgR derivative of CF lung isolate K2156 was engineered using the pEX18Tc::ΔamgR plasmid pCG005 (Table 1) and a previously described protocol (60). A RIF-resistant rpoB derivative of P. aeruginosa PAO1 strain K767, K3696, was selected by plating an overnight culture (100 μl) on L agar containing 32 μg/ml of RIF (2× MIC) and picking a colony that grew up overnight at 37°C. The rpoB gene was amplified in two parts from the mutant using Phusion DNA polymerase (New England BioLabs, Ltd., Pickering, ON, Canada) and primer pairs rpoBFor1 (5′-GAGTGGGCAATGCAGGCC-3′) and rpoBRev1 (5′-CTGCTTCGGCGACACGTC-3′) and rpoBFor2 (5′-GAAGGGTCAACTGGTGGACG-3′) and rpoBRev2 (5′-CAAGGCCTTTCCTCCTCACG-3′). Reaction mixtures (50 μl) contained 1 μg of chromosomal P. aeruginosa K767 DNA as the template, 0.5 μM each primer, 0.2 mM each deoxynucleoside triphosphate (dNTP), and 1 U of Phusion DNA polymerase in 1× Phusion GC (part 1) or HF (part 2) buffer. Following an initial denaturation step at 98°C for 30 s, the mixture was subjected to 30 cycles of heating at 98°C for 30 s, 65°C for 30 s, and 72°C for 60 s, before finishing with a 5-min incubation at 72°C. The amplified gene was then sequenced using a variety of custom primers, and a single mutation, yielding a D521Y substitution, was identified in rpoB. D521 mutations are seen in several highly RIF-resistant Pseudomonas spp., including P. aeruginosa, with D521Y seen in RIF-resistant Pseudomonas putida (63).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Descriptiona | Reference |
|---|---|---|
| E. coli strains | ||
| DH5α | ϕ80d lacZΔM15 endA1 recA1 hsdR17 (rK− mK+) supE44 thi-1 gyrA96 relA1 F−Δ(lacZYA-argF) U169 | 84 |
| S17-1 | thi pro hsdR recA Tra+ | 85 |
| P. aeruginosa strains | ||
| K767 | PAO1 prototroph | 86 |
| K3159 | K767 ΔamgR | 60 |
| K1525 | K767 ΔmexXY | 87 |
| K3162 | K767 ΔPA2798 | 60 |
| K3696 | K767 rpoBD521Y (Rifr) | This study |
| K3249 | K767 amgSR182C | 62 |
| K3260 | K767 amgSV121G | 62 |
| K3584 | K3260 ΔamgR | 62 |
| K3585 | K3249 ΔamgR | 62 |
| K3589 | K767 ΔyccA | 61 |
| K3590 | K767 ΔhtpX | 61 |
| K3591 | K767 ΔPA5528 | 61 |
| K3593 | K767 ΔyccA ΔhtpX | 61 |
| K3594 | K767 ΔhtpX ΔPA5528 | 61 |
| K3595 | K767 ΔPA5528 ΔyccA | 61 |
| K3596 | K767 ΔhtpX ΔPA5528 ΔyccA | 61 |
| K2154 | Aminoglycoside-resistant CF isolate | 68 |
| K2156 | Aminoglycoside-resistant CF isolate | 68 |
| K2157 | Aminoglycoside-resistant CF isolate | 68 |
| K2158 | Aminoglycoside-resistant CF isolate | 68 |
| K2162 | Aminoglycoside-resistant CF isolate | 68 |
| K3630 | K2156 ΔamgR | This study |
| K2167 | K2156 ΔmexXY | 68 |
| Plasmids | ||
| pEX18Tc | Broad-host-range gene replacement vector; sacB Tcr | 88 |
| pCG005 | pEX18Tc::ΔamgR | 60 |
RIFr, rifampin resistant; Tcr, tetracycline resistant.
Compound library screen for potentiators of aminoglycoside activity.
Wild-type Pseudomonas aeruginosa PAO1 strain K767 was screened against the Pharmakon-1600 library (Microsource Discovery Systems, Inc.) for agents that rendered the organism sensitive to one-quarter MIC of the AG paromomycin (PAR; 64 μg/ml) according to CLSI guidelines. Duplicate screens were carried out in 100 μl of Mueller-Hinton broth in 96-well microtiter plates, with a Biomek FX liquid handler (Beckman Coulter Inc.) used to dispense both PAR and the library compounds (dissolved in dimethyl sulfoxide [DMSO]; 10 μM final concentration). Following compound addition, K767 was added to all wells and the plates were incubated for 18 h at 37°C. To control for potential inhibitory effects of the compounds on their own, each was tested as a single agent against strain K767, in duplicate. Background controls, containing only broth and DMSO, and growth controls, containing broth, DMSO, and K767 (8 wells/plate each), were also tested. Using an EnVision plate reader (PerkinElmer), the optical density at 600 nm (OD600) was then determined for each well and the percentage growth for each test well (compound + PAR) was calculated as (OD600 for test − mean OD600 for background control)/(mean OD600 for growth control − mean OD600 for background control) × 100. Following normalization to the percent growth determined for a Pharmakon-1600 compound alone, a growth ratio was obtained. A ratio of <1.0 is suggestive of compound potentiation of PAR activity, although a hit cutoff of 0.6 was chosen to identify PAR potentiators.
Antibiotic susceptibility testing.
The susceptibility of P. aeruginosa to antimicrobial agents was assessed using the 2-fold serial microtiter broth dilution method described previously (64), with an inoculum of ∼5 × 105 cells per ml. MICs were recorded as the lowest concentration of antibiotic inhibiting visible growth after 18 h of incubation at 37°C.
qRT-PCR.
RNA was isolated from log phase cells exposed or not to 1× MIC PAR (K767, 256 μg/ml; K2154, 512 μg/ml; added at early log phase, 90 min prior to harvesting of cells for RNA isolation) and converted to cDNA as described previously (65). Expression of mexX (65), htpX (62), armZ (PA5471) (65), and rpoD (62) was assessed using a previously detailed quantitative real-time PCR (qRT-PCR) protocol (65). Expression of yccA was also determined as described previously (65) using the primer pair yccA-F (5′-GTTCTGCGCAATACCTACGG-3′) and yccA-R (5′-GAACACGTTCGGATAGGGC-3′), which had an amplification efficiency of 95% (r2 = 0.994). In some experiments, P. aeruginosa was treated with rifampin (8 μg/ml) for 30 min prior to PAR addition (or prior to RNA isolation in instances where cells were not exposed to PAR).
Membrane depolarization assay.
A previously described fluorometric assay (60), involving the membrane potential-sensitive dye bis-(1,3-dibutylbarbituric acid) trimethine oxonol (DiBAC4 [3]), was employed to measure the degree of cytoplasmic membrane depolarization promoted by AG treatment of P. aeruginosa and the impact of RIF on this. Briefly, early-logarithmic-phase (OD600 = 0.3 to 0.5) L broth subcultures of P. aeruginosa were treated with either of the AGs gentamicin (GEN; final concentration, 2 or 5 μg/ml) and PAR (final concentration, 256 or 640 μg/ml). Samples (5 ml) of the AG-treated and untreated control cultures were taken immediately and then hourly over 3 h and exposed to DiBAC4 (3) (Invitrogen) at 37°C for 5 min in the dark at a final concentration of 10 μg/ml. Bacteria were then pelleted and resuspended in phosphate-buffered saline (66) to a final OD600 of 0.1. Membrane depolarization-dependent fluorescence emitted by cells was then measured using a Varian (now Agilent) Cary Eclipse fluorescent spectrophotometer with excitation and emission wavelengths of 490 and 518 nm, respectively. To assess the impact of chloramphenicol (CAM) or RIF on AG-promoted membrane depolarization, P. aeruginosa was pretreated with CAM (128 μg/ml for 15 min [61]) or RIF (8 μg/ml for 30 min) prior to the addition of the AGs.
RESULTS
Rifampin potentiates AG activity against P. aeruginosa dependent on AmgRS.
A number of chromosomal genes contribute to aminoglycoside (AG) resistance in P. aeruginosa, including the uncharacterized regulatory gene pair PA2797-PA2798 (60), the AmgRS two-component system (TCS) (59, 60) and the MexXY-OprM multidrug efflux system (57, 60, 67), all of which have been shown to contribute to AG resistance in clinical isolates (60, 62, 68, 69). In an effort to identify inhibitors of these AG resistance determinants, and so improve AG activity against P. aeruginosa, a preliminary screen of a limited compound library was undertaken, looking initially for potentiators of AG activity. Paromomycin (PAR) was chosen as a representative AG since it demonstrated the greatest difference in MIC between strains carrying versus lacking the aforementioned AG resistance determinants (60) and, thus, provided a suitably sensitive screen for AG potentiators that targeted these determinants. Using wild-type P. aeruginosa PAO1 strain K767, a 1,600-compound library was screened for agents that rendered K767 susceptible to one-quarter MIC of PAR for this strain but had no intrinsic antimicrobial activity (at the concentrations being screened). A number of putative AG-potentiating compounds were identified, including the rifamycin-related compound rifaximin. We subsequently tested the related rifamycin compound, rifampin (RIF), and demonstrated that it, too, potentiated PAR activity, increasing the PAR susceptibility of strain K767 8-fold (Table 2).
TABLE 2.
AmgRS-dependent rifampin potentiation of paromomycin activity against P. aeruginosaa
| Strain | Relevant genotype | Rifampin | Paromomycin MIC (μg/ml) |
|---|---|---|---|
| K767 | Wild type | − | 256 |
| + | 32 | ||
| K3159 | ΔamgR | − | 32 |
| + | 32 | ||
| K1525 | ΔmexXY | − | 16 |
| + | 8 | ||
| K3162 | ΔPA2798 | − | 16 |
| + | 4 | ||
| K3249 | amgSR182C | − | 512 |
| + | 32 | ||
| K3584 | amgSR182C ΔamgR | − | 32 |
| + | 32 | ||
| K3260 | amgSV121G | − | 512 |
| + | 128 | ||
| K3585 | amgSV121G ΔamgR | − | 32–64 |
| + | 32 | ||
| K3696 | rpoBD521Y (RIFr) | − | 256 |
| + | 256 | ||
| K3590 | ΔhtpX | − | 512 |
| + | 64 | ||
| K3589 | ΔyccA | − | 128–256 |
| + | 64 | ||
| K3591 | ΔPA5528 | − | 128 |
| + | 64 | ||
| K3593 | ΔyccA ΔhtpX | − | 64 |
| + | 16 | ||
| K3594 | ΔhtpX ΔPA5528 | − | 512 |
| + | 32 | ||
| K3595 | ΔPA5528 ΔyccA | − | 64 |
| + | 32 | ||
| K3596 | ΔhtpX ΔPA5528 ΔyccA | − | 8 |
| + | 8–16 |
The paromomycin MICs were determined for the indicated P. aeruginosa strains in the absence (−) and presence (+) of one-half MIC of rifampin (RIF) for each strain (8 μg/ml). While the RIF MIC for the rpoB mutant K3696 was >1,024 μg/ml, RIF was also used at 8 μg/ml for this strain in order to assess whether its potentiation of PAR in wild-type strain K767 was via RpoB or independent of this RIF target. RIFr, RIF resistant.
To ascertain whether RIF acted via one of the above-mentioned AG resistance determinants that are linked to resistance in clinical isolates, deletion strains individually lacking these resistance determinants were assessed for RIF potentiation of PAR activity. The mutants lacking mexXY or PA2797-PA2798, as expected, showed increased susceptibility to PAR, consistent with their known contributions to intrinsic AG resistance (Table 2). Nonetheless, RIF reduced PAR MICs (2- to 4-fold) in these mutants (Table 2), an indication that it was still potentiating PAR activity in the absence of these resistance determinants and, so, not targeting them. In contrast, while the ΔamgR mutant also showed enhanced PAR susceptibility, consistent with its known contribution to intrinsic AG resistance, RIF did not impact the PAR MIC for this strain (Table 2), suggesting that RIF acts via AmgRS in its potentiation of PAR activity. Significantly, the PAR MIC for the ΔamgR mutant was the same as that observed for wild-type strain K767 treated with RIF (Table 2), an indication that RIF was effectively blocking the contribution of this TCS to AG resistance in wild-type P. aeruginosa. RIF also potentiated PAR activity against derivatives of K767 harboring AmgRS-activating amgS gain-of-function mutations and showing elevated AG resistance, in one instance (K3249) rendering the mutant as susceptible as an amgR knockout strain (Table 2), evidence that it was fully reversing the AmgRS-promoted increase in PAR resistance in this mutant. As expected, loss of amgR in the amgS mutants reduced PAR MICs, and RIF failed to potentiate PAR activity in these AmgR− derivatives (Table 2, strains K3584 and K3585), confirmation that RIF was targeting AmgRS in potentiating PAR activity against the mutants. RIF potentiation of PAR was lost in a RIF-resistant P. aeruginosa stain (K3696) harboring a mutation in the rpoB gene that encodes the RNA polymerase β subunit, which is the target of RIF (70) (Table 2), an indication that its PAR-potentiating activity is dependent on its interaction with and disruption of RNA polymerase and not some other possible activity of this compound.
Rifampin specifically potentiates the activity of 4,5-linked aminoglycosides.
Having confirmed AmgRS-dependent RIF potentiation of PAR in P. aeruginosa, it was of interest to assess whether this was, as expected, limited to AGs (AmgRS is linked to AG resistance only). Surprisingly, in screening a number of AGs and non-AGs, RIF showed an ability to potentiate a limited subset of AGs (and no non-AGs) that included only neomycin (NEO) and ribostamycin in addition to PAR (Table 3), while failing to potentiate the more traditional antipseudomonal AGs such as gentamicin and amikacin (Table 3). Intriguingly, PAR, NEO, and ribostamycin are examples of 4,5-linked AGs whose structures differ substantially from those of the other AGs listed in Table 3, which are all 4,6-linked AGs (except streptomycin, which has its own unique structure) (71). These results suggest that the 4,5-linked AGs have some unique effects on P. aeruginosa relative to the 4,6-linked AGs and that the mechanism of AmgRS protection against the effects of the 4,5-linked AGs is unique and specifically targeted by RIF.
TABLE 3.
Rifampin potentiates the activity of 4,5-linked aminoglycosides against wild-type P. aeruginosaa
| Antimicrobial | MIC (μg/ml) |
|
|---|---|---|
| −Rifampin | +Rifampin | |
| Paromomycin | 256 | 32 |
| Neomycin | 32 | 4 |
| Ribostamycin | 1,024 | 256 |
| Gentamicin | 2 | 2 |
| Amikacin | 2 | 2 |
| Kanamycin | 64 | 64 |
| Streptomycin | 32 | 32 |
| Erythromycin | 512 | 512 |
| Chloramphenicol | 32 | 16–32 |
| Nalidixic acid | 128 | 128 |
| Carbenicillin | 64 | 32–64 |
The MICs for the indicated antimicrobials were determined for wild-type P. aeruginosa PAO1 strain K767 in the absence (−) and presence (+) of one-half MIC of rifampin (8 μg/ml).
Rifampin targeting of AmgRS does not impact the membrane-protective role of this TCS.
AmgRS was previously shown to protect P. aeruginosa from membrane damage resultant from exposure to AGs and the resultant production of membrane-damaging aberrant polypeptides (61). Thus, one possibility to explain the RIF potentiation of AG activity via AmgRS was that it compromised this TCS's membrane protective activities but only for the 4,5-linked subset of AGs. The earlier studies showing AmgRS-mediated protection against AGs examined the 4,6-linked AGs gentamicin and tobramycin only. Thus, to assess whether 4,5-linked AGs such as PAR also promote membrane damage and whether this is ameliorated by AmgRS and exacerbated by RIF, PAR-promoted membrane damage was first assessed using a membrane depolarization assay. As seen in Fig. 1A, exposure of wild-type P. aeruginosa K767 to PAR at 1× MIC resulted in a time-dependent increase in membrane damage, as was seen previously with gentamicin- and tobramycin-treated cells (61). As reported previously for gentamicin-exposed cells (61), too, PAR-promoted membrane damage was abrogated when P. aeruginosa was first treated with CAM to block translation and, so, production of membrane-perturbing aberrant mistranslated polypeptides (Fig. 1A), consistent with PAR treatment ultimately generating these membrane-damaging polypeptides. Strikingly, however, loss of amgR in mutant strain K3159 did not increase PAR-promoted membrane damage (Fig. 1B), in contrast to previous results with GEN, where GEN-promoted membrane damage was enhanced in the absence of this TCS (61). For the 4,5-linked AG, PAR, then, AmgRS-mediated protection is not manifest via a membrane-protective mechanism, in contrast with the 4,6-linked AGs. Not surprisingly, RIF treatment did not enhance PAR-promoted membrane damage and, indeed, was seen to reduce PAR-promoted membrane damage (Fig. 1C). A similar result was seen for the 4,6-linked GEN (Fig. 1C). This reduction in AG-promoted membrane damage likely reflects the noted connection between transcription and translation and the potential, therefore, for perturbation of the former to compromise the latter. Indeed, RIF has been shown to limit protein as well as RNA synthesis (72). Therefore, its ability to reduce AG-promoted membrane damage may simply reflect a RIF-driven reduction in the synthesis of membrane-damaging AG-generated mistranslation products. In any case, it is apparent that AmgRS controls some additional AG-protective activity independent of membrane damage and that this is related to some unique effect(s) of the 4,5-linked AGs.
FIG 1.

Impact of chloramphenicol and rifampin on paromomycin-promoted cytoplasmic membrane depolarization in wild-type P. aeruginosa. Cytoplasmic membrane depolarization, as assessed by DiBAC (4) fluorescence, was measured over time following exposure of P. aeruginosa to various antimicrobials added at 0 h. (A) Wild-type P. aeruginosa K767 exposed to 1× MIC of paromomycin (PAR; 256 μg/ml) in the absence or presence of 128 μg/ml of chloramphenicol (CAM). Untreated K767 was included as a control. (B) P. aeruginosa K767 and its amgR deletion derivative exposed to 1× MIC of paromomycin (PAR; 256 μg/ml). Untreated controls are also shown. (C) P. aeruginosa K767 exposed to 1× MIC of paromomycin (PAR; 256 μg/ml) or 1× MIC of gentamicin (GEN; 2 μg/ml) in the absence or presence of 8 μg/ml of rifampin (RIF). Untreated K767 was included as a control. The data are means ± standard errors of the means (SEMs) from 3 independent experiments.
Role of AmgRS target genes in rifampin potentiation of AG activity.
A possible explanation for RIF's potentiation of 4,5-linked AGs is that it compromises AmgRS activity or its activation by these AGs and, ultimately, expression of an AmgRS-dependent 4,5-linked AG resistance mechanism. A number of genes, including htpX, whose homologue in E. coli encodes a cytoplasmic membrane-associated protease (72), are AG inducible, dependent on AmgRS (59, 61). Therefore, htpX expression is a good measure of AmgRS activation or activity. Consistent with previous results, exposure of K767 to PAR increased htpX expression (3-fold) Fig. 2A). Strikingly, this was largely absent in K767 treated with RIF (Fig. 2A), an indication that RIF was interfering with PAR-mediated AmgRS activation. To ensure that this was not a general effect of this RNA polymerase inhibitor, which might be expected to reduce global gene expression, the impact of RIF on expression of another AG-inducible but AmgRS-independent gene, armZ (61, 73), was also assessed. As seen in Fig. 2E, armZ was strongly PAR inducible, and this was unaffected by RIF treatment. Thus, RIF was specifically compromising the PAR-induced expression of the AmgRS-regulated gene htpX. In light of this result, it was possible that the loss of htpX expression was responsible for the increased PAR and NEO susceptibility of RIF-treated cells and that this AmgRS target gene was, thus, specifically responsible for AmgRS-promoted resistance to this AG subset. Still, loss of htpX in strain K3590 did not render P. aeruginosa as susceptible to PAR as an amgR knockout, and RIF still showed potentiation of PAR activity in strain K3590 (Table 2), an indication that RIF was not acting solely on this AmgRS target in potentiating PAR activity. A previous study (59) identified 3 AmgRS-dependent genes as the major contributors to AmgRS-promoted AG resistance, htpX, PA5528, and yccA, and it may be that collectively, these are responsible for the AmgRS-dependent 4,5-linked AG resistance that is compromised by RIF. The yccA homologue in E. coli encodes a modulator of the FtsH protease that is implicated in membrane protein quality control (74), while PA5528 encodes a predicted cytoplasmic membrane-associated protein of unknown function. A mutant stain lacking these three genes, K3596, showed the expected substantial increase in PAR susceptibility, and RIF had no additional effect on PAR susceptibility in this mutant (Table 2). Mutant strains individually lacking yccA (K3589) or PA5528 (K3591), like the htpX mutant, still showed RIF potentiation of PAR activity, as did strains lacking any 2 of these (Table 2, strains K3593 to K3595), further support for htpX, yccA, and PA5528 contributing collectively to RIF-targeted AG resistance. In examining the impact of RIF on PAR induction of PA5528 and yccA in strain K767, however, RIF did not compromise PAR-inducible PA5528 expression (Fig. 2B), and while it did not fully block PAR induction of yccA (Fig. 2C), PAR-induced yccA levels in RIF-treated K767 were markedly below that for RIF-untreated K767 (Fig. 2C), an indication that RIF was adversely impacting yccA expression. Interestingly, PAR-inducible expression of mexXY, shown previously to be dependent on htpX, PA5528, and yccA (61), was also somewhat compromised by RIF treatment (Fig. 2D). Taken together, these results are consistent with RIF somehow disrupting AmgRS activation and/or operation.
FIG 2.
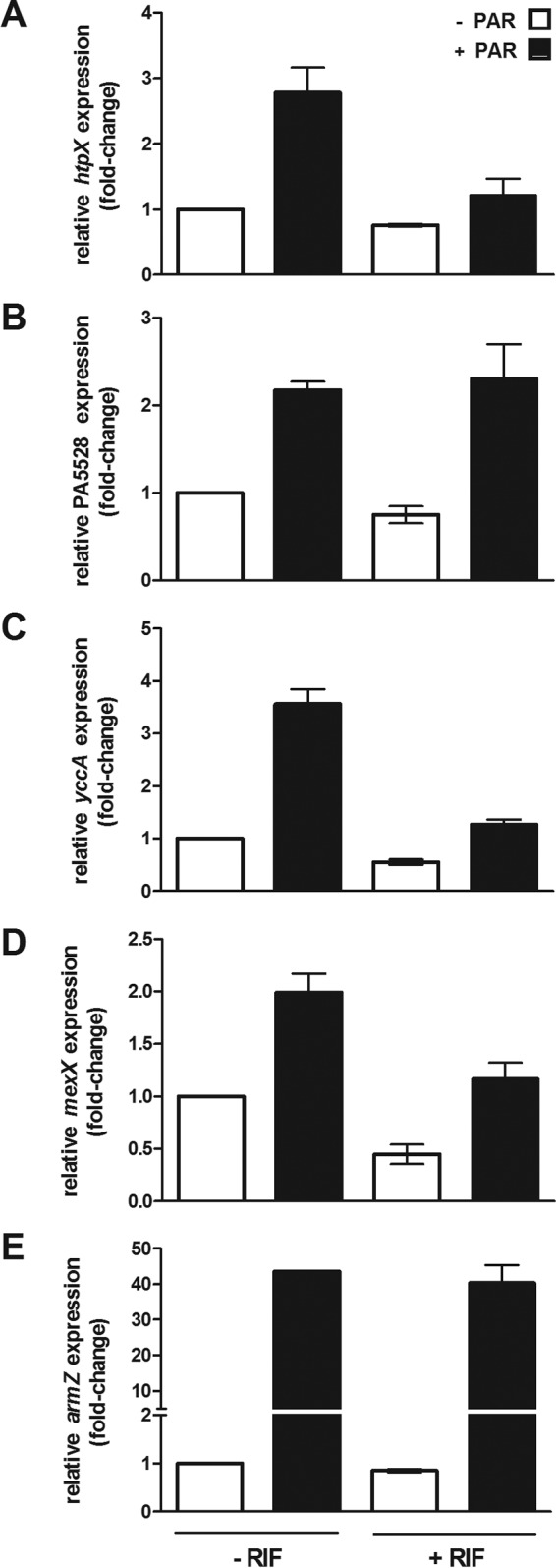
Impact of rifampin on aminoglycoside-promoted expression of AmgRS-regulated genes in wild-type P. aeruginosa. The impact of paromomycin (256 μg/ml) on expression of htpX (A), PA5528 (B), yccA (C), mexXY (D), and armZ (E) was assessed in log-phase cultures of P. aeruginosa strain K767 with or without prior exposure to rifampin (8 μg/ml) using quantitative real-time PCR. Expression was normalized to rpoD and is reported relative to that for the wild-type P. aeruginosa strain K767 (fold change). Values are means ± SEMs from at least three independent determinations, each performed in triplicate.
Pan-aminoglycoside potentiation by rifampin in clinical isolates of P. aeruginosa.
Clinical isolates of P. aeruginosa, particularly CF lung isolates, often exhibit elevated resistance to multiple AGs. It was of interest, therefore, to assess whether RIF might potentiate AG activity against such isolates and whether such potentiation would also be limited to the 4,5-linked AGs. Five previously studied CF isolates showing elevated pan-AG resistance (68) all showed markedly enhanced susceptibility (8- to 16-fold) to the 4,5-linked AGs PAR and NEO in the presence of one-half MIC RIF (Table 4). Unexpectedly, while some of the isolates showed minimal RIF potentiation of a representative 4,6-linked AG, amikacin (2-fold) (Table 4), consistent with what was seen with the wild-type laboratory strain K767, RIF significantly increased amikacin susceptibility (4- to 16-fold) in two isolates, K2154 and K2156 (Table 4). RIF also substantially increased the susceptibility of these isolates to additional 4,6-linked AGs (KAN and GEN; 8-fold) as well as streptomycin (4- to 32-fold) (Table 4). To ascertain whether the ability of RIF to potentiate a broad range of AGs in K2154 and K2156 was still via an action on AmgRS, deletions of amgR were engineered into these strains and the impact of RIF on AG MICs was again determined. Despite repeated attempts, an amgR knockout could not be obtained in K2154, consistent with previous failed attempts to generate deletions in this clinical isolate (68). An amgR deletion was, however, successfully engineered into K2156. The K2156 AmgR− derivative showed the expected increase in susceptibility to 4,5- and 4,6-linked AGs (Table 5), consistent with AmgRS contributing to the broad-range AG resistance of K2156, although RIF did not further influence AG MICs for this strain (Table 5), an indication that RIF potentiation of AGs in K2156 was via AmgRS. Significantly, the impact of the amgR deletion on AG MICs was nominally equivalent to the impact of RIF treatment of K2156, further support for RIF acting via this TCS in promoting enhanced AG susceptibility.
TABLE 4.
Rifampin potentiation of aminoglycoside activity against aminoglycoside-resistant clinical isolates of P. aeruginosaa
| Strain | RIF | MIC (μg/ml) forb,c: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PAR | NEO | AMI | KAN | GEN | STR | CAM | ERY | NAL | CAR | SPC | NOR | ||
| K2154 | − | 512 | 128 | 16 | 256 | 512 | 4,096 | 256 | 256 | 2,048 | >4,096 | >4,096 | 64 |
| + | 64 | 16 | 4 | 64 | 64 | 1,024 | 256 | 256 | 2,048 | >4,096 | 1,024 | 64 | |
| K2156 | − | 512 | 64 | 16 | 128 | 8 | 256 | 128 | 256 | 2,048 | 1,024 | 1,024 | 4 |
| + | 32 | 8 | 1 | 32 | 1 | 8 | 16 | 256 | 2,048 | 512 | 32 | 4 | |
| K2157 | − | 2,048 | 256 | 64 | — | — | — | — | — | — | — | — | — |
| + | 256 | 32 | 32 | — | — | — | — | — | — | — | — | — | |
| K2158 | − | 2,048 | 256 | 64 | — | — | — | — | — | — | — | — | — |
| + | 256 | 32 | 32 | — | — | — | — | — | — | — | — | — | |
| K2162 | − | >4,096 | 512 | 256 | — | — | — | — | — | — | — | — | — |
| + | 2048 | 32 | 128 | — | — | — | — | — | — | — | — | — | |
The MICs for the indicated antimicrobials were determined for the indicated clinical (cystic fibrosis) isolates of P. aeruginosa in the absence (−) and presence (+) of one-half MIC of rifampin (RIF) for each strain (8 μg/ml for strains K2154, K2156, and K2162 and 16 μg/ml for strains K2157 and K2158).
PAR, paromomycin; NEO, neomycin; AMI, amikacin; KAN, kanamycin; GEN, gentamicin; STR, streptomycin; CAM, chloramphenicol; ERY, erythromycin; NAL, nalixidic acid; CAR, carbenicillin; SPC, spectinomycin; NOR, norfloxacin.
—, not determined.
TABLE 5.
AmgRS- and MexXY-dependent rifampin potentiation of aminoglycoside activity against an aminoglycoside-resistant clinical isolate of P. aeruginosaa
| Strain | Genotype | RIF | MIC (μg/ml) forb: |
|||||
|---|---|---|---|---|---|---|---|---|
| PAR | NEO | AMI | KAN | GEN | STR | |||
| K2156 | Wild type | − | 512 | 64 | 16 | 128 | 8 | 256 |
| + | 32 | 8 | 1 | 32 | 1 | 8 | ||
| K3630 | ΔamgR | − | 16 | 8 | 1 | 16 | 1 | 16 |
| + | 16 | 8 | 1 | 16 | 1 | 16 | ||
| K2167 | ΔmexXY | − | 32 | 16 | 1 | 32 | 2 | 4 |
| + | 16 | 8 | 1 | 32 | 1 | 2 | ||
The MICs for the indicated antimicrobials were determined for the clinical (cystic fibrosis) P. aeruginosa isolate K2156 and its AmgR− and MexXY− derivatives in the absence (−) and presence (+) of one-half MIC of rifampin (RIF) for each strain (8 μg/ml).
PAR, paromomycin; NEO, neomycin; AMI, amikacin; KAN, kanamycin; GEN, gentamicin; STR, streptomycin.
Surprisingly, in examination of the AG specificity of RIF's potentiation activity in K2154 and K2156, RIF was shown to also potentiate CAM and spectinomycin (SPC) activity in K2156, though not the other non-AGs that were tested (Table 4). AmgRS is a membrane-protective TCS that contributes to resistance to mistranslation-causing AGs but not CAM and SPC, which do not promote mistranslation. Indeed, the antimicrobials impacted by RIF in K2156 are those typically associated with the MexXY-OprM multidrug efflux system. In light of the recent report that AG-promoted or mutational activation of AmgRS can drive mexXY expression, it may be that AmgRS somehow promotes mexXY expression in K2156, with MexXY-OprM responsible for much of the AG (and CAM and SPC) resistance of this isolate that is, in turn, negatively impacted by RIF. Consequently, RIF interference with AmgRS would compromise mexXY expression and, so, resistance to AGs as well as CAM and SPC. In agreement with this, mexXY expression in strain K2156 was elevated markedly relative to the K767 wild-type laboratory strain and was reduced by RIF exposure K2156 (Fig. 3). RIF potentiation of AGs was largely absent in a mexXY deletion derivative of K2156 (Table 5), further support for RIF ultimately acting on this efflux system in potentiation of AG activity in strain K2156. Notably, with the exception of STR, the impact of the loss of mexXY was nominally the same as that of loss of amgR, consistent with AG resistance in K2156 being mediated by MexXY-OprM but dependent on AmgRS. Consistent with this, the elevated expression of mexXY seen in K2156 was lost upon deletion of amgR (Fig. 3). That RIF treatment had a much more modest negative effect on mexXY expression than the amgR knockout likely reflects the failure of RIF inactivation of AmgRS to eliminate those AmgRS-dependent mexXY transcripts formed prior to RIF treatment. If one assumes that the steady-state mexXY transcript levels reflect a balance between new synthesis and turnover of existing transcripts, RIF treatment of strain K2156 would only limit new synthesis, with mexXY transcript turnover responsible for the observed decline in mexXY expression. In any case, MexXY-OprM-mediated high-level pan-AG resistance in K2156 is clearly linked to AmgRS and, therefore, is inhibited by RIF. Sequencing of the amgRS genes failed to identify any mutations in this TCS operon, however, an indication that other mutations impact mexXY expression in K2156, in a manner that is dependent on AmgRS.
FIG 3.
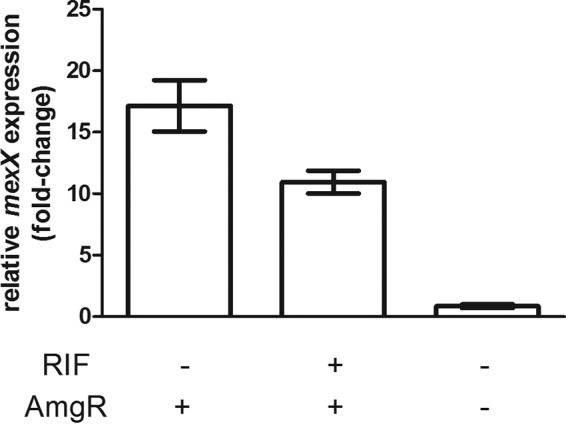
Impact of rifampin and amgR loss on mexXY expression in an aminoglycoside-resistant clinical isolate of P. aeruginosa. mexX expression was measured in log-phase cells of clinical strain K2156 without or with RIF exposure (8 μg/ml for 30 min) and log-phase cells of its ΔamgR derivative, K3630, using quantitative real-time PCR. Expression was normalized to rpoD and is reported relative to that for the wild-type P. aeruginosa strain K767 (fold change). Values are means ± SEMs from at two independent determinations, each performed in triplicate.
DISCUSSION
RIF potentiation of AG activity in P. aeruginosa is dependent on the presence of AmgRS, an indication that it acts on this envelope stress response TCS. In agreement with this, RIF was shown to compromise expression of the AmgRS target genes htpX and yccA. While expression of a third AmgRS target, PA5528, was not influenced by RIF treatment, it may be that this reflected some AmgRS-independent compensatory impact on PA5528 expression masking the possibly inhibitory effect of RIF inhibition on AmgRS and, ultimately, PA5528. Certainly, the impact of an htpX-PA5528-yccA triple deletion on AG susceptibility was greater than that seen for an amgRS knockout, an indication that one or more of htpX, PA5528, and yccA is expressed and contributes to AG resistance independent of AmgRS. Moreover, deletion of htpX in P. aeruginosa has been shown to increase PA5528 expression more than 3-fold (C. H.-F. Lau, unpublished data), a result that is suggestive of PA5528 expression responding to defects in other AmgRS targets. Of note, deletion of PA5528 did not impact htpX expression (Lau, unpublished), an indication that htpX does not respond in a similarly compensatory fashion: i.e., this is possibly unique to PA5528. Thus, perturbation of AmgRS and/or its targets may stimulate a compensatory increase in PA5528 that is AmgRS independent, although the latter has yet to be tested. The observation that mexXY expression was compromised by RIF is also consistent with this agent somehow perturbing the operation of AmgRS and its AG resistance-promoting targets HtpX, PA5528, and YccA; AG-inducible expression of this multidrug efflux operon is dependent on AmgRS, specifically the htpX, PA5528, and yccA targets of this TCS. This effect of RIF on mexXY expression likely explains the more modest effect of RIF on AG MICs in a ΔmexXY strain (2-fold) versus that in the wild type (8-fold), since part of the effect of RIF on AG susceptibility in K767 is likely due to the loss of AmgRS-promoted mexXY expression.
The observation that RIF potentiates only 4,5-linked AGs in wild-type strain K767 is striking and speaks to some unique property of this class of AG. Possibly, 4,5-linked AG perturbation of the ribosome generates some unique (relative to 4,6-linked AGs) cell-damaging products that can both activate AmgRS and be “neutralized” by the products of AmgRS-regulated AG resistance genes such as htpX, PA5528 and yccA. Of note, 4,5-linked AGs are better activators of AmgRS (as determined by the level of induction of htpX and PA5528) (62), suggesting that these unique products are better sensed by and/or are preferred targets of the AmgRS TCS and its AG resistance determinants. It may be, therefore, that RIF acts by somehow limiting the 4,5-linked AG generation of these deleterious products, thereby compromising the activation of AmgRS and the resultant recruitment of its AG resistance determinants. Consequently, resistance to these AGs but not the 4,6-linked AGs would be compromised by RIF. Consistent with the two classes of AGs having different detrimental effects on P. aeruginosa that could be differentially sensed by AmgRS, loss of the TCS increases susceptibility to both 4,5- and 4,6-linked AGs but increases only membrane damage caused by the 4,6-linked AGs. Differences between 4,5- and 4,6-linked AGs have also been noted in terms of their impact on ribosome conformation (75), which might manifest as different downstream effects and, so, different products of ribosome perturbation. Presumably, since a RIF-promoted reduction in AmgRS-activating, 4,5-linked AG-generated deleterious products might be expected to enhance and not reduce AG resistance, 4,5-linked AGs must have additional deleterious effects on the cell that are ameliorated as a result of AmgRS activation. Therefore, RIF limitation of 4,5-linked AG products that activate this TCS would still render cells sensitive to 4,5-linked AGs.
How RIF might limit production of AG-generated AmgRS-activating products is unclear, though it must do so ultimately as a downstream effect of its action on its known target, RNA polymerase, since it failed to potentiate AG activity in a RIF-resistant rpoB mutant. The noted connection between transcription and translation, with the rate of one impacting the rate of the other (76), does suggest, however, that RIF may ultimately impact ribosome function. Indeed, RIF has been shown to reduce protein as well as RNA synthesis (77), and RIF treatment of E. coli is known to alter ribosome structure and composition (78). How this might manifest specifically in fewer products of 4,5-linked AG perturbation of ribosomes is unknown. It is likely, however, that these products act on AmgRS and are substrates for the AmgRS-regulated htpX, yccA, and PA5528 gene products. These gene products are collectively required for AG induction of mexXY expression, a result interpreted as their activities, two of three of which are linked to proteolysis, ultimately generating the mexXY inducing signal(s) (61). The observation that RIF fails to block PAR-inducible PA5528 expression in wild-type P. aeruginosa even as it compromises PAR-inducible mexXY expression argues that PA5528 function must somehow be perturbed by RIF. Since there is no reason to suspect that RIF specifically hampers PA5528 translation or operation, apparent loss of PA5528 function might simply reflect a RIF-promoted lack of 4,5-linked AG-generated substrates on which PA5528 (and HtpX and YccA) must apparently act in yielding the mexXY inducer molecule(s) (61).
Apparent synergy between AGs and RIF has been noted previously (79–81) and in one case was attributed to RIF suppressing what was then known as AG adaptive resistance (81). Still, in these instances synergy was demonstrated with 4,6-linked AGs, in contrast to results shown here. The link to adaptive AG resistance is, however, interesting, inasmuch as we now recognize this to involve the AG-inducible MexXY multidrug efflux system (82), which has been shown to be AmgRS dependent (61) and whose expression is, as we show here, limited to some extent by RIF. It may be that in these earlier studies RIF was, in fact, targeting AmgRS and this was compromising mexXY expression such that resistance to 4,6-linked AGs was affected (resistance to 4,5-linked was not examined). The genetic background and mexXY status of the earlier P. aeruginosa strains are unknown, though all were clinical strains and, so, not wild type. As such, they may have resembled our clinical isolate K2156, in which the AmgRS-dependent upregulation of MexXY was promoting resistance to both classes of AG, with RIF targeting of AmgRS ultimately compromising MexXY-mediated AG resistance. In any case, while the results of the current study suggest that in wild-type laboratory strains, RIF may be of limited utility since it only potentiated lesser-used 4,5-linked AGs, it substantially potentiated the more commonly used 4,6-linked AGs (e.g., amikacin and gentamicin) in some AG-resistant clinical isolates, an indication that it might prove to be of some use in the clinic. Indeed, a randomized trial involving the treatment of P. aeruginosa bacteremias with an AG-penicillin combination with or without RIF showed a significantly increased rate of bacteriologic cure in the arm with RIF and a marked reduction in relapsing bacteremias (83).
ACKNOWLEDGMENTS
This work was supported by operating grants from Cystic Fibrosis Canada to K.P. and to E.D.B. E.D.B. was also supported by a salary award from the Canada Research Chairs program.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control Hosp Epidemiol 29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 2.de Vrankrijker AM, Wolfs TF, van der Ent CK. 2010. Challenging and emerging pathogens in cystic fibrosis. Paediatr Respir Rev 11:246–254. [DOI] [PubMed] [Google Scholar]
- 3.Brugha RE, Davies JC. 2011. Pseudomonas aeruginosa in cystic fibrosis: pathogenesis and new treatments. Br J Hosp Med (Lond) 72:614–619. doi: 10.12968/hmed.2011.72.11.614. [DOI] [PubMed] [Google Scholar]
- 4.Olivares J, Bernardini A, Garcia-Leon G, Corona F, Sanchez MB, Martinez JL. 2013. The intrinsic resistome of bacterial pathogens. Front Microbiol 4:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breidenstein EB, de la Fuente-Nunez C, Hancock RE. 2011. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front Microbiol 2:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliver A, Mulet X, Lopez-Causape C, Juan C. 2015. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist Updat 21-22:41–59. [DOI] [PubMed] [Google Scholar]
- 8.Ciofi Degli Atti M, Bernaschi P, Carletti M, Luzzi I, Garcia-Fernandez A, Bertaina A, Sisto A, Locatelli F, Raponi M. 2014. An outbreak of extremely drug-resistant Pseudomonas aeruginosa in a tertiary care pediatric hospital in Italy. BMC Infect Dis 14:494. doi: 10.1186/1471-2334-14-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buhl M, Peter S, Willmann M. 2015. Prevalence and risk factors associated with colonization and infection of extensively drug-resistant Pseudomonas aeruginosa: a systematic review. Expert Rev Anti Infect Ther 13:1159–1170. doi: 10.1586/14787210.2015.1064310. [DOI] [PubMed] [Google Scholar]
- 10.Dhariwal AK, Tullu MS. 2013. Colistin: re-emergence of the ‘forgotten’ antimicrobial agent. J Postgrad Med 59:208–215. doi: 10.4103/0022-3859.118040. [DOI] [PubMed] [Google Scholar]
- 11.Sánchez A, Gattarello S, Rello J. 2011. New treatment options for infections caused by multiresistant strains of Pseudomonas aeruginosa and other nonfermenting gram-negative bacilli. Semin Respir Crit Care Med 32:151–158. doi: 10.1055/s-0031-1275527. [DOI] [PubMed] [Google Scholar]
- 12.Boyd N, Nailor MD. 2011. Combination antibiotic therapy for empiric and definitive treatment of gram-negative infections: insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy 31:1073–1084. doi: 10.1592/phco.31.11.1073. [DOI] [PubMed] [Google Scholar]
- 13.Tamma PD, Cosgrove SE, Maragakis LL. 2012. Combination therapy for treatment of infections with gram-negative bacteria. Clin Microbiol Rev 25:450–470. doi: 10.1128/CMR.05041-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Souza BB, Padmaraj SR, Rekha PD, Tellis RC, Prabhu S, Pothen P. 2014. In vitro synergistic activity of colistin and ceftazidime or ciprofloxacin against multidrug-resistant clinical strains of Pseudomonas aeruginosa. Microb Drug Resist 20:550–554. doi: 10.1089/mdr.2014.0006. [DOI] [PubMed] [Google Scholar]
- 15.Berditsch M, Jager T, Strempel N, Schwartz T, Overhage J, Ulrich AS. 2015. Synergistic effect of membrane-active peptides polymyxin B and gramicidin S on multidrug-resistant strains and biofilms of Pseudomonas aeruginosa. Antimicrob Agents Chemother 59:5288–5296. doi: 10.1128/AAC.00682-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Safarika A, Galani I, Pistiki A, Giamarellos-Bourboulis EJ. 2015. Time-kill effect of levofloxacin on multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii: synergism with imipenem and colistin. Eur J Clin Microbiol Infect Dis 34:317–323. doi: 10.1007/s10096-014-2231-7. [DOI] [PubMed] [Google Scholar]
- 17.Samonis G, Maraki S, Karageorgopoulos DE, Vouloumanou EK, Falagas ME. 2012. Synergy of fosfomycin with carbapenems, colistin, netilmicin, and tigecycline against multidrug-resistant Klebsiella pneumoniae, Escherichia coli, and Pseudomonas aeruginosa clinical isolates. Eur J Clin Microbiol Infect Dis 31:695–701. doi: 10.1007/s10096-011-1360-5. [DOI] [PubMed] [Google Scholar]
- 18.Dundar D, Otkun M. 2010. In-vitro efficacy of synergistic antibiotic combinations in multidrug resistant Pseudomonas aeruginosa strains. Yonsei Med J 51:111–116. doi: 10.3349/ymj.2010.51.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sader HS, Jones RN. 2005. Comprehensive in vitro evaluation of cefepime combined with aztreonam or ampicillin/sulbactam against multi-drug resistant Pseudomonas aeruginosa and Acinetobacter spp. Int J Antimicrob Agents 25:380–384. doi: 10.1016/j.ijantimicag.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 20.Chen YH, Peng CF, Lu PL, Tsai JJ, Chen TP. 2004. In vitro activities of antibiotic combinations against clincal isolates of Pseudomonas aeruginosa. Kaohsiung J Med Sci 20:261–267. doi: 10.1016/S1607-551X(09)70116-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kmeid JG, Youssef MM, Kanafani ZA, Kanj SS. 2013. Combination therapy for Gram-negative bacteria: what is the evidence? Expert Rev Anti Infect Ther 11:1355–1362. doi: 10.1586/14787210.2013.846215. [DOI] [PubMed] [Google Scholar]
- 22.Traugott KA, Echevarria K, Maxwell P, Green K, Lewis JS II. 2011. Monotherapy or combination therapy? The Pseudomonas aeruginosa conundrum. Pharmacotherapy 31:598–608. [DOI] [PubMed] [Google Scholar]
- 23.Young DC, Zobell JT, Stockmann C, Waters CD, Ampofo K, Sherwin CM, Spigarelli MG. 2013. Optimization of anti-pseudomonal antibiotics for cystic fibrosis pulmonary exacerbations: V. Aminoglycosides. Pediatr Pulmonol 48:1047–1061. doi: 10.1002/ppul.22813. [DOI] [PubMed] [Google Scholar]
- 24.McKeage K. 2013. Tobramycin inhalation powder: a review of its use in the treatment of chronic Pseudomonas aeruginosa infection in patients with cystic fibrosis. Drugs 73:1815–1827. doi: 10.1007/s40265-013-0141-0. [DOI] [PubMed] [Google Scholar]
- 25.Janknegt R. 1990. Aminoglycoside therapy. Current use and future prospects. Pharm Weekbl Sci 12:81–90. [DOI] [PubMed] [Google Scholar]
- 26.Baltch AL, Smith RP. 1985. Combinations of antibiotics against Pseudomonas aeruginosa. Am J Med 79:8–16. [DOI] [PubMed] [Google Scholar]
- 27.Levy J, Baran D, Klastersky J. 1982. Comparative study of the antibacterial activity of amikacin and tobramycin during Pseudomonas pulmonary infection in patients with cystic fibrosis. J Antimicrob Chemother 10:227–234. [DOI] [PubMed] [Google Scholar]
- 28.Piccoli L, Guerrini M, Felici A, Marchetti F. 2005. In vitro and in vivo synergy of levofloxacin or amikacin both in combination with ceftazidime against clinical isolates of Pseudomonas aeruginosa. J Chemother 17:355–360. doi: 10.1179/joc.2005.17.4.355. [DOI] [PubMed] [Google Scholar]
- 29.Kresken M, Korber-Irrgang B, Lauffer J, Decker-Burgard S, Davies T. 2011. In vitro activities of ceftobiprole combined with amikacin or levofloxacin against Pseudomonas aeruginosa: evidence of a synergistic effect using time-kill methodology. Int J Antimicrob Agents 38:70–75. doi: 10.1016/j.ijantimicag.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 30.Plotz PH, Davis BD. 1962. Synergism between streptomycin and penicillin: a proposed mechanism. Science 135:1067–1068. doi: 10.1126/science.135.3508.1067. [DOI] [PubMed] [Google Scholar]
- 31.Moellering RC Jr, Weinberg AN. 1971. Studies on antibiotic syngerism against enterococci. II. Effect of various antibiotics on the uptake of 14C-labeled streptomycin by enterococci. J Clin Invest 50:2580–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller MH, el-Sokkary MA, Feinstein SA, Lowy FD. 1986. Penicillin-induced effects on streptomycin uptake and early bactericidal activity differ in viridans group and enterococcal streptococci. Antimicrob Agents Chemother 30:763–768. doi: 10.1128/AAC.30.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller MH, Feinstein SA, Chow RT. 1987. Early effects of β-lactams on aminoglycoside uptake, bactericidal rates, and turbidimetrically measured growth inhibition in Pseudomonas aeruginosa. Antimicrob Agents Chemother 31:108–110. doi: 10.1128/AAC.31.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montgomery AB, Rhomberg PR, Abuan T, Walters KA, Flamm RK. 2014. Potentiation effects of amikacin and fosfomycin against selected amikacin-nonsusceptible Gram-negative respiratory tract pathogens. Antimicrob Agents Chemother 58:3714–3719. doi: 10.1128/AAC.02780-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Díez-Aguilar M, Morosini MI, Tedim AP, Rodriguez I, Aktas Z, Canton R. 2015. Antimicrobial activity of fosfomycin and tobramycin combination against Pseudomonas aeruginosa isolates assessed by time-kill assays and mutant prevention concentrations. Antimicrob Agents Chemother 59:6039–6045. doi: 10.1128/AAC.00822-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warchol ME. 2010. Cellular mechanisms of aminoglycoside ototoxicity. Curr Opin Otolaryngol Head Neck Surg 18:454–458. doi: 10.1097/MOO.0b013e32833e05ec. [DOI] [PubMed] [Google Scholar]
- 37.Wargo KA, Edwards JD. 2014. Aminoglycoside-induced nephrotoxicity. J Pharm Pract 27:573–577. doi: 10.1177/0897190014546836. [DOI] [PubMed] [Google Scholar]
- 38.Durante-Mangoni E, Grammatikos A, Utili R, Falagas ME. 2009. Do we still need the aminoglycosides? Int J Antimicrob Agents 33:201–205. doi: 10.1016/j.ijantimicag.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Sens MA, Hennigar GR, Hazen-Martin DJ, Blackburn JG, Sens DA. 1988. Cultured human proximal tubule cells as a model for aminoglycoside nephrotoxicity. Ann Clin Lab Sci 18:204–214. [PubMed] [Google Scholar]
- 40.Anniko M. 1983. Aspects on the ototoxic potential of netilmicin. Acta Otolaryngol 96:75–89. [DOI] [PubMed] [Google Scholar]
- 41.Allison KR, Brynildsen MP, Collins JJ. 2011. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lebeaux D, Chauhan A, Letoffe S, Fischer F, de Reuse H, Beloin C, Ghigo JM. 2014. pH-mediated potentiation of aminoglycosides kills bacterial persisters and eradicates in vivo biofilms. J Infect Dis 210:1357–1366. doi: 10.1093/infdis/jiu286. [DOI] [PubMed] [Google Scholar]
- 43.Bazzaz BS, Lavaei S, Hosseinzadeh H. 2012. Interaction of methylxanthines and gentamicin against Staphylococcus aureus and Pseudomonas aeruginosa: role of phosphodiesterase inhibition. Acta Microbiol Immunol Hung 59:13–20. doi: 10.1556/AMicr.59.2012.1.2. [DOI] [PubMed] [Google Scholar]
- 44.Charles BG, Rawal BD. 1973. Synergistic effect of methyl-substituted xanthines and neomycin sulphate on Staphylococcus aureus and Pseudomonas aeruginosa in vitro. Lancet i:971–973. [DOI] [PubMed] [Google Scholar]
- 45.Pan J, Bahar AA, Syed H, Ren D. 2012. Reverting antibiotic tolerance of Pseudomonas aeruginosa PAO1 persister cells by (Z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one. PLoS One 7:e45778. doi: 10.1371/journal.pone.0045778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Busetti A, Shaw G, Megaw J, Gorman SP, Maggs CA, Gilmore BF. 2015. Marine-derived quorum-sensing inhibitory activities enhance the antibacterial efficacy of tobramycin against Pseudomonas aeruginosa. Mar Drugs 13:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brackman G, Cos P, Maes L, Nelis HJ, Coenye T. 2011. Quorum sensing inhibitors increase the susceptibility of bacterial biofilms to antibiotics in vitro and in vivo. Antimicrob Agents Chemother 55:2655–2661. doi: 10.1128/AAC.00045-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng Y, Lim A, Lee J, Chen S, An S, Dong YH, Zhang LH. 2014. Diffusible signal factor (DSF) quorum sensing signal and structurally related molecules enhance the antimicrobial efficacy of antibiotics against some bacterial pathogens. BMC Microbiol 14:51. doi: 10.1186/1471-2180-14-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reznick Y, Banin E, Lipovsky A, Lubart R, Polak P, Zalevsky Z. 2013. The synergistic effect of visible light and gentamycin on Pseudomona aeruginosa microorganisms. J Vis Exp 2013(77):e430. doi: 10.3791/4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poole K. 2005. Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 49:479–487. doi: 10.1128/AAC.49.2.479-487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi K, Caldwell SJ, Fong DH, Berghuis AM. 2013. Prospects for circumventing aminoglycoside kinase mediated antibiotic resistance. Front Cell Infect Microbiol 3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Labby KJ, Garneau-Tsodikova S. 2013. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med Chem 5:1285–1309. doi: 10.4155/fmc.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shawar RM, MacLeod DL, Garber RL, Burns JL, Stapp JR, Clausen CR, Tanaka SK. 1999. Activities of tobramycin and six other antibiotics against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob Agents Chemother 43:2877–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henrichfreise B, Wiegand I, Pfister W, Wiedemann B. 2007. Resistance mechanisms of multiresistant Pseudomonas aeruginosa strains from Germany and correlation with hypermutation. Antimicrob Agents Chemother 51:4062–4070. doi: 10.1128/AAC.00148-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Islam S, Oh H, Jalal S, Karpati F, Ciofu O, Hoiby N, Wretlind B. 2009. Chromosomal mechanisms of aminoglycoside resistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Clin Microbiol Infect 15:60–66. doi: 10.1111/j.1469-0691.2008.02097.x. [DOI] [PubMed] [Google Scholar]
- 56.Lau CH, Hughes D, Poole K. 2014. MexY-promoted aminoglycoside resistance in Pseudomonas aeruginosa: involvement of a putative proximal binding pocket in aminoglycoside recognition. mBio 5:e01068. doi: 10.1128/mBio.01068-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aires JR, Kîhler T, Nikaido H, Plesiat P. 1999. Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob Agents Chemother 43:2624–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mine T, Morita Y, Kataoka A, Mitzushima T, Tsuchiya T. 1999. Expression in Escherichia coli of a new multidrug efflux pump, MexXY, from Pseudomonas aeruginosa. Antimicrob Agents Chemother 43:415–417. doi: 10.1093/jac/43.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee S, Hinz A, Bauerle E, Angermeyer A, Juhaszova K, Kaneko Y, Singh PK, Manoil C. 2009. Targeting a bacterial stress response to enhance antibiotic action. Proc Natl Acad Sci U S A 106:14570–14575. doi: 10.1073/pnas.0903619106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krahn T, Gilmour C, Tilak J, Fraud S, Kerr N, Lau CH, Poole K. 2012. Determinants of intrinsic aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:5591–5602. doi: 10.1128/AAC.01446-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lau CH, Krahn T, Gilmour C, Mullen E, Poole K. 2015. AmgRS-mediated envelope stress-inducible expression of the mexXY multidrug efflux operon of Pseudomonas aeruginosa. Microbiologyopen 4:121–135. doi: 10.1002/mbo3.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lau CH, Fraud S, Jones M, Peterson SN, Poole K. 2013. Mutational activation of the AmgRS two-component system in aminoglycoside-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:2243–2251. doi: 10.1128/AAC.00170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jatsenko T, Tover A, Tegova R, Kivisaar M. 2010. Molecular characterization of Rifr mutations in Pseudomonas aeruginosa and Pseudomonas putida. Mutat Res 683:106–114. doi: 10.1016/j.mrfmmm.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 64.Jo JT, Brinkman FS, Hancock RE. 2003. Aminoglycoside efflux in Pseudomonas aeruginosa: involvement of novel outer membrane proteins. Antimicrob Agents Chemother 47:1101–1111. doi: 10.1128/AAC.47.3.1101-1111.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lau CHF, Fraud S, Jones M, Peterson SN, Poole K. 2012. Reduced expression of the rplU-rpmA ribosomal protein operon in mexXY-expressing pan-aminoglycoside-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:5171–5179. doi: 10.1128/AAC.00846-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nehme D, Li XZ, Elliot R, Poole K. 2004. Assembly of the MexAB-OprM multidrug efflux system of Pseudomonas aeruginosa: identification and characterization of mutations in mexA compromising MexA multimerization and interaction with MexB. J Bacteriol 186:2973–2983. doi: 10.1128/JB.186.10.2973-2983.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Masuda N, Sakagawa E, Ohya S, Gotoh N, Tsujimoto H, Nishino T. 2000. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-OprM efflux pumps in Pseudomonas aeruginosa. Antimicrob Agents Chemother 44:3322–3327. doi: 10.1128/AAC.44.12.3322-3327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sobel ML, McKay GA, Poole K. 2003. Contribution of the MexXY multidrug transporter to aminoglycoside resistance in Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother 47:3202–3207. doi: 10.1128/AAC.47.10.3202-3207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guénard S, Muller C, Monlezun L, Benas P, Broutin I, Jeannot K, Plesiat P. 2014. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother 58:221–228. doi: 10.1128/AAC.01252-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. 2001. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104:901–912. doi: 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- 71.Chittapragada M, Roberts S, Ham YW. 2009. Aminoglycosides: molecular insights on the recognition of RNA and aminoglycoside mimics. Perspect Medicin Chem 3:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sakoh M, Ito K, Akiyama Y. 2005. Proteolytic activity of HtpX, a membrane-bound and stress-controlled protease from Escherichia coli. J Biol Chem 280:33305–33310. doi: 10.1074/jbc.M506180200. [DOI] [PubMed] [Google Scholar]
- 73.Hay T, Fraud S, Lau CH, Gilmour C, Poole K. 2013. Antibiotic inducibility of the mexXY multidrug efflux operon of Pseudomonas aeruginosa: involvement of the MexZ anti-repressor ArmZ. PLoS One 8:e56858. doi: 10.1371/journal.pone.0056858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Stelten J, Silva F, Belin D, Silhavy TJ. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753–756. doi: 10.1126/science.1172221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang L, Pulk A, Wasserman MR, Feldman MB, Altman RB, Cate JH, Blanchard SC. 2012. Allosteric control of the ribosome by small-molecule antibiotics. Nat Struct Mol Biol 19:957–963. doi: 10.1038/nsmb.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Proshkin S, Rahmouni AR, Mironov A, Nudler E. 2010. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science 328:504–508. doi: 10.1126/science.1184939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reid P, Speyer J. 1970. Rifampicin inhibition of ribonucleic acid and protein synthesis in normal and ethylenediaminetetraacetic acid-treated Escherichia coli. J Bacteriol 104:376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blundell MR, Wild DG. 1971. Altered ribosomes after inhibition of Escherichia coli by rifampicin. Biochem J 121:391–398. doi: 10.1042/bj1210391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Valdes JM, Baltch AL, Smith RP, Hammer MC, Ritz WJ. 1990. The effect of rifampicin on the in-vitro activity of cefpirome or ceftazidime in combination with aminoglycosides against Pseudomonas aeruginosa. J Antimicrob Chemother 25:575–584. doi: 10.1093/jac/25.4.575. [DOI] [PubMed] [Google Scholar]
- 80.Barclay ML, Begg EJ, Chambers ST, Peddie BA. 1996. The effect of aminoglycoside-induced adaptive resistance on the antibacterial activity of other antibiotics against Pseudomonas aeruginosa in vitro. J Antimicrob Chemother 38:853–858. doi: 10.1093/jac/38.5.853. [DOI] [PubMed] [Google Scholar]
- 81.Xiong YQ, Caillon J, Drugeon H, Potel G, Baron D. 1996. The effect of rifampicin on adaptive resistance of Pseudomonas aeruginosa to aminoglycosides. J Antimicrob Chemother 37:993–998. doi: 10.1093/jac/37.5.993. [DOI] [PubMed] [Google Scholar]
- 82.Hocquet D, Vogne C, El Garch F, Vejux A, Gotoh N, Lee A, Lomovskaya O, Plesiat P. 2003. MexXY-OprM efflux pump is necessary for adaptive resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob Agents Chemother 47:1371–1375. doi: 10.1128/AAC.47.4.1371-1375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Korvick JA, Peacock JE Jr, Muder RR, Wheeler RR, Yu VL. 1992. Addition of rifampin to combination antibiotic therapy for Pseudomonas aeruginosa bacteremia: prospective trial using the Zelen protocol. Antimicrob Agents Chemother 36:620–625. doi: 10.1128/AAC.36.3.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (ed). 1992. Short protocols in molecular biology, 2nd ed John Wiley & Sons, Inc, New York, NY. [Google Scholar]
- 85.Simon R, Priefer U, Puehler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Biotechnology 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 86.Masuda N, Ohya S. 1992. Cross-resistance to meropenem, cephems, and quinolones in Pseudomonas aeruginosa. Antimicrob Agents Chemother 36:1847–1851. doi: 10.1128/AAC.36.9.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Kievit TR, Parkins MD, Gillis RJ, Srikumar R, Ceri H, Poole K, Iglewski BH, Storey DG. 2001. Multidrug efflux pumps: expression patterns and contribution to antibiotic resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 45:1761–1770. doi: 10.1128/AAC.45.6.1761-1770.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]