

Abstract
A series of new selenocyanates and diselenides bearing interesting bioactive scaffolds (quinoline, quinoxaline, acridine, chromene, furane, isosazole, etc.) was synthesized, and their in vitro leishmanicidal activities against Leishmania infantum amastigotes along with their cytotoxicities in human THP-1 cells were determined. Interestingly, most tested compounds were active in the low micromolar range and led us to identify four lead compounds (1h, 2d, 2e, and 2f) with 50% effective dose (ED50) values ranging from 0.45 to 1.27 μM and selectivity indexes of >25 for all of them, much higher than those observed for the reference drugs. These active derivatives were evaluated against infected macrophages, and in order to gain preliminary knowledge about their possible mechanism of action, the inhibition of trypanothione reductase (TryR) was measured. Among these novel structures, compounds 1h (3,5-dimethyl-4-isoxazolyl selenocyanate) and 2d [3,3′-(diselenodiyldimethanediyl)bis(2-bromothiophene)] exhibited good association between TryR inhibitory activity and antileishmanial potency, pointing to 1h, for its excellent theoretical ADME (absorption, distribution, metabolism, and excretion) properties, as the most promising lead molecule for leishmancidal drug design.
INTRODUCTION
Leishmaniasis is an infectious poverty-associated disease caused by protozoan parasites of the genus Leishmania. In fact, this term includes a wide spectrum of vector-borne diseases with great epidemiological and clinical diversity. Even though exact statistical data are lacking (1, 2), within the 350 million people that live in areas where leishmaniasis is endemic, approximately 12 million people get infected per year. There are three major clinical types: cutaneous (CL), mucocutaneous (MCL), and visceral leishmaniasis (VL; also known as kala-azar), which differ in their immunopathologies and degrees of morbidity and mortality (3). Among the different manifestations, VL is the most severe form, with nearly 200,000 to 400,000 new cases, causing more than 20,000 deaths, per year. Left untreated, it is usually fatal within 2 years.
The efficacy of the different drugs available seems to vary according to the Leishmania species, and the current chemotherapy is far from being satisfactory. Furthermore, they present several problems, including toxicity, many adverse side effects, and high costs. The most relevant problem is related to the fact that many of these drugs were developed many years ago, and currently, there are resistant strains (4).
Since their discovery in the 1940s, the toxic pentavalent antimony [Sb(V)] compounds have been the mainstay of treatment against all forms of leishmaniasis through parenteral administration, and their efficacy is progressively decreasing owing to the development of resistance (5). For this reason, in the last decades several drugs, such as amphotericin B and miltefosine (6), paromomycin and pentamidine (7), sitamaquine (8), and edelfosine (9), have been used in the treatment of leishmaniasis. Nevertheless, their high cost and therapeutic complications limit their use. Nowadays, several other drugs based on natural products show promising antileishmanial activity, but despite significant progress, an ideal drug is still awaited (10).
The development of new antiparasitic drugs has not been much of a priority for the pharmaceutical industry because many of the parasitic diseases occur in poor countries where the populations cannot afford to pay a high price for the drugs. Thus, although important initiatives, such as the Drugs for Neglected Diseases Initiative (DNDi), are attracting more interest in these neglected pathologies, an investment in drug development against parasitic diseases is needed.
The incorporation of different functionalities bearing the Se atom (i.e., methylseleno, selenocyanate, and diselenide) onto organic scaffolds can be considered a promising rational design to achieve potent and selective cytotoxic compounds (11). Several reports have shown vast and miscellaneous types of structures applying this approach, resulting in very promising antitumoral compounds in preclinical models (12, 13). Recently, our research group has been using this rational design in order to obtain new derivatives with potent and selective antileishmanicidal activity. Continuing with these efforts, herein we have designed novel Se compounds which gather two different chemical entities: the selenium entity on its selenocyanate and diselenide forms; and different carbo- and heterocyclic entities with proven leishmanicidal activity. Below in this section, a brief description with several reported data that supports the selection for each of these subunits can be found.
During the last years, various reports have shown recognition of an increase in plasma selenium levels as a new defensive strategy against Leishmania infection (14, 15). The choice of the chemical form for the selenium derivatives can modulate the level of this element on the basis of several metabolic routes (16). The mechanism of action for selenium is unknown, though some enzymatic pathways, such as mitochondrial peroxiredoxins (17), selenophosphate synthetases (18), or ascorbate peroxidases (19), could be implicated. On the other hand, the incorporation of selenium into novel nanomaterials has demonstrated effectiveness in the treatment of leishmaniasis (20). We have reported (21–24) new selenium compounds with potent in vitro antiparasitic activity against Leishmania infantum and Leishmania major, and selectivity indexes higher than those observed for the reference drugs miltefosine, edelfosine, and paromomycin. Additionally, some of them induced nitric oxide production and alterations in gene expression profiling related to proliferation (PCNA), treatment resistance (ABC-transporter and α-tubulin), and virulence (QDPR) (23). Among the various antileishmanial scaffolds containing selenium earlier reported by us, selenocyanate and diselenide showed promising activity against Leishmania parasites (24).
We have payed special attention to quinoline, which constitutes the central nucleus of sitamaquine (25, 26), acridine (27, 28), quinoxaline (29–31), and coumarins (32, 33). On the other hand, nitrofuran compounds (34, 35), the most relevant registered as nifurtimox, and derivatives of the benzodioxol core (36) have been selected. In addition, substituted five-membered heterocyclic rings such as isoxazol (37) and thiophenyl (38) or pirrol (39) have been tested as leishmanicidal agents. Furthermore, related to heterocycles derivatives, some fused aryl azo and triazo molecules have been described (34, 40). Finally, some carbocycles, such as ones with an adamantane ring (41) or anthraquinone structure (42, 43), have been described.
Among the potential molecular targets for the treatment of leihshmaniasis, trypanothione reductase (TryR) is considered an ideal enzyme, since it is involved in the unique thiol-based metabolism observed in the Trypanosomatidae family and is a validated target in the search for drugs against members of this family. TryR catalyzes the reduction of trypanothione disulfide to trypanothione (44). Therefore, during recent years a great number of inhibitors of this key enzyme have been reported (45–47). Based on the chemical analogy of sulfur and selenium, we decided to explore the relevance of this trace element to generate new TryR inhibitors.
In summary, as a continuation of an ongoing program aiming to find new structural leaders with potential leishmanicidal activity, we have constructed a new class of seleno derivatives. They were designed by incorporating selenocyanate or diselenide moieties onto other bioactive carbo- or heterocycles selected on the basis of the above-mentioned findings. In this work, we present the synthesis of 23 new Se compounds (Fig. 1) and their leishmanicidal activities against the amastigote form of L. infantum. In parallel, the cytotoxicities of these newly synthesized molecules were assessed. Moreover, the leishmanicidal activities of the most active compounds were evaluated in L. infantum-infected macrophages. Finally, in order to elucidate a preliminary mechanism of action, their inhibitory activities against trypanothione reductase were determined.
FIG 1.

General structure of new pharmacophoric Se compounds.
MATERIALS AND METHODS
Chemistry.
Melting points (mp) were determined with a Mettler FP82+FP80 apparatus (Greifense, Switzerland) and are not corrected. The 1H nuclear magnetic resonance (NMR) and 13C NMR spectra were recorded on a Bruker 400 Ultrashield spectrometer (Rheinstetten, Germany) using TMS (tetramethylsilane) as the internal standard. The infrared (IR) spectra were obtained on a Thermo Nicolet FT-IR Nexus spectrophotometer with KBr pellets. Mass spectrometry was carried out on an ICP-MS (inductively induced plasma mass spectrometer), Agilent system MSD/DS 5973N (G2577A). Elemental microanalyses were carried out on vacuum-dried samples using a LECO CHN-900 elemental analyzer. Silica gel 60 (0.040 to 0.063 mm) 1.09385.2500 (Merck KGaA, Darmstadt, Germany) was used for column chromatography, and Alugram SIL G/UV254 (layer: 0.2 mm) (Macherey-Nagel GmbH & Co. KG, Düren, Germany) was used for thin-layer chromatography (TLC). Chemicals were purchased from E. Merck (Darmstadt, Germany), Scharlau (F.E.R.O.S.A., Barcelona, Spain), Panreac Química S.A. (Montcada i Reixac, Barcelona, Spain), Sigma-Aldrich Química, S.A. (Alcobendas, Madrid, Spain), Acros Organics (Janssen Pharmaceuticalaan 3a, Geel, Belgium) and Lancaster (Bischheim-Strasbourg, France).
General procedure for the synthesis of compounds 1a to 1o.
The synthesis of compounds 1a to 1o was carried out according to the procedure described in the literature (48–50), with a few modifications. Briefly, KSeCN (4 mmol) was added to a solution of the appropriate halyl derivative (4 mmol) in acetone (50 ml) and the mixture was heated under reflux for 2 to 4 h. The resulting precipitate (KBr) was filtered off. The filtrate was evaporated under vacuum and the residue was treated with water (2 × 50 ml) and dried. The target compounds were obtained with a high degree of purity.
(Quinolin-8-yl)methyl selenocyanate (1a).
Compound 1a was from 8-bromomethylquinoline and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). Brown solid. Yield: 71.5%. mp: 49.5 to 50.5°C. IR (KBr) cm−1: 2,138 (s, C N); 1,593 (f, C=N).1H NMR (400 MHz, DMSO-d6) δ: 4.89 (s, 2H, CH2-Se); 7.58 to 7.63 (m, 2H, H6 + H7); 7.88 (m, 1H, H3); 7.97 (dd, 1H, H5, J5-6 = 8.1 Hz, J5-7 = 1.6 Hz); 8.44 (dd, 1H, H4, J4-3 = 8.4 Hz, J4-2 = 2.2 Hz); 8.94 (dd, 1H, H2, J2-3 = 4.3 Hz, J2-4 = 2.2 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 29.5 (CH2-Se); 106.1 (CN); 123.6 (C3); 127.3 (C7); 129.5 (C5, C6); 131.7 (C8); 136.0 (C9); 138.1 (C4); 146.9 (C2); 151.4 (C10). MS (m/z [percent abundance]): 222 (58); 158 (35); 142 (100); 130 (13); 115 (18); 89 (8); 63 (6). Elemental analysis for C11H8N2Se, calculated/found (percent): C, 53.44/53.35; H, 3.33/3.49; N, 11.33/11.06.
N); 1,593 (f, C=N).1H NMR (400 MHz, DMSO-d6) δ: 4.89 (s, 2H, CH2-Se); 7.58 to 7.63 (m, 2H, H6 + H7); 7.88 (m, 1H, H3); 7.97 (dd, 1H, H5, J5-6 = 8.1 Hz, J5-7 = 1.6 Hz); 8.44 (dd, 1H, H4, J4-3 = 8.4 Hz, J4-2 = 2.2 Hz); 8.94 (dd, 1H, H2, J2-3 = 4.3 Hz, J2-4 = 2.2 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 29.5 (CH2-Se); 106.1 (CN); 123.6 (C3); 127.3 (C7); 129.5 (C5, C6); 131.7 (C8); 136.0 (C9); 138.1 (C4); 146.9 (C2); 151.4 (C10). MS (m/z [percent abundance]): 222 (58); 158 (35); 142 (100); 130 (13); 115 (18); 89 (8); 63 (6). Elemental analysis for C11H8N2Se, calculated/found (percent): C, 53.44/53.35; H, 3.33/3.49; N, 11.33/11.06.
(Quinolin-2-yl)methyl selenocyanate (1b).
Compound 1b was from 2-chloromethylquinoline hydrochloride and potassium selenocyanate. First, quinoline hydrochloride was treated with an aqueous solution of NaOH (1 N) in water-methanol (40:20) for 15 min in order to obtain 2-chloromethylquinoline. The white powder obtained was washed with water (4 × 25 ml) and dried. The compound was washed with ethyl ether (2 × 50 ml). Brown solid. Yield: 27.5%. mp: 83 to 84°C. IR (KBr) cm−1: 2,142 (m, CN); 1,591 (m, C=N). 1H NMR (400 MHz, DMSO-d6) δ: 4.73 (s, 4H, 2CH2); 7.62 (t, 2H, H6 + H7, J6-5 = J7-8 = 11.0 Hz); 7.79 (d, 1H, H3, J3-4 = 9.4 Hz); 7.98 (dd, 2H, H5 + H8, J5-6 = J8-7 = 11.0 Hz, J5-7 = J8-6 = 9.4 Hz); 8.41 (d, 1H, H4, J4-3 = 9.4 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 35.8 (CH2-Se); 105.9 (CN); 122.6 (C3); 127.3 (C5,C7); 129.0 (C6,C9); 131.7 (C8); 138.2 (C4); 147.8 (C10); 158.0 (C2). MS (m/z [percent abundance]): 248 (25); 142 (100); 115 (35); 89 (8); 63 (5); 51 (4). Elemental analysis for C11H8N2Se, calculated/found (percent): C, 53.44/53.52; H, 3.33/3.62; N, 11.33/11.10.
Acridin-9-ylmethyl selenocyanate (1c).
Compound 1c was from 9-(bromomethyl)acridine and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). Yellow solid. Yield: 92.3%. mp: 137 to 138°C. IR (KBr) cm−1: 2,145 (m, CN); 1,625 (d, C=N).1H NMR (400 MHz, DMSO-d6) δ: 5.47 (s, 2H, CH2-Se); 7.69 (t, 2H, H2 + H7, J2-1
= J7-8 = 9.3 Hz); 7.87 (t, 2H, H3 + H6, J3-4
= J6-5 = 9.1 Hz); 8.19 (d, 2H, H4 + H5, J4-3
= J5-6 = 9.1 Hz); 8.58 (d, 2H, H1 + H8, J1-2
= J8-7 = 9.3 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 24.4 (CH2-Se); 104.0 (CN); 124.9 (C12, C14); 125.3 (C1, C11); 127.0 (C2, C10); 130.7 (C4, C8); 132.2 (C3, C9); 142.1 (C13); 149.4 (C5, C7). MS (m/z [percent abundance]): 204 (12); 193 (100); 177 (5); 165 (9); 87 (7); 63 (4). Elemental analysis for C15H10N2Se, calculated/found (percent): C, 60.60/60.74; H, 3.36/3.36; N, 9.42/9.13.
Quinoxalin-2,3-diyldimethanediyl bisselenocyanate (1d).
Compound 1d was from 2,3-bis(bromomethyl)quinoxaline and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). Brown solid. Yield: 46.5%. mp: 153 to 154°C. IR (KBr) cm−1: 2,153 (s, CN); 1,608 (m, C=N). 1H NMR (400 MHz, DMSO-d6) δ: 4.91 (s, 4H, CH2-Se); 7.87 to 7.89 (m, 2H, H6 + H7); 8.06 to 8.08 (m, 2H, H5 + H8). 13C NMR (100 MHz, DMSO-d6) δ: 32.3 (CH2-Se); 105.0 (CN); 129.9 (C6, C9); 132.2 (C7, C8); 141.3 (C5, C10); 151.0 (C2, C3). MS (m/z [abundance]): 262 (100); 235 (44); 156 (72); 129 (27); 102 (21); 76 (20). Elemental analysis for C12H8N4Se2, calculated/found (percent): C, 39.34/39.16; H, 2.18/2.17; N, 15.30/15.06.
(6,7-Dimethoxy-2-oxo-2H-chromen-4-yl)methyl selenocyanate (1e).
Compound 1e was from 6,7-dimethoxy-4-bromomethyl-2H-chromen-2-one and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). Yellow solid. Yield: 28.1%. mp: 197 to 199°C. IR (KBr) cm−1: 2,150 (m, CN); 1,709 (s, C=O). 1H NMR (400 MHz, DMSO-d6) δ: 3.83 (s, 3H, OCH3); 3.87 (s, 3H, OCH3); 4.42 (s, 2H, CH2-Se); 6.33 (s, 1H, CH-CO); 7.10 (s, 1H, H5); 7.43 (s, 1H, H8).13C NMR (100 MHz, DMSO-d6) δ: 29.5 (CH2-Se); 56.3 (OCH3); 58.7 (OCH3); 100.1 (C9); 102.7 (CN); 107.8 (C6); 110.0 (C3); 114.6 (C5); 147.2 (C10); 150.3 (C7); 152.2 (C8); 154.1 (C4); 161.5 (CO). MS (m/z [abundance]): 325 (59); 219 (73); 191 (100); 163 (12); 147 (25); 119 (7); 69 (8). Elemental analysis for C13H11NO4Se, calculated/found (percent): C, 48.15/48.02; H, 3.40/3.40; N, 4.32/4.20.
(5-Nitrofuran-2-yl)methyl selenocyanate (1f).
Compound 1f was from 5-nitro-2-bromomethylfurane and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). Yellow solid. Yield: 42%. mp: 87 to 88°C. IR (KBr) cm−1: 2,152 (m, CN).1H NMR (400 MHz, DMSO-d6) δ: 4.45 (s, 2H, CH2-Se); 6.80 (d, 1H, H3, J3-4
= 7.8 Hz); 7.70 (d, 1H, H4, J4-3
= 7.8 Hz). 13C NMR (100 MHz, DMSO-d6) 23.0 (CH2-Se); 101.1 (CN); 109.4 (C3); 111.2 (C4); 150.0 (C5); 156.3 (C2). MS (m/z [abundance]): 126 (100); 113 (85). Elemental analysis for C6H4N2O3Se, calculated/found (percent): C, 31.17/31.28; H, 1.73/2.02; N, 12.12/11.72.
(6-Bromo-1,3-benzodioxol-5-yl)methyl selenocyanate (1g).
Compound 1g was from 5-bromo-6-(bromomethyl)-1,3-benzodioxole and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). White solid. Yield: 78.6%. mp: 110 to 111°C. IR (KBr) cm−1: 2150 (s, CN); 1033 (s, C-Br).1H NMR (400 MHz, DMSO-d6) δ: 4.36 (s, 2H, CH2-Se); 6.10 (s, 2H, O-CH2-O); 7.11 (s. 1H, H6); 7.27 (s, 1H, H3).13C NMR (100 MHz, DMSO-d6) δ: 34.3 (CH2-Se); 103.5 (O-CH2-O); 105.9 (CN); 111.3 (C6); 113.8 (C4); 115.1 (C3); 130.6 (C5); 148.2 (C2); 149.4 (C1). MS (m/z [abundance]): 213 (100); 157 (7); 75 (19); 50 (15). Elemental analysis for C9H6BrNO2Se, calculated/found (percent): C, 33.86/34.02; H, 1.88/2.05; N, 4.39/4.13.
3,5-Dimethyl-4-isoxazolyl selenocyanate (1h).
Compound 1h was from 4-chloromethyl-3,5-dimethylisoxazole and potassium selenocyanate. The brown oil obtained after washed with water was extracted with dichloromethane (3 × 50 ml). The organic layer was dried with Na2SO4. The dichloromethane was removed under vacuum, and the residue was treated with ethyl ether (3 × 25 ml); a clear brown powder was obtained. Yield: 21.3%. mp: 69 to 70°C. IR (KBr) cm−1: 2,143 (s, CN); 1,628 (s, C=N). 1H NMR (400 MHz, DMSO-d6) δ: 2.23 (s, 3H, CH3-C5); 2.40 (s, 3H, CH3-C3); 4.19 (s, 2H, CH2-Se). 13C NMR (100 MHz, DMSO-d6) δ: 10.6 (C3-CH3); 11.8 (C5-CH3); 21.4 (-CH2-Se-); 105.9 (C4); 112.3 (CN); 159.9 (C3); 168.2 (C5). MS (m/z [abundance]): 156 (4); 110 (100); 68 (89); 52 (5); 43 (45). Elemental analysis for C7H8N2OSe, calculated/found (percent): C, 39.08/39.14; H, 3.75/3.88; N, 13.02/12.89.
(2-Bromothiophene-3-yl)methyl selenocyanate (1i).
Compound 1i was from 2-bromo-3-bromomethylthiophene and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). Brown solid. Yield: 88%. mp: 53 to 55°C. IR (KBr) cm−1: 2,148 (m, CN).1H NMR (400 MHz, DMSO-d6) δ: 4.27 (s, 2H, CH2-Se); 7.09 (d, 1H, H2, J1-2 = 5.7 Hz); 7.63 (d, 1H, H1, J2-1 = 5.7 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 26.5 (CH2); 105.3 (CN); 112.5 (C2); 128.4 (C5); 129.7 (C4); 138.3 (C3). MS (m/z [abundance]): 281 (M+, 3); 175 (100). Elemental analysis for C6H4BrNSSe, calculated/found (percent): C, 25.62/25.44; H, 1.42/1.38; N, 4.98/4.59.
(2-Chlorothiophene-5-yl)methyl selenocyanate (1j).
Compound 1j was from 5-bromo-2-chloro-methylthiophene and potassium selenocyanate. The brown oil obtained after washed with water was extracted with ethyl ether (3 × 50 ml). The organic layer was washed with water (3 × 50 ml) and dried with Na2SO4. The ethyl ether was removed under vacuum, and a brown solid was obtained. Brown solid. Yield: 60%. mp: 37 to 39°C. IR (KBr) cm−1: 2,149 (m, CN). 1H NMR (400 MHz, DMSO-d6) δ: 4.52 (s, 2H, CH2-Se); 6.98 (d, 1H, H3, J3-4
= 7.8 Hz); 7.00 (d, 1H, H4, J4-3 = 7.8 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 28.6 (CH2); 106.5 (CN); 127.5 (C4); 128.4 (C3); 129.3 (C2); 141.4 (C5). MS (m/z [abundance]): 131 (100); 95 (7); 87 (5); 69 (5); 45 (5). Elemental analysis for C6H4ClNSSe, calculated/found (percent): C, 30.44/30.04; H, 1.69/1.82; N, 5.92/5.80.
4-(1H-Pyrrol-1-yl)benzyl selenocyanate (1k).
Compound 1k was from 1-[4-(bromomethyl)phenyl]-1H-pyrrole and potassium selenocyanate. The solid obtained after the treatment with water was solved in THF (tetrahydrofuran), and the insoluble fraction was rejected. The THF was removed under vacuum, and the residue was washed with water (3 × 25 ml) and with hexane (3 × 25 ml). Brown solid. Yield: 73.3%. mp: 154 to 155°C. IR (KBr) cm−1: 2,145 (m, CN). 1H NMR (400 MHz, DMSO-d6) δ: 4.35 (s, 2H, CH2-Se); 6.27 (t, 2H, H3 + H4, J3-2 = J4-5 = 8.5 Hz); 7.38 (t, 2H, H2 + H5, J2-3 = J5-4 = 8.5 Hz); 7.45 (d, 2H, H3′ + H5′, J3′-2′ = J5′-6′ = 8.5 Hz); 7.58 (d, 2H, H2′ + H6′, J2′-3′ = J6′-5′ = 8.5 Hz. 13C NMR (100 MHz, DMSO-d6) δ: 33.1(-CH2-Se-); 105.8 (CN); 111.5 (C3, C4); 119.8 (C2, C5); 120.1 (C2′, C6′); 131.1 (C3′, C5′); 136.1 (C4′); 140.2 (C1′). MS (m/z [abundance]): 262 (M + 1+, 3); 156 (100); 128 (17); 89 (8); 78 (4). Elemental analysis for C12H10N2Se, calculated/found (percent): C, 55.18/55.59; H, 3.86/4.01; N, 10.73/10.07.
1H-Benzotriazol-1-ylmethyl selenocyanate (1l).
Compound 1l was from 1-chloromethyl-1H-benzotriazole and potassium selenocyanate. The orange oil obtained after washed with water was extracted with dichloromethane (3 × 50 ml). The organic phase was washed with water (3 × 20 ml) and dried over Na2SO4. The dichloromethane was removed under vacuum, and the residue was recrystallized from ethanol to give an orange solid. Yield: 19.7%. mp: 158 to 160°C. IR (KBr) cm−1: 2,151 (m, CN). 1H NMR (400 MHz, DMSO-d6) δ: 6.51 (s, 2H, CH2-Se); 7.49 (t, 1H, H2, J2-1 = 7.5 Hz, J2-3 = 7.6 Hz); 7.66 (t, 1H, H3, J3-4 = 7.5 Hz, J3-2 = 7.6 Hz); 8.02 (d, 1H, H1, J1-2 = 7.5 Hz); 8.12 (d, 1H, H4, J4-3 = 7.5 Hz).13C NMR (100 MHz, DMSO-d6) δ: 45.4 (CH2); 105.3 (CN); 112.3 (C8); 120.4 (C5); 125.6 (C6); 128.8 (C7); 132.9 (C9); 146.41 (C4). MS (m/z [abundance]): 132 (62); 77 (100). Elemental analysis for C8H6N4Se, calculated/found (percent): C, 40.51/40.47; H, 2.53/2.75; N, 23.63/23.95.
2-Adamant-1-yl-2-oxoethyl selenocyanate (1m).
Compound 1m was from 1-adamant-1-yl-2-bromoethanone and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). Yellow solid. Yield: 77%. mp: 108 to 110°C. IR (KBr) cm−1: 2,149 (s, CN); 1678 (s, C=O). 1H NMR (400 MHz, DMSO-d6) δ: 1.63 to 1.67 (m, 6H, CH2-CH); 1.80-1.81 (m, 6H, CH2-C-CO); 1.99 (s, 3H, CH); 4.63 (s, 2H, CH2-Se). 13C NMR (100 MHz, DMSO-d6) δ: 27.1 (CH2-Se); 29.0 (CH); 36.7 (CH2-CH); 38.4 (CH2-C-CO); 47.1 (C-CO); 104.5 (CN); 210.2 (C=O). MS (m/z [abundance]): 163 (4); 135 (100); 107 (8); 93 (17); 79 (18); 67 (7). Elemental analysis for C13H17NOSe, calculated/found (percent): C, 55.31/55.38; H, 6.03/6.19; N, 4.96/4.70.
(9,10-Dioxo-9,10-dihydroanthracen-2-yl)methyl selenocyanate (1n).
Compound 1n was from 2-chloromethylanthraquinone and potassium selenocyanate. The compound was washed with ethyl ether (2 × 50 ml). Yellow solid. Yield: 75.3%. mp: 168 to 169°C. IR (KBr) cm−1: 2,145 (m, CN); 1,674 (s, C=O). 1H NMR (400 MHz, DMSO-d6) δ: 4.51 (s, 2H, CH2-Se); 7.90 (dd, 1H, H3, J3-4 = 8.1 Hz, J3-1 = 2.5 Hz); 7.93 to 7.96 (m, 2H, H1 + H4); 8.22-8.25 (m, 4H, H5 + H6 + H7 + H8).13C NMR (100 MHz, DMSO-d6) δ: 33.3 (CH2-Se); 106.5 (CN); 128.5 (C1, C4, C8, C11); 133.2 (C3); 134.1 (C9, C10); 135.0 (C5, C7, C12, C14); 146.9 (C2); 183.4 (CO). MS (m/z [abundance]): 256 (9); 221 (100); 207 (4); 165 (22); 139 (5); 76 (4); 63 (4). Elemental analysis for C16H9NO2Se, calculated/found (percent): C, 58.89/58.74; H, 2.76/2.86; N, 4.29/4.25.
(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl selenocyanate (1o).
Compound 1o was from N-bromomethylphthalimide and potassium selenocyanate. The oil obtained after washed with water was extracted with dichloromethane (3 × 50 ml). The organic phase was washed with water (3 × 20 ml) and dried over Na2SO4. The dichloromethane was removed under vacuum, and the residue was recrystallized from ethanol to give a white-pink solid. Yield: 50.4%. mp: 162 to 164°C. IR (KBr) cm−1: 2,155 (m, CN); 1,778 (CO); 1,718 (CO).1H NMR (400 MHz, DMSO-d6) δ: 5.28 (s, 2H, CH2); 7.89-7.98 (m, 4H, H3 + H4 + H5 + H6).13C NMR (100 MHz, DMSO-d6) δ: 35.0 (CH2); 105.2 (CN); 124.5 (C4); 132.2 (C3); 136.0 (C5); 166.8 (C2). MS (m/z [abundance]): 160 (100); 104 (22). Elemental analysis for C10H6N2O2Se, calculated/found (percent): C: 45.29/44.93; H: 2.26/2.49; N: 10.57/10.37.
General procedure for the synthesis of compounds 2a to 2g.
The appropriate selenocyanate derivative (3 mmol) was solved in absolute ethanol (40 ml), and NaBH4 (6.2 mmol) was added in small portions with caution to the solution. To obtain compound 2g, NaBH3CN (6.2 mmol) was used. The mixture was stirred at room temperature for 2 h. The solvents were removed under vacuum by rotary evaporation, and the residue was treated with water (50 ml) and purified in order to obtain the target compounds.
8,8′-(Diselenodiyldimethanediyl)diquinoline (2a).
Compound 2a was from (quinolin-8-yl)methyl selenocyanate (1a) and sodium borohydride. The resultant solid was washed with ethyl ether (3 × 50 ml) and recrystallized from ethanol to give a yellow solid. Yield: 60.3%. mp: 103 to 104°C. IR (KBr) cm−1: 1591 (s, C=C); 790 (s, Se-Se). 1H NMR (400 MHz, DMSO-d6) δ: 4.58 (s, 4H, 2 CH2-Se); 7.43 to 7.48 (m, 4H, H6 + H7, H6′ + H7′); 7.57 (dd, 2H, H3, H3′, J3-4 = 8.1 Hz, J3-2 = 4.6 Hz); 7.89 (dd, 2H, H5, H5′, J5-6 = 8.1 Hz, J5-7 = 2.5 Hz); 8.38 (dd, 2H, H4, H4′, J4-3 = 8.1 Hz, J4-2 = 2.5 Hz); 8.97 (dd, 2H, H2, H2′, J2-3 = 4.6 Hz, J2-4 = 2.5 Hz).13C NMR (100 MHz, DMSO-d6) δ: 30.2 (CH2-Se); 122.5 (C3, C3′); 127.1 (C7, C7′); 128.0 (C5, C6, C5′, C6′); 130.7 (C8,C8′); 137.9 (C9, C9′); 138.4 (C4, C4′); 146.2 (C2, C2′); 150.1 (C10, C10′). MS (m/z [percent abundance]): 442 (M+·, 5); 222 (75); 142 (100); 130 (10); 115 (17); 89 (7); 63 (5). Elemental analysis for C20H16N2Se2, calculated/found (percent): C, 54.30/54.80; H, 3.62/4.00; N, 6.33/6.10.
9,9′-(Diselenodiyldimethanediyl)diacridine (2b).
Compound 2b was from acridin-9-ylmethyl selenocyanate (1c) and sodium borohydride. The resultant solid was washed with ethyl ether (3 × 50 ml) and recrystallized from ethanol to give an orange solid. Yield: 45%. mp: 108 to 109°C. IR (KBr) cm−1: 1552 (s, C=C); 752 (s, Se-Se). 1H NMR (400 MHz, DMSO-d6) δ: 5.22 (s, 4H, CH2-Se, CH2′-Se); 7.63 (t, 4H, H2 + H7, J2-3 = J7-6 = 9.0 Hz); 7.84 (t, 4H, H3 + H6, J3-2 = J6-7 = 9.0 Hz); 8.14 (d, 4H, H4 + H5, J4-3 = J5-6 = 9.0 Hz); 8.41 (d, 4H, H1 + H8, J1-2 = J8-7 = 9.1 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 26.3 (CH2-Se); 123.1 (C12, C14,C12′, C14′); 125.0 (C1, C11, C1′, C11′); 128.6 (C2, C10, C2′, C10′); 130.0 (C4, C8, C4′, C8′); 133.4 (C3, C9, C3′, C9′); 141.8 (C13, C13′); 149.9 (C5, C7, C5′, C7′). MS (m/z [percent abundance]): 204 (100); 165 (47); 63 (6). Elemental analysis for C28H20N2Se2, calculated/found (percent): C, 61.99/61.62; H, 3.69/4.03; N, 5.16/5.12.
5,5′-(Diselenodiyldimethanediyl)bis(6-bromo-1,3-benzodioxole) (2c).
Compound 2c was from (6-bromo-1,3-benzodioxol-5-yl)methyl selenocyanate (1g) and sodium borohydride. The resultant solid was washed with ethyl ether (3 × 50 ml) and recrystallized from ethanol to give a yellow solid. Yield: 70.3%. mp: 90 to 91°C. IR (KBr) cm−1: 766 (s, Se-Se). 1H NMR (400 MHz, DMSO-d6) δ: 4.03 (s, 4H, CH2-Se, CH2′-Se); 6.00 (s, 4H, O-CH2-O, O-CH2′-O); 6.77 (s, 2H, H6, H6′); 7.01 (s, 2H, H3, H3′). 13C NMR (100 MHz, DMSO-d6) δ: 33.3 (CH2-Se); 103.1 (O-CH2-O); 111.6 (C6,C6′); 113.7 (C4′, C4′); 115.2 (C3, C3′); 132.9 (C5, C5′); 147.0 (C2, C2′); 148.8 (C1, C1′). MS (m/z [percent abundance]): 213 (100); 157 (9); 135 (9); 75 (10); 50 (5). Elemental analysis for C16H12Br2O4Se2, calculated/found (percent): C, 32.76/32.99; H, 2.04/2.02.
3,3′-(Diselenodiyldimethanediyl)bis(2-bromothiophene) (2d).
Compound 2d was from (2-bromothiophene-3-yl)methyl selenocyanate (1i) and sodium borohydride. The mixture was extracted with ethyl ether (3 × 50 ml). The organic phase was washed with water (3 × 50 ml) and dried with anhydrous Na2SO4. The ethyl ether was removed under vacuum, and a yellow powder was obtained. Yield: 29%. mp: 49 to 50°C. IR (KBr) cm−1: 3,099 (w, C-H); 722 (s, Se-Se). 1H NMR (400 MHz, DMSO-d6) δ: 4.04 (s, 4H, CH2-Se, CH2′-Se); 7.00 (d, 2H, H2 + H2′, J1-2 = 5.6 Hz); 7.58 (d, 2H, H1 + H1′, J2-1 = 5.6 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 26.5 (CH2-Se); 111.1 (C2, C2′); 128.4 (C4′, C4′); 130.6 (C5, C5′); 140.0 (C3,C3′). MS (m/z [percent abundance]): 510 (M+., 3); 175 (100); 96 (19); 69 (9); 45 (8). Elemental analysis for C10H8Br2S2Se2, calculated/found (percent): C, 23.55/23.49; H, 1.58/1.52.
1,1′-(Diselenodiyldimethanediyl)bis(1H-benzotriazole) (2e).
Compound 2e was from 1H-benzotriazol-1-ylmethyl selenocyanate (1l) and sodium borohydride. The mixture was extracted with dichloromethane (3 × 50 ml). The organic phase was washed with water (3 × 50 ml) and dried with anhydrous Na2SO4. The dichloromethane was removed under vacuum, and a white powder was obtained. Yield: 25%. mp: 197 to 199°C. IR (KBr) cm−1: 744 (s, Se-Se). 1H NMR (400 MHz, DMSO-d6) δ: 6.19 (s, 4H, N-CH2-Se, N-CH2′-Se); 7.44 (t, 2H, H3 + H3′,J3-2 = 8.2 Hz, J3-4 = 8.2 Hz); 7.57 (t, 2H, H2 + H2′, J2-1 = 8.2 Hz, J2-3 = 8.2 Hz); 7.87 (d, 2H, H4 + H4′, J3-4 = 8.2 Hz); 8.07 (d, 2H, H1 + H1′, J1-2 = 8.2 Hz). 13C NMR (100 MHz, DMSO-d6) δ: 43.6 (CH2-Se); 112.2 (C8, C14,C12′, C14′); 120.2 (C5,); 125.3 (C6); 128.3 (C7); 133.0 (C9); 146 (C4). MS (m/z [percent abundance]): 132 (86); 77 (100). Elemental analysis for C14H12N6Se2·H2O, calculated/found (percent): C, 38.80/38.40; H, 2.77/2.74; N, 19.61/19.39.
2,2′-(Diselenodiyldimethanediyl)di(9,10-dihydroanthracene-9,10-dione) (2f).
Compound 2f was from (9,10-dioxo-9,10-dihydroanthracen-2-yl)methyl selenocyanate (1n) and sodium borohydride. The resultant solid was washed with ethyl ether (3 × 50 ml) and recrystallized from ethanol to give a yellow solid. Yield: 52%. mp: 186 to 187°C. IR (KBr) cm−1: 1,666 (CO); 710 (m, Se-Se). 1H NMR (400 MHz, DMSO-d6) δ: 4.21 (s, 4H, CH2-Se, CH2′-Se); 7.70 (d, 2H, H3, H3′, J3-4 = 8.1 Hz); 7.86-7.89 (m, 4H, H5 + H8, H5′ + H8′); 7.95 (s, 2H, H1, H1′); 8.06 (d, 2H, H4, H4′, J4-3 = 8.1 Hz); 8.09 to 8.11 (m, 4H, H6 + H7, H6′ + H7′). 13C NMR (100 MHz, DMSO-d6) δ: 31.4 (CH2-Se); 127.6 (C1, C4, C8, C11, C1′, C4′, C8′, C11′); 133.2 (C3, C3′); 134.1 (C9, C10,C9′, C10′); 135.3 (C5, C7, C12, C14,C5′, C7′, C12′, C14′); 148.0 (C2,C2′); 183.2 (CO). MS (m/z [percent abundance]): 177 (100); 149 (13); 96 (22); 69 (17); 51 (8). Elemental analysis for C30H18O4Se2·½H2O, calculated/found (percent): C, 59.11/59.12; H, 3.12/3.44.
2,2′-(Diselenodiyldimethanediyl)bis(1H-isoindole-1,3(2H)-dione) (2g).
Compound 2g was from (1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl selenocyanate and sodium cyaneborohydride. A white solid was obtained. Yield: 28%. mp: 162 to 164°C. IR (KBr) cm−1: 1,774 and 1,715 (vs, C=O); 719 (m, Se-Se). 1H NMR (400 MHz, DMSO-d6) δ: 5.07 (s, 4H, N-CH2-Se, N-CH2′-Se); 7.86 (s, 8H, H3 + H3′ + H4 + H4′ + H5 + H5′ + H6 + H6′).13C NMR (100 MHz, DMSO-d6) δ: 34.0 (CH2-Se); 124.2 (C4); 132.3 (C3); 135.6 (C5); 167.3 (CO). MS (m/z [percent abundance]): 478 (M+·, 2); 160 (100). Elemental analysis for C18H12N2O4Se2, calculated/found (percent): C, 45.19/45.25; H, 2.51/2.70; N, 5.86/5.64.
2,2′-(Selenodiyldimethanediyl)bis(1H-isoindole-1,3(2H)-dione) (3).
Compound 3 was from (1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl selenocyanate (1o) (0.75 mmol) and sodium borohydride (0.75 mmol). Yield: 47%. mp: 230 to 232°C. IR (KBr) cm−1: 1,772 and 1,718 (vs, C=O). 1H NMR (400 MHz, DMSO-d6) δ: 5.13 (s, 4H, N-CH2-Se, N-CH2′-Se); 7.87 to 7.89 (m, 8H, H3 + H3′ + H4 + H4′ + H5 + H5′ + H6 + H6′).13C NMR (100 MHz, DMSO-d6) δ: 31.5 (CH2-Se); 124.2 (C4); 132.5 (C3); 135.6 (C5); 167.8 (CO). MS (m/z [percent abundance]): 400 (M + 1+, 2); 160 (100). Elemental analysis for C18H12N2O4Se, calculated/found (percent): C, 54.13/54.13; H, 3.00/2.79; N, 7.02/7.07.
Biological evaluation. (i) Cells and culture conditions.
L. infantum promastigotes (MCAN/ES/89/IPZ229/1/89) were grown in RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% heat-inactivated fetal calf serum (FCS), antibiotics, and 25 mM HEPES (pH 7.2) at 26°C.
L. infantum axenic amastigotes were obtained by incubation of 4.5 × 106 late-logarithmic-phase promastigotes in 5 ml of M199 medium (Invitrogen, Leiden, The Netherlands) supplemented with 10% heat-inactivated FCS, 1 g liter−1 of β-alanine, 100 mg liter−1 of l-asparagine, 200 mg liter−1 of sucrose, 50 mg liter−1 of sodium pyruvate, 320 mg liter−1 of malic acid, 40 mg liter−1 of fumaric acid, 70 mg liter−1 of succinic acid, 200 mg liter−1 of α-ketoglutaric acid, 300 mg liter−1 of citric acid, 1.1 g liter−1 of sodium bicarbonate, 5 g liter−1 of morpholineethanesulfonic acid (MES), 0.4 mg liter−1 of hemin, and 10 mg liter−1 of gentamicin, pH 5.4, at 37°C. After 48 h of incubation, all parasites had a rounded morphology without a flagellum and divided during several weeks under the described conditions.
THP-1 cells were grown in RPMI 1640 medium (Gibco, Leiden, The Netherlands) supplemented with 10% heat-inactivated FCS, antibiotics, 1 mM HEPES, 2 mM glutamine, and 1 mM sodium pyruvate, pH 7.2, at 37°C and 5% CO2.
(ii) Leishmanicidal activity and cytotoxicity assays.
Drug treatment of amastigotes was performed during the logarithmic growth phase at a concentration of 2 × 106 parasites/ml at 26°C or 1 × 106 parasites/ml at 37°C for 24 h, respectively. Drug treatment of THP-1 cells was performed during the logarithmic growth phase at a concentration of 4 × 105 cells/ml at 37°C and 5% CO2 for 24 h. The percentage of living cells was evaluated by flow cytometry by the propidium iodide (PI) exclusion method (51). Drug concentrations ranged from 0.2 μM to 25 μM.
(iii) Leishmania infection assay.
Human THP-1 monocytic cells were seeded at 1.2 × 105 cells/ml in 24-multidish plates (Nunc, Roskilde, Denmark) and differentiated to macrophages for 24 h in 1 ml of RPMI 1640 medium containing 10 ng/ml of phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, St. Louis, MO). Medium culture was removed, and 1.2 × 106 Leishmania amastigotes in 1 ml of THP-1 medium were added to each well. Four hours later, all medium with noninfective amastigotes was removed, washed 3 times with 1× phosphate-buffered saline (PBS), and replaced with new THP-1 medium and corresponding treatment. After 48 h of treatment, medium was removed; THP-1 cells were washed 3 times with 1× PBS and detached with TrypLE Express (Invitrogen, Leiden, The Netherlands) according to the manufacturer's indications. Infection quantification was measured by flow cytometry. Drug concentrations ranged from 0.2 μM to 25 μM.
(iv) Trypanothione reductase assay.
Oxidoreductase activity was determined according to the method described by Toro et al. (52). Briefly, reactions were carried out at 26°C in 250 μl of 40 mM HEPES buffer (pH 8.0) containing 1 mM EDTA, 150 μM NADPH, 30 μM NADP+, 25 μM DTNB [5,5′-dithiobis-(2-nitrobenzoic acid) or Ellman's reagent], 1 μM T[S]2, 0.02% glycerol, 1.5% dimethyl sulfoxide (DMSO), and 7 nM recombinant Li-TryR. Enzyme activity was monitored by the increase in absorbance at 412 nm for 1 h at 26°C in a VERSAmax microplate reader (Molecular Devices, CA). All the assays were conducted in triplicate in at least three independent experiments. Data were analyzed using a nonlineal regression model with Grafit6 software (Erithacus, Horley, Surrey, United Kingdom).
Drug likeness parameters.
The drug likeness and drug score values along with the topological polar surface area (TPSA) values and the properties described in the Lipinski's rule of five (molecular mass ≤ 500 Da, log P ≤ 5, H-bond donors [HBD] ≤ 5, and H-bond acceptors [HBA] ≤ 10) were calculated using Osiris (53) and Molinspiration property calculation programs (54), respectively. Topological polar surface area was used to calculate the percentage of absorption (ABS) according to the equation 109 − (0.345 × TPSA) (55).
RESULTS
Chemistry.
The synthetic approaches adopted to obtain the target compounds are depicted in Fig. 2. The selenocyanate derivatives (compounds 1a to 1o) were obtained in variable yields (27 to 92%) by reaction between the commercially available haloalkyl carbo or heterocyclic reactives with potassium selenocyanate in the molar ratio 1.1 in acetone under reflux over 3 to 4 h (24). The subsequent reduction of compounds 1a to 1o with sodium borohydride or sodium cyanoborohydride in ethanol afforded derivatives 2a to f and 2g (24) in yields ranging from 25 to 70%. Unfortunately, for some selenocyanates (1a, 1d, 1e, 1f, 1 h, 1i, 1k, and 1m) several difficulties were found, and this procedure failed to afford the expected products. This prompted us to seek alternative routes to prepare the corresponding diselenides. Surprisingly, modification of the reaction conditions (temperature, solvents, or molar ratio) resulted in decomposition of the starting materials by rupture of the bond between the heterocycle and methylene. This hypothesis was confirmed by the disappearance of the signal corresponding to the methylene group in 1H NMR. Additionally, an undesired mixture of side compounds was identified in TLC. The alternative strategy employing 100% hydrazine hydrate and sodium hydroxide in DMF (N,N-dimethylformamide) to reduce elemental selenium and generate sodium diselenide followed by reaction with the corresponding haloalkyl reactives (56) did not allow the synthesis of the corresponding diselenides. These results can be explained by the greater steric hindrance so as the quick degradation of the starting materials in the reduction process.
FIG 2.

Synthesis of compounds 1a to 1o, 2a to 2g, and 3.
Finally, and contrary to our expectations, the reduction of 1o with NaBH4 in ethanol yielded compound 3, an unexpected compound instead of the corresponding diselenide that was obtained by reaction with NaBH3CN.
All of the compounds prepared during the course of these investigations are stable. Their purity was assessed by TLC and elemental analyses, and their structures were identified from spectroscopic data. IR, 1H NMR, 13C NMR, mass spectrometry, and elementary analysis methods were used for structure elucidation (Fig. 2).
The IR spectra of compounds 1a to 1o illustrate sharp peaks around 2,138 to 2,155 cm−1 due to a CN group. Derivatives 2a to 2g showed multiple bands in the range of 710 to 790 cm−1 attributable to the Se-Se group and lacked the CN band, confirming the reduction. In NMR all the signals were fully consistent with proposed structures. Copies of the registered 1H and 13C NMR spectra for the selected compounds (1h, 2d, and 2e) can be found in the supplemental material.
Biological evaluation. (i) Activity in amastigotes and cytotoxic activity in human cells.
Compounds 1a to 1o, 2a to 2g, and 3 were tested for their antiprotozoal activity against amastigotes of the pathogenic Leishmania infantum using miltefosine and edelfosine as standard drugs according to a previously described procedure (57). Although most of the studies on the in vitro biological activity of new compounds against Leishmania spp. are performed on promastigote forms, this assay must be considered preliminary because this stage of parasite is significantly more susceptible to drug-induced effects than the amastigote form. Moreover, promastigotes are not the developed forms of the parasite in vertebrate hosts so the evaluations made with them are merely indicative of the potential leishmanicidal activity of the compounds tested. Accordingly, because amastigotes are responsible for all clinical manifestations in humans, the intracellular amastigote model has been cited as the “gold standard” for in vitro Leishmania drug discovery research. Taking this into account, all the analyses were carried out in the amastigote form with a minimum of three independent experiments and the results are expressed as 50% effective dose (ED50) values. In addition, for a compound to be a candidate for antileishmanial drug, it is required to have both high leishmanicidal activity and low cytotoxicity. Cytotoxicity for the THP-1 cell line was evaluated for all compounds in order to identify drugs with low toxicity in human cells and as a prelude to selecting drugs for in vitro assay on the relevant clinical Leishmania amastigote stage. The selectivity index (SI) of the compounds is expressed by the ratio between cytotoxicity (ED50 value on THP-1 cells) and activity (ED50 value on L. infantum amastigotes).
Table 1 shows the ED50 values obtained after 24 h of exposure to the compounds in L. infantum axenic amastigotes. Values for the reference drugs miltefosine and edelfosine are included in all cases for comparison. Biological data evidenced that most of the screened compounds (15 out of 23) showed high bioactivity (ED50 ≤ 2.53 μM) against L. infantum, being more potent than miltefosine (ED50 = 2.84 μM). In addition, under our experimental conditions, seven compounds (1d, 1e, 1h, 1m, 1n, 2e, and 2f) displayed in vitro potency comparable to or higher than that of edelfosine (ED50 = 0.82 μM).
TABLE 1.
ED50 values for compounds on amastigotes and cytotoxic activity in the THP-1 cell line
| Compound | R (substituted group) | ED50 (mean ± SEM), μM |
SIa | |
|---|---|---|---|---|
| Amastigotes | THP-1 | |||
| 1a | Quinol-8-yl | 4.49 ± 0.21 | 14.48 ± 0.37 | 3.2 |
| 1b | Quinol-2-yl | 1.76 ± 0.04 | 14.91 ± 0.92 | 8.5 |
| 1c | Acridin-9-yl | 7.40 ± 0.60 | 15.01 ± 0.68 | 2.0 |
| 1d | Quinoxalin-2,3-diylmethanediyl | 0.69 ± 0.06 | 13.83 ± 1.59 | 20.0 |
| 1e | 6,7-Dimethoxy-2-oxo-2H-chromen-4-yl | 0.82 ± 0.07 | 15.95 ± 1.54 | 19.4 |
| 1f | 5-Nitrofur-2-yl | 1.99 ± 0.23 | 3.95 ± 0.29 | 2.0 |
| 1g | 6-Bromo-1,3-benzodioxol-5-yl | 10.10 ± 1.81 | 15.26 ± 1.19 | 1.5 |
| 1h | 3,5-Dimethylisoxazol-4-yl | 0.73 ± 0.10 | 21.82 ± 2.40 | 29.9 |
| 1i | 2-Bromothien-3-yl | 2.86 ± 0.29 | 19.54 ± 0.52 | 6.8 |
| 1j | 5-Chlorothien-2-yl | 1.85 ± 0.33 | 21.02 ± 0.52 | 11.4 |
| 1k | N-Phenylpyrrol-4-yl | 8.87 ± 1.32 | 23.74 ± 0.48 | 2.7 |
| 1l | Benzotriazol-1-yl | 1.11 ± 0.21 | 22.00 ± 1.20 | 19.8 |
| 1m | 2-Adamant-1-yl-2-oxoethyl | 0.83 ± 0.03 | 19.68 ± 1.98 | 23.7 |
| 1n | 9,10-Dioxo-9,10-dihydroanthracen-2-yl | 0.74 ± 0.18 | 6.05 ± 1.19 | 8.2 |
| 1o | Phthalimidyl | 2.53 ± 0.32 | 22.50 ± 1.63 | 8. 9 |
| 2a | Quinol-8-yl | 2.05 ± 0.24 | 8.60 ± 1.10 | 5.6 |
| 2b | Acridin-9-yl | 5.46 ± 0.01 | 3.76 ± 0.07 | <1 |
| 2c | 6-Bromo-1,3-benzodioxol-5-yl | 3.99 ± 0.62 | >25 | >6.3 |
| 2d | 2-Bromothien-3-yl | 1.20 ± 0.03 | 30.9 ± 0.02 | 25.8 |
| 2e | Benzotriazol-1-yl | 0.45 ± 0.03 | >25 | >55.5 |
| 2f | 9,10-Dioxo-9,10-dihydroanthracen-2-yl | 0.68 ± 0.30 | >25 | 36.8 |
| 2g | Phthalimidyl | 1.35 ± 0.17 | 9.26 ± 0.31 | 6.8 |
| 3 | Phthalimidyl | >25 | >25 | |
| Edelfosine | 0.82 ± 0.13 | 4.96 ± 0.16 | 6.0 | |
| Miltefosine | 2.84 ± 0.10 | 18.50 ± 0.60 | 7.0 | |
Selectivity index (SI) is the ratio of ED50 values of compounds against THP-1 cells relative to their corresponding ED50 against L. infantum amastigotes.
Different authors have claimed that compounds having SI values greater than 20 can be considered ideal candidates for further development as leishmanicidal drugs (58). However, in this study and with rigorous criteria, we have considered the SI threshold of >25 for further analysis of their activity in amastigote-infected THP-1 cells. This requirement is satisfied by compounds 1h, 2d, 2e, and 2f considering them as the lead ones in this series due to their excellent biological behavior.
(ii) Leishmanicidal activity in infected macrophages.
According to their activity and selectivity, four compounds (1h, 2d, 2e, and 2f) were advanced for testing leishmanicidal activity in amastigote-infected THP-1 cells. Compound 2f (ED50 = 0.68 μM; SI = 36.8) was not further tested due to the reproducibility issues showed by this derivative pertaining to its lack of solubility under the assay conditions. The ED50 values for the other selected derivatives (1h, 2d, and 2e) were calculated and summarized in Table 2. The potency of the analogues was compared with that of edelfosine, a current antileishmanial agent (ED50 = 3.1 ± 0.1 μM). These compounds reduced the parasite load of the cells, exhibiting ED50 values of 23.2, 14.0, and 14.4 μM, respectively, with the members of diselenide family being the most potent compounds.
TABLE 2.
ED50 values for compounds in amastigote-infected THP-1 cells
| Compound | ED50 (mean ± SEM), μM |
|---|---|
| 1h | 23.2 ± 4.3 |
| 2d | 14.0 ± 2.1 |
| 2e | 14.4 ± 2.6 |
| Edelfosine | 3.1 ± 0.1 |
(iii) Inhibition of L. infantum trypanothione reductase activity.
In an attempt to investigate a possible mechanism of action, the ability to inhibit the trypanothione reductase activity for the most active compounds was first screened at six different concentrations between 0.1 and 75 μM. Mepacrine, a well-known TryR inhibitor, was used as positive control (59) and DMSO as a vehicle. The 50% inhibitory concentrations (IC50s) obtained are shown in Table 3.
TABLE 3.
IC50s for the selected compounds against TryR inhibition
| Compound | IC50 (mean ± SEM), μM |
|---|---|
| 1h | 0.46 ± 0.01 |
| 2d | 6.85 ± 0.49 |
| 2e | >75 |
| Mepacrine | 16.99 ± 1.18 |
The compounds 1h and 2d were able to inhibit TryR with IC50s of 0.46 and 6.85 μM, respectively. It is remarkable that derivative 1 h was 37-fold more active than the positive control. 1 h and 2d exhibited good association between TryR inhibitory activity and antileishmanial potency (intracellular forms of the parasite). The results for 2e, which did not show inhibitory activity, suggest an alternative mechanism of action for its potent leishmanicidal activity. Compound 1h, as well as compound 2d, can be considered promising antileishmanial lead candidates because they show a strong inhibitory activity against axenic amastigotes (IC50s of 0.73 and 1.27 μM), excellent SI (29.9 and 25.8), and a marked inhibitory activity against TryR.
Drug likeness properties.
Employing the Molinspiration (54) and Osiris (53) software, the selected compounds (1h, 2d, and 2e) were subjected to Lipinski's rule of five analyses (drug likeness), which helps to predict and explain the biological behavior of small molecules. This preliminary analysis allows prediction of the physicochemical properties related to their absorption and bioavailability. We have found that the derivatives 1h and 2e show no violations of Lipinski's rule of five (Table 4). Among leishmanicidal drugs available on the market, only miltefosine does not violate Lipinski's rule of five; all other drugs have at least 1 violation (edelfosine). It has been well established that optimal lipophilicity range along with low log P (<5) and low topological polar surface area (TPSA) are the major driving forces that lead to good absorption, including intestinal absorption, bioavailability, Caco-2 cell permeability, and blood-brain barrier penetration. Molecules with a TPSA of <140 Å2 are indicative of excellent bioavailability (60). According to the theoretical study carried out by de Toledo et al. (61), the TPSA of most leishmanicidal drugs currently on the market is higher than this limit, which probably restricts their absorption and bioavailability. The log P and TPSA values for compounds 1h, 2d, and 2e range from 0.75 to 4.68 and 0 to 61.44, respectively, suggesting that these compounds are potentially able to cross cell membranes in a permeation process, which could explain the ability to reach the amastigotes inside the phagolysosome.
TABLE 4.
Theoretical ADME properties for lead compounds
| Compound | Molinspiration calculationsa |
Osiris calculations |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| MW | miLogP | TPSA | nON | nOHNH | NV | Vol | Drug likeness | Drug score | |
| 1h | 215.1 | 1.2 | 49.8 | 3 | 0 | 0 | 151.7 | −6.8 | 0.5 |
| 2d | 510.0 | 5.8 | 0.00 | 0 | 0 | 2 | 253.2 | −7.5 | 0.1 |
| 2e | 422.2 | 3.6 | 61.4 | 6 | 0 | 0 | 278.1 | −8.0 | 0.1 |
| Edelfosine | 523.7 | 0.7 | 77.1 | 7 | 0 | 1 | 550.9 | −58.2 | 0.3 |
MW, molecular weight; miLogP, octanol-water partition coefficient; TPSA, topological polar surface area; nON, hydrogen bond acceptors; nOHNH, hydrogen bond donors; NV, number of violations; Vol, volume.
The drug score uses and relates other molecular parameters, such as drug likeness, log P, molecular weight, and toxicity risks, and can be considered a convenient value that may be used to judge the overall potential of a compound to become a drug. A value of 0.5 or more is indicative of a promising lead for future development of a safe and efficient drug (62). Compound 1h possess the maximum drug score (0.5) for the selected compounds, whereas edelfosine presented a drug score of 0.3.
DISCUSSION
There are many available antileishmanial agents, but the drug of choice is still awaited because of several limitations of current drugs, such as high cost, poor compliance, drug resistance, low efficacy, and poor safety. The high prevalence and severity of this illness justify the urgency for the discovery of new drugs. In the last 2 decades, several interesting drug targets have been proposed, including many proteins and enzymes that differ from mammalian counterparts which can interfere with the redox system. Among the promising targets that the scientific community considers for the design of useful therapies, enzymes (trypanothione reductase, proteinases, superoxide dismutase, dihydrofolate reductase, metacaspase, topoisomerase, kinases, sirtuins, etc.) are one of the classes with the most representation. In this context, we notice that selenium plays an important role in medicinal chemistry, particularly in antioxidant, antitumoral, chemopreventive, or antiparasitic agents. Thus, we have described here the synthesis and leishmanicidal activity of novel selenocyanate and diselenide compounds.
For the novel seleno derivatives presented in this work, there seems to exist a tendency suggesting that analogues with the diselenide unit were more active than those with the selenocyanate moiety (1a versus 2a, 1c versus 2b, 1g versus 2c, 1i versus 2d, 1l versus 2e, and 1o versus 2g) against L. infantum amastigotes. Regarding the selectivity index, the addition of the diselenide scaffold clearly improved the selectivity, for example, in compounds 1g versus 2c, 1i versus 2d, 1l versus 2e, and 1n versus 2f. In general, it was observed that tricyclic nitrogenated rings, such as acridine (1c and 2b), are detrimental to the biological activity and selectivity compared with bicyclic nitrogenated rings (1b, 1d, 1l, 1o, 2e, and 2g). Furthermore, no regular order of decrease or increase in the activity among the rest derivatives can be concluded.
Taking into account the results related to activity and selectivity and considering our exigent criteria for both parameters (ED50 of <2.5 μM and SI of >25), four derivatives, one selenocyanate, 1h, and two diselenides (2d and 2e) were selected for further studies. Despite the fact that compound 2f fulfilled these criteria, it could not be tested due to solubility problems. When we performed intracellular form tests, these derivatives did not improve the activity compared to that of edelfosine. However, their lack of toxicity against THP-1 cells (Table 1) represents a remarkable advantage over the reference drug.
We hypothesized that TryR inhibition may be related with the leishmanicidal activity observed for seleno derivatives. The results for in vitro assays revealed that compounds 1h and 2d exhibited a good correlation between leishmanicidal activity and TryR inhibition, confirming our previous proposal. On the other hand, 2e did not show inhibitory activity, suggesting that not only is this enzyme involved in its potent leishmanicidal activity but also other mechanisms can be implicated.
Finally, in silico prediction studies were performed in order to determine the drug like properties for the lead compounds. Considering these properties, derivatives 1h and 2e were shown to meet the Lipinski's rule of five, indicating favorable properties for drug development. The in silico toxicity profile, drug likeness, and drug score (0.5) data for compound 1h make it a promising leader for future development of safer and more efficient leishmanicidal drugs.
In conclusion, the present study reports the synthesis of new selenocyanates and diselenides bearing interesting bioactive scaffolds (quinoline, quinoxaline, acridine, chromene, furane, and isosazole) and their in vitro leishmanicidal activities against L. infantum amastigotes along with their cytotoxicities in THP-1 cells. Fifteen such compounds exhibited better potency against axenic amastigotes than the standard drug miltefosine. Based on their antiparasitic activities and low toxicity in THP-1 cells, compounds 1h, 2d, 2e, and 2f were identified as the best candidates for further studies with infected macrophages. Although their potency against intracellular amastigotes is lower than that observed for the reference drug, these compounds combined a potent leishmanicidal activity with excellent selectivity index (>25), resulting in promising therapeutic utility. In order to get further insight into their putative mechanism of action, their activity against L. infantum TryR was determined. A clear correlation between enzyme inhibition and antiparasitic activity was observed for compounds 1h and 2d, which may be considered evidence for one of their many possible mechanisms of action. No correlation was detected for 2e, which suggest the existence of different targets in this family of compounds. The ADME (absorption, distribution, metabolism, and excretion) parameters calculated for derivatives 1h and 2e predict a good bioavailability. In silico ADME profiling and drug score results along with in vitro leishmanicidal activity, cytotoxicity, and TryR inhibitory activity make 1h a promising lead compound for the development of more potent antiparasitary drugs. A graphical summary of the conclusions drawn from this work is depicted in Fig. 3.
FIG 3.
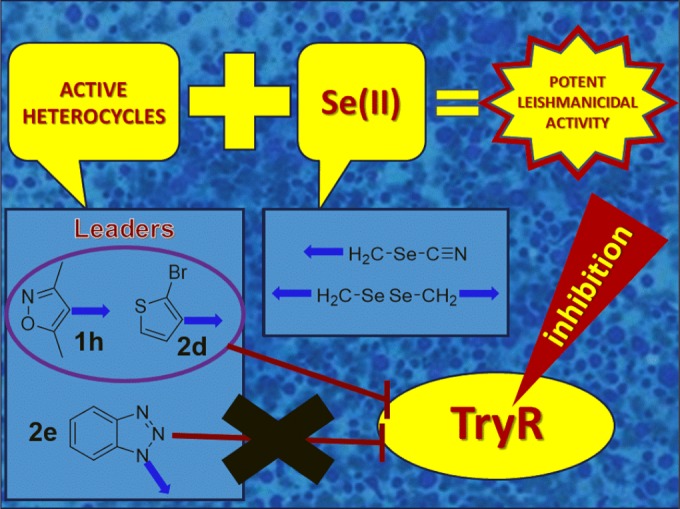
Schematic illustration of conclusions.
Moreover, to the best of our knowledge, this is the first study that reports new selenoderivatives as leishmanicidal and TryR inhibitors and opens new possibilities in the field of neglected diseases.
Supplementary Material
ACKNOWLEDGMENTS
We express our gratitude to the Foundation for Applied Medical Investigation (FIMA), University of Navarra. We also acknowledge financial support from the Ministerio de Educación y Ciencia, Spain (grant SAF2012-39760-C02-02).
We have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in this article apart from those disclosed.
Funding Statement
The authors express their gratitude to the Foundation for Applied Medical Investigation (FIMA), University of Navarra. The authors also acknowledge financial support from the Ministerio de Educación y Ciencia, Spain (grant SAF2012-39760-C02-02). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02529-15.
REFERENCES
- 1.Louzir H, Aoun K, Spath GF, Laouini D, Prina E, Victoir K, Bouratbine A. 2013. Leishmania epidemiology, diagnosis, chemotherapy and vaccination approaches in the international network of Pasteur Institutes. Med Sci (Paris) 29:1151–1160. doi: 10.1051/medsci/20132912020. [DOI] [PubMed] [Google Scholar]
- 2.McGwire BS, Satoskar AR. 2014. Leishmaniasis: clinical syndromes and treatment. QJM 107:7–14. doi: 10.1093/qjmed/hct116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundar S, Chakravarty J. 2013. Leishmaniasis: an update of current pharmacotherapy. Expert Opin Pharmacother 14:53–63. doi: 10.1517/14656566.2013.755515. [DOI] [PubMed] [Google Scholar]
- 4.Yasinzai M, Khan M, Nadhman A, Shahnaz G. 2013. Drug resistance in leishmaniasis: current drug-delivery systems and future perspectives. Future Med Chem 5:1877–1888. doi: 10.4155/fmc.13.143. [DOI] [PubMed] [Google Scholar]
- 5.Fernandes FR, Ferreira WA, Campos MA, Ramos GS, Kato KC, Almeida GG, Correa JDJ, Melo MN, Demicheli C, Frezard F. 2013. Amphiphilic antimony(V) complexes for oral treatment of visceral leishmaniasis. Antimicrob Agents Chemother 57:4229–4236. doi: 10.1128/AAC.00639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solomon M, Pavlotzky F, Barzilai A, Schwartz E. 2013. Liposomal amphotericin B in comparison to sodium stibogluconate for Leishmania braziliensis cutaneous leishmaniasis in travelers. J Am Acad Dermatol 68:284–289. doi: 10.1016/j.jaad.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 7.Seifert K, Munday J, Syeda T, Croft SL. 2011. In vitro interactions between sitamaquine and amphotericin B, sodium stibogluconate, miltefosine, paromomycin and pentamidine against Leishmania donovani. J Antimicrob Chemother 66:850–854. doi: 10.1093/jac/dkq542. [DOI] [PubMed] [Google Scholar]
- 8.Loiseau PM, Cojean S, Schrevel J. 2011. Sitamaquine as a putative antileishmanial drug candidate: from the mechanism of action to the risk of drug resistance. Parasite 18:115–119. doi: 10.1051/parasite/2011182115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varela-M RE, Villa-Pulgarin JA, Yepes E, Muller I, Modolell M, Munoz DL, Robledo SM, Muskus CE, Lopez-Aban J, Muro A, Velez ID, Mollinedo F. 2012. In vitro and in vivo efficacy of ether lipid edelfosine against Leishmania spp. and SbV-resistant parasites. PLoS Negl Trop Dis 6:e1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh N, Mishra BB, Bajpai S, Singh RK, Tiwari VK. 2014. Natural product based leads to fight against leishmaniasis. Bioorg Med Chem 22:18–45. doi: 10.1016/j.bmc.2013.11.048. [DOI] [PubMed] [Google Scholar]
- 11.Arsenyan P, Paegle E, Domracheva I, Gulbe A, Kanepe-Lapsa I, Shestakova I. 2014. Selenium analogues of raloxifene as promising antiproliferative agents in treatment of breast cancer. Eur J Med Chem 87:471–483. doi: 10.1016/j.ejmech.2014.09.088. [DOI] [PubMed] [Google Scholar]
- 12.Fernández-Herrera MA, Sandoval-Ramirez J, Sanchez-Sanchez L, Lopez-Munoz H, Escobar-Sanchez ML. 2014. Probing the selective antitumor activity of 22-oxo-26-selenocyanocholestane derivatives. Eur J Med Chem 74:451–460. doi: 10.1016/j.ejmech.2013.12.059. [DOI] [PubMed] [Google Scholar]
- 13.Gowda R, Madhunapantula SV, Desai D, Amin S, Robertson GP. 2013. Simultaneous targeting of COX-2 and AKT using selenocoxib-1-GSH to inhibit melanoma. Mol Cancer Ther 12:3–15. doi: 10.1158/1535-7163.MCT-12-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Araújo AP, Rocha OG, Mayrink W, Machado-Coelho GL. 2008. The influence of copper, selenium and zinc on the response to the Montenegro skin test in subjects vaccinated against American cutaneous leishmaniasis. Trans R Soc Trop Med Hyg 102:64–69. doi: 10.1016/j.trstmh.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 15.da Silva MT, Silva-Jardim I, Thiemann OH. 2014. Biological implications of selenium and its role in trypanosomiasis treatment. Curr Med Chem 21:1772–1780. doi: 10.2174/0929867320666131119121108. [DOI] [PubMed] [Google Scholar]
- 16.Weekley CM, Harris HH. 2013. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem Soc Rev 42:8870–8894. [DOI] [PubMed] [Google Scholar]
- 17.Castro H, Teixeira F, Romao S, Santos M, Cruz T, Florido M, Appelberg R, Oliveira P, Ferreira-da-Silva F, Tomas AM. 2011. Leishmania mitochondrial peroxiredoxin plays a crucial peroxidase-unrelated role during infection: insight into its novel chaperone activity. PLoS Pathog 7:e1002325. doi: 10.1371/journal.ppat.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faim LM, Rosa e Silva I, Bertacine Dias MV, D'Muniz Pereira H, Brandao-Neto J, Alves da Silva MT, Thiemann OH. 2013. Crystallization and preliminary X-ray diffraction analysis of selenophosphate synthetases from Trypanosoma brucei and Leishmania major. Acta Crystallogr Sect F Struct Biol Cryst Commun 69:864–867. doi: 10.1107/S1744309113014632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pal S, Dolai S, Yadav RK, Adak S. 2010. Ascorbate peroxidase from Leishmania major controls the virulence of infective stage of promastigotes by regulating oxidative stress. PLoS One 5:e11271. doi: 10.1371/journal.pone.0011271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beheshti N, Soflaei S, Shakibaie M, Yazdi MH, Ghaffarifar F, Dalimi A, Shahverdi AR. 2013. Efficacy of biogenic selenium nanoparticles against Leishmania major: in vitro and in vivo studies. J Trace Elem Med Biol 27:203–207. doi: 10.1016/j.jtemb.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Moreno D, Plano D, Baquedano Y, Jimenez-Ruiz A, Palop JA, Sanmartin C. 2011. Antileishmanial activity of imidothiocarbamates and imidoselenocarbamates. Parasitol Res 108:233–239. doi: 10.1007/s00436-010-2073-x. [DOI] [PubMed] [Google Scholar]
- 22.Baquedano Y, Moreno E, Espuelas S, Nguewa P, Font M, Gutierrez KJ, Jimenez-Ruiz A, Palop JA, Sanmartin C. 2014. Novel hybrid selenosulfonamides as potent antileishmanial agents. Eur J Med Chem 74:116–123. doi: 10.1016/j.ejmech.2013.12.030. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez-Rubio C, Campbell D, Vacas A, Ibanez E, Moreno E, Espuelas S, Calvo A, Palop JA, Plano D, Sanmartin C, Nguewa PA. 6 July 2015. Exploring leishmanicidal activities of novel methylseleno imidocarbamates. Antimicrob Agents Chemother doi: 10.1128/aac.00997-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plano D, Baquedano Y, Moreno-Mateos D, Font M, Jimenez-Ruiz A, Palop JA, Sanmartin C. 2011. Selenocyanates and diselenides: a new class of potent antileishmanial agents. Eur J Med Chem 46:3315–3323. doi: 10.1016/j.ejmech.2011.04.054. [DOI] [PubMed] [Google Scholar]
- 25.Gopinath VS, Pinjari J, Dere RT, Verma A, Vishwakarma P, Shivahare R, Moger M, Kumar Goud PS, Ramanathan V, Bose P, Rao MV, Gupta S, Puri SK, Launay D, Martin D. 2013. Design, synthesis and biological evaluation of 2-substituted quinolines as potential antileishmanial agents. Eur J Med Chem 69:527–536. doi: 10.1016/j.ejmech.2013.08.028. [DOI] [PubMed] [Google Scholar]
- 26.Bompart D, Nunez-Duran J, Rodriguez D, Kouznetsov VV, Melendez Gomez CM, Sojo F, Arvelo F, Visbal G, Alvarez A, Serrano-Martin X, Garcia-Marchan Y. 2013. Anti-leishmanial evaluation of C2-aryl quinolines: mechanistic insight on bioenergetics and sterol biosynthetic pathway of Leishmania braziliensis. Bioorg Med Chem 21:4426–4431. doi: 10.1016/j.bmc.2013.04.063. [DOI] [PubMed] [Google Scholar]
- 27.Di Giorgio C, Shimi K, Boyer G, Delmas F, Galy JP. 2007. Synthesis and antileishmanial activity of 6-mono-substituted and 3,6-di-substituted acridines obtained by acylation of proflavine. Eur J Med Chem 42:1277–1284. doi: 10.1016/j.ejmech.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Carole DG, Michel DM, Julien C, Florence D, Anna N, Severine J, Gerard D, Pierre TD, Jean-Pierre G. 2005. Synthesis and antileishmanial activities of 4,5-di-substituted acridines as compared to their 4-mono-substituted homologues. Bioorg Med Chem 13:5560–5568. doi: 10.1016/j.bmc.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 29.Lizarazo-Jaimes EH, Reis PG, Bezerra FM, Rodrigues BL, Monte-Neto RL, Melo MN, Frezard F, Demicheli C. 2014. Complexes of different nitrogen donor heterocyclic ligands with SbCl3 and PhSbCl2 as potential antileishmanial agents against SbIII-sensitive and -resistant parasites. J Inorg Biochem 132:30–36. doi: 10.1016/j.jinorgbio.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Villalobos-Rocha JC, Sanchez-Torres L, Nogueda-Torres B, Segura-Cabrera A, Garcia-Perez CA, Bocanegra-Garcia V, Palos I, Monge A, Rivera G. 2014. Anti-Trypanosoma cruzi and anti-leishmanial activity by quinoxaline-7-carboxylate 1,4-di-N-oxide derivatives. Parasitol Res 113:2027–2035. doi: 10.1007/s00436-014-3850-8. [DOI] [PubMed] [Google Scholar]
- 31.Ronga L, Del Favero M, Cohen A, Soum C, Le Pape P, Savrimoutou S, Pinaud N, Mullie C, Daulouede S, Vincendeau P, Farvacques N, Agnamey P, Pagniez F, Hutter S, Azas N, Sonnet P, Guillon J. 2014. Design, synthesis and biological evaluation of novel 4-alkapolyenylpyrrolo[1,2-a]quinoxalines as antileishmanial agents—part III. Eur J Med Chem 81:378–393. doi: 10.1016/j.ejmech.2014.05.037. [DOI] [PubMed] [Google Scholar]
- 32.Arango V, Robledo S, Seon-Meniel B, Figadere B, Cardona W, Saez J, Otalvaro F. 2010. Coumarins from Galipea panamensis and their activity against Leishmania panamensis. J Nat Prod 73:1012–1014. doi: 10.1021/np100146y. [DOI] [PubMed] [Google Scholar]
- 33.Vila-Nova NS, de Morais SM, Falcao MJ, Alcantara TT, Ferreira PA, Cavalcanti ES, Vieira IG, Campello CC, Wilson M. 2013. Different susceptibilities of Leishmania spp. promastigotes to the Annona muricata acetogenins annonacinone and corossolone, and the Platymiscium floribundum coumarin scoparone. Exp Parasitol 133:334–338. doi: 10.1016/j.exppara.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tahghighi A, Razmi S, Mahdavi M, Foroumadi P, Ardestani SK, Emami S, Kobarfard F, Dastmalchi S, Shafiee A, Foroumadi A. 2012. Synthesis and anti-leishmanial activity of 5-(5-nitrofuran-2-yl)-1,3,4-thiadiazol-2-amines containing N-[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] moieties. Eur J Med Chem 50:124–128. doi: 10.1016/j.ejmech.2012.01.046. [DOI] [PubMed] [Google Scholar]
- 35.Upadhayaya RS, Dixit SS, Foldesi A, Chattopadhyaya J. 2013. New antiprotozoal agents: their synthesis and biological evaluations. Bioorg Med Chem Lett 23:2750–2758. doi: 10.1016/j.bmcl.2013.02.054. [DOI] [PubMed] [Google Scholar]
- 36.Parise-Filho R, Pasqualoto KF, Magri FM, Ferreira AK, da Silva BA, Damiao MC, Tavares MT, Azevedo RA, Auada AV, Polli MC, Brandt CA. 2012. Dillapiole as antileishmanial agent: discovery, cytotoxic activity and preliminary SAR studies of dillapiole analogues. Arch Pharm (Weinheim) 345:934–944. doi: 10.1002/ardp.201200212. [DOI] [PubMed] [Google Scholar]
- 37.Suryawanshi SN, Tiwari A, Chandra N, Ramesh Gupta S. 2012. Chemotherapy of leishmaniasis. Part XI: synthesis and bioevaluation of novel isoxazole containing heteroretinoid and its amide derivatives. Bioorg Med Chem Lett 22:6559–6562. [DOI] [PubMed] [Google Scholar]
- 38.Marrapu VK, Mittal M, Shivahare R, Gupta S, Bhandari K. 2011. Synthesis and evaluation of new furanyl and thiophenyl azoles as antileishmanial agents. Eur J Med Chem 46:1694–1700. doi: 10.1016/j.ejmech.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 39.Baiocco P, Poce G, Alfonso S, Cocozza M, Porretta GC, Colotti G, Biava M, Moraca F, Botta M, Yardley V, Fiorillo A, Lantella A, Malatesta F, Ilari A. 2013. Inhibition of Leishmania infantum trypanothione reductase by azole-based compounds: a comparative analysis with its physiological substrate by X-ray crystallography. ChemMedChem 8:1175–1183. doi: 10.1002/cmdc.201300176. [DOI] [PubMed] [Google Scholar]
- 40.Fernandes MC, Da Silva EN, Pinto AV, De Castro SL, Menna-Barreto RF. 2012. A novel triazolic naphthofuranquinone induces autophagy in reservosomes and impairment of mitosis in Trypanosoma cruzi. Parasitology 139:26–36. doi: 10.1017/S0031182011001612. [DOI] [PubMed] [Google Scholar]
- 41.Papanastasiou I, Prousis KC, Georgikopoulou K, Pavlidis T, Scoulica E, Kolocouris N, Calogeropoulou T. 2010. Design and synthesis of new adamantyl-substituted antileishmanial ether phospholipids. Bioorg Med Chem Lett 20:5484–5487. doi: 10.1016/j.bmcl.2010.07.078. [DOI] [PubMed] [Google Scholar]
- 42.Tavares J, Ouaissi A, Kong Thoo Lin P, Loureiro I, Kaur S, Roy N, Cordeiro-da-Silva A. 2010. Bisnaphthalimidopropyl derivatives as inhibitors of Leishmania SIR2 related protein 1. ChemMedChem 5:140–147. doi: 10.1002/cmdc.200900367. [DOI] [PubMed] [Google Scholar]
- 43.Stec J, Huang Q, Pieroni M, Kaiser M, Fomovska A, Mui E, Witola WH, Bettis S, McLeod R, Brun R, Kozikowski AP. 2012. Synthesis, biological evaluation, and structure-activity relationships of N-benzoyl-2-hydroxybenzamides as agents active against P. falciparum (K1 strain), trypanosomes, and Leishmania. J Med Chem 55:3088–3100. doi: 10.1021/jm2015183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stump B, Kaiser M, Brun R, Krauth-Siegel RL, Diederich F. 2007. Betraying the parasite's redox system: diaryl sulfide-based inhibitors of trypanothione reductase: subversive substrates and antitrypanosomal properties. ChemMedChem 2:1708–1712. doi: 10.1002/cmdc.200700172. [DOI] [PubMed] [Google Scholar]
- 45.Braga SF, Alves EV, Ferreira RS, Fradico JR, Lage PS, Duarte MC, Ribeiro TG, Junior PA, Romanha AJ, Tonini ML, Steindel M, Coelho EF, de Oliveira RB. 2014. Synthesis and evaluation of the antiparasitic activity of bis-(arylmethylidene) cycloalkanones. Eur J Med Chem 71:282–289. doi: 10.1016/j.ejmech.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 46.Colotti G, Baiocco P, Fiorillo A, Boffi A, Poser E, Chiaro FD, Ilari A. 2013. Structural insights into the enzymes of the trypanothione pathway: targets for antileishmaniasis drugs. Future Med Chem 5:1861–1875. doi: 10.4155/fmc.13.146. [DOI] [PubMed] [Google Scholar]
- 47.Bernardes LS, Zani CL, Carvalho I. 2013. Trypanosomatidae diseases: from the current therapy to the efficacious role of trypanothione reductase in drug discovery. Curr Med Chem 20:2673–2696. doi: 10.2174/0929867311320210005. [DOI] [PubMed] [Google Scholar]
- 48.Müller J, Terfort A. 2006. Synthesis of pure aromatic, aliphatic, and araliphatic diselenides. Inorganica Chim Acta 359:4821–4827. doi: 10.1016/j.ica.2006.05.032. [DOI] [Google Scholar]
- 49.Shaaban S, Arafat MA, Hamama WS. 2014. Vistas in the domain of organoselenocyanates. Arkivoc 2014:470–505. doi: 10.3998/ark.5550190.p008.763. [DOI] [Google Scholar]
- 50.Shaaban S, Negm A, Sobh MA, Wessjohann LA. 2015. Organoselenocyanates and symmetrical diselenides redox modulators: design, synthesis and biological evaluation. Eur J Med Chem 97:190–201. doi: 10.1016/j.ejmech.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 51.Alzate JF, Arias AA, Moreno-Mateos D, Alvarez-Barrientos A, Jimenez-Ruiz A. 2007. Mitochondrial superoxide mediates heat-induced apoptotic-like death in Leishmania infantum. Mol Biochem Parasitol 152:192–202. doi: 10.1016/j.molbiopara.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 52.Toro MA, Sanchez-Murcia PA, Moreno D, Ruiz-Santaquiteria M, Alzate JF, Negri A, Camarasa MJ, Gago F, Velazquez S, Jimenez-Ruiz A. 2013. Probing the dimerization interface of Leishmania infantum trypanothione reductase with site-directed mutagenesis and short peptides. Chembiochem 14:1212–1217. doi: 10.1002/cbic.201200744. [DOI] [PubMed] [Google Scholar]
- 53.Sander T, Freyss J, von Korff M, Reich JR, Rufener C. 2009. OSIRIS, an entirely in-house developed drug discovery informatics system. J Chem Infect Model 49:232–246. doi: 10.1021/ci800305f. [DOI] [PubMed] [Google Scholar]
- 54.Agnihotri S, Narula R, Joshi K, Rana S, Singh M. 2012. In silico modeling of ligand molecule for non structural 3 (NS3) protein target of flaviviruses. Bioinformation 8:123–127. doi: 10.6026/97320630008123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahsan MJ, Samy JG, Khalilullah H, Nomani MS, Saraswat P, Gaur R, Singh A. 2011. Molecular properties prediction and synthesis of novel 1,3,4-oxadiazole analogues as potent antimicrobial and antitubercular agents. Bioorg Med Chem Lett 21:7246–7250. doi: 10.1016/j.bmcl.2011.10.057. [DOI] [PubMed] [Google Scholar]
- 56.Bhasin KK, Singh J. 2002. A novel and convenient synthesis towards 2-pyridylselenium compounds: X-ray crystal structure of 4,4′-dimethyl-2,2′-dipyridyl diselenide and tris(2-pyridylseleno)methane. J Organomet Chem 658:71–76. doi: 10.1016/S0022-328X(02)01627-3. [DOI] [Google Scholar]
- 57.Alzate JF, Alvarez-Barrientos A, Gonzalez VM, Jimenez-Ruiz A. 2006. Heat-induced programmed cell death in Leishmania infantum is reverted by Bcl-XL expression. Apoptosis 11:161–171. doi: 10.1007/s10495-006-4570-z. [DOI] [PubMed] [Google Scholar]
- 58.Nwaka S, Hudson A. 2006. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov 5:941–955. doi: 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- 59.Eberle C, Burkhard JA, Stump B, Kaiser M, Brun R, Krauth-Siegel RL, Diederich F. 2009. Synthesis, inhibition potency, binding mode, and antiprotozoal activities of fluorescent inhibitors of trypanothione reductase based on mepacrine-conjugated diaryl sulfide scaffolds. ChemMedChem 4:2034–2044. doi: 10.1002/cmdc.200900327. [DOI] [PubMed] [Google Scholar]
- 60.Hundsdorfer C, Hemmerling HJ, Hamberger J, Le Borgne M, Bednarski P, Gotz C, Totzke F, Jose J. 2012. Novel indeno[1,2-b]indoloquinones as inhibitors of the human protein kinase CK2 with antiproliferative activity towards a broad panel of cancer cell lines. Biochem Biophys Res Commun 424:71–75. doi: 10.1016/j.bbrc.2012.06.068. [DOI] [PubMed] [Google Scholar]
- 61.de Toledo JS, Junior PE, Manfrim V, Pinzan CF, de Araujo AS, Cruz AK, Emery FS. 2013. Synthesis, cytotoxicity and in vitro antileishmanial activity of naphthothiazoles. Chem Biol Drug Des 81:749–756. doi: 10.1111/cbdd.12123. [DOI] [PubMed] [Google Scholar]
- 62.Ali AR, El-Bendary ER, Ghaly MA, Shehata IA. 2013. Novel acetamidothiazole derivatives: synthesis and in vitro anticancer evaluation. Eur J Med Chem 69:908–919. doi: 10.1016/j.ejmech.2013.08.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.