

Abstract
Naloxegol, a peripherally acting μ‐opioid receptor antagonist for the treatment of opioid‐induced constipation, is a substrate for cytochrome P450 (CYP) 3A4/3A5 and the P‐glycoprotein (P‐gp) transporter. By integrating in silico, preclinical, and clinical pharmacokinetic (PK) findings, minimal and full physiologically based pharmacokinetic (PBPK) models were developed to predict the drug‐drug interaction (DDI) potential for naloxegol. The models reasonably predicted the observed changes in naloxegol exposure with ketoconazole (increase of 13.1‐fold predicted vs. 12.9‐fold observed), diltiazem (increase of 2.8‐fold predicted vs. 3.4‐fold observed), rifampin (reduction of 76% predicted vs. 89% observed), and quinidine (increase of 1.2‐fold predicted vs. 1.4‐fold observed). The moderate CYP3A4 inducer efavirenz was predicted to reduce naloxegol exposure by ∼50%, whereas weak CYP3A inhibitors were predicted to minimally affect exposure. In summary, the PBPK models reasonably estimated interactions with various CYP3A modulators and can be used to guide dosing in clinical practice when naloxegol is coadministered with such agents.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Naloxegol is a substrate of CYP3A and P‐gp, as confirmed by clinical pharmacology DDI studies with strong, moderate, and weak inhibitors (ketoconazole, diltiazem, and quinidine) and an inducer (rifampin) of CYP3A and P‐gp. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ PBPK modeling was applied to mechanistically predict the effect of CYP3A modulators likely to be coadministered with naloxegol on its PK. Effects of CYP3A/P‐gp modulators that were commonly coadministered in the phase III program of naloxegol were predicted to help guide naloxegol dosing in clinical practice. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ PBPK modeling extended findings from the clinical DDI studies to predict the effects of moderate or weak CYP3A inhibitors and a moderate CYP3A inducer on the PK of naloxegol. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ PBPK models qualified from clinical trial results can be used to predict the effects of specific CYP3A modulators with naloxegol and to guide the dosing of naloxegol.
Naloxegol is an oral, peripherally acting μ‐opioid receptor antagonist approved by the US Food and Drug Administration (FDA) for the treatment of opioid‐induced constipation in patients with noncancer pain.1, 2, 3 The metabolic stability of naloxegol studied in vitro in the presence of individual cytochrome P450 (CYP) enzymes and human liver microsomes indicated naloxegol is metabolized primarily by CYP3A and not by the other CYP enzymes.4 In vitro studies also indicated naloxegol is a substrate for the P‐glycoprotein (P‐gp) transporter.4 Therefore, the metabolism, distribution (including brain penetration), and disposition of naloxegol may be altered by drugs that interfere with CYP3A and P‐gp. After administration of 27 mg as an oral solution to healthy male volunteers, [14C]naloxegol was absorbed rapidly, and clearance was primarily metabolic.5 Naloxegol metabolites were formed by partial loss of the polyethylene glycol chain, demethylation and oxidation, and dealkylation.5 Naloxegol and its metabolites were primarily eliminated in feces.5 Renal elimination was a minor pathway; parent drug excreted in urine accounted for <6% of the dose.5
The effects of CYP3A and P‐gp modulators on the pharmacokinetics (PKs) of naloxegol were previously characterized in a series of drug‐drug interaction (DDI) studies,6, 7 the findings of which informed current FDA labeling for naloxegol.3 These DDI trials also confirmed that naloxegol is a sensitive substrate of CYP3A and suggest that at the recommended dose of 25 mg naloxegol, CYP3A modulation has a greater impact on the exposure of naloxegol than P‐gp modulation.6, 7 Specifically, ketoconazole (400 mg once daily [q.d.] for 5 days), a strong inhibitor of CYP3A and an inhibitor of P‐gp, increased the area under the curve (AUC) of naloxegol (coadministered on day 4) by 12.9‐fold.6 Diltiazem (240 mg q.d. for 5 days), a moderate inhibitor of CYP3A and an inhibitor of P‐gp, increased the AUC of naloxegol (coadministered on day 4) by 3.4‐fold.6 Quinidine (600 mg, single dose), a weak inhibitor of CYP3A and a strong inhibitor of P‐gp, increased the AUC of naloxegol by 1.4‐fold with coadministration.7 Rifampin (600 mg q.d. for 10 days), a strong inducer of CYP3A and an inducer of P‐gp, reduced the AUC of naloxegol (coadministered on day 10) by almost 90%.6 The objectives of the current analysis, therefore, were: (1) to develop a physiologically based pharmacokinetic (PBPK) model for naloxegol by incorporating physicochemical, absorption, distribution, and elimination properties of naloxegol; (2) investigate changes in systemic exposure of naloxegol after oral coadministration with other CYP3A and P‐gp modulators; and (3) predict clinical DDIs between naloxegol and other CYP3A modulators likely to be used concomitantly with naloxegol.
METHODS
Clinical studies
Clinical DDI study results of orally administered naloxegol 25 mg and various CYP3A and P‐gp inhibitors (ketoconazole, diltiazem, and quinidine) and an inducer (rifampin) were used as comparisons for qualification during the PBPK model development.6, 7 All studies were performed in healthy volunteers using a crossover design, with an appropriate washout period after initial naloxegol dosing.6, 7 For the naloxegol and ketoconazole interaction study (n = 22), a single dose of naloxegol was administered alone, and then in combination with ketoconazole 400 mg q.d. (5 days dosing, naloxegol coadministered on day 4).6 For the naloxegol and diltiazem interaction study (n = 43), a single dose of naloxegol was administered alone, and then in combination with diltiazem 240 mg q.d. (5 days dosing, naloxegol coadministered on day 4).6 For the naloxegol and rifampin interaction study (n = 22), a single dose of naloxegol was administered alone, followed by coadministration with rifampin 600 mg q.d. (10 days dosing, naloxegol coadministered on day 10).6 For the naloxegol and quinidine interaction study (n = 38), a single dose of naloxegol was administered in combination with a single oral dose of quinidine 600 mg.7 Study designs are detailed in Supplementary Table S1.
These studies were conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation/Good Clinical Practice, applicable regulatory requirements, and the AstraZeneca policy on Bioethics. The final protocol and informed consent form were approved by the institutional review boards at the respective study sites. Informed consent was obtained from all volunteers before any study procedures were conducted.
Model development
Model development and simulation were performed with the population‐based absorption, distribution, metabolism, and excretion simulator Simcyp, version 12 (Simcyp, Sheffield, UK).8 First, a minimal PBPK model with a single adjustable compartment,9 which considers both liver and intestinal metabolism, was developed for naloxegol. The single adjustable compartment is a nonphysiologic compartment that allows distribution kinetics in other tissues to be modeled and combines all tissues, except for the liver and portal vein. Distribution was defined by first‐order rate constants and volume of distribution for the single adjustable compartment, an apparent volume of distribution (L/kg). The model was then expanded to a full PBPK model by including additional tissues (e.g., adipose, brain, bone, heart, lung, muscle, and skin) and incorporating the P‐gp transporter contribution at both intestinal epithelia and hepatocytes to evaluate naloxegol absorption and biliary excretion, as well as its interaction with P‐gp inhibitors/inducers. The P‐gp contributions at kidney proximal tubules and the blood‐brain barrier were not considered in the current model.
The final input parameters to simulate the kinetics of naloxegol included molecular weight, compound type, absorption rate constant, blood/plasma ratio, unbound fraction in plasma, apparent permeability coefficient (Papp) in Caco‐2 cells, predicted volume of distribution at steady state, total clearance after an oral dose, renal clearance, and others (Table 1). The volume of distribution at steady state was predicted to be 2.0 L/kg, based on the Rodgers and Rowland method.10 The in vivo total clearance after an oral dose value of 150 L/h observed in the ketoconazole DDI study and the renal clearance value of 4.74 L/h from the renal impairment study11 were used to define metabolic clearance of naloxegol with the retrograde method, which is a feature included in the Simcyp software. The retrograde method estimated an intrinsic metabolic clearance value of 36.6 μL/min/mg. As CYP3A was the only CYP isoform identified to metabolize naloxegol in the in vitro experiment, the percentage of naloxegol metabolized by CYP3A4 was set at 100% (fraction metabolized = 1) and the in vitro transporter‐mediated intrinsic clearance value of 0.267 μL/min/pmol for CYP3A4 was assigned.
Table 1.
Input parameter values used to simulate the kinetics of naloxegol
| Parameter | Value | Assumptions and References |
|---|---|---|
| Molecular weight | 652 | Experimental data |
| Compound type | Diprotic base | Experimental data |
| pKa | 8.45, 9.48 | Experimental data |
| Log P | 1.43 | Experimental data |
| B/P | 1 | Assumed |
| fu | 0.958 | Experimental data |
| fumic | 1 | Software default value |
| fuGut | 1 | Software default value |
| fa | 0.649 | Predicted by Simcyp |
| ka (h−1) | 0.338 | Predicted by Simcyp |
| Papp Caco‐2 (10−6 cm/s) | 4.55 | Experimental data |
| Predicted Vss (L/kg) | 2.0 | Predicted using Simcyp Rodgers and Rowland method |
| VSAC (L/kg) | 1.33 | Estimated to fit targeted profile |
| CLpo (L/h) | 150 | Clinical observation6 |
| CYP3A4 CLint (μL/min/pmol protein) | 0.267 | Retrograde calculation |
| CLR (L/h) | 4.74 | Clinical observation11 |
| CLint, T (μL/min) | 2.5 | Assumed to be the same as digoxin |
B/P, blood to plasma ratio; CLint, intrinsic clearance; CLint, T, in vitro transporter‐mediated intrinsic clearance; CLpo, total clearance after an oral dose; CLR, renal clearance; CYP3A4, cytochrome P450 3A4; fa, fraction of absorbed dose that escapes gut wall metabolism; fu, unbound fraction in plasma; fuGut, unbound fraction in gut; fumic, unbound fraction in microsomes; ka, absorption rate constant; Log P, partition coefficient; Papp Caco‐2, apparent permeability coefficient in Caco‐2 cells; pKa, negative logarithm of the dissociation constant, Ka; VSAC, volume of distribution for the single adjustable compartment; Vss, volume of distribution at steady state.
As there was no in vitro P‐gp transporter study specifically designed to capture certain parameters, assumptions were made to obtain a reasonable PK profile of naloxegol. The model assumed hepatic perfusion‐limited diffusion by assigning the default value of passive diffusion clearance of 0.1 mL/min per million cells at the sinusoidal side for hepatocytes to determine the role of transporters on the hepatic uptake of naloxegol. Nor were the kinetic parameters (rate constant for the elimination of a metabolite and maximal flux value) of P‐gp‐mediated intestinal efflux available regarding naloxegol. However, as naloxegol and digoxin demonstrated similar efflux results using the same Caco‐2 transport assay, the reported in vitro transporter‐mediated intrinsic clearance value for digoxin (2.5 μL/min) was assigned.12 A sensitivity analysis was applied to demonstrate that this was a reasonable assumption value for naloxegol. Because the incorporation of the efflux transporter, P‐gp, in the system is expected to restrict drug absorption in the gastrointestinal tract, the Papp in humans value in the duodenum, jejunum I, and jejunum II was increased to 3.5 × 10−4 cm/s, whereas the Papp in humans value was retained at the original estimation of 0.83 × 10−4 cm/s for the other segments in the advanced dissolution, absorption, and metabolism model. The use of the original Papp in humans value estimated from Papp Caco‐2 to all segments would underestimate naloxegol exposure.
Model simulation
Comparative simulations
Simulations were conducted during the PBPK model development as a qualification step to ensure appropriate parameter estimations. Default model library files for the CYP3A/P‐gp modulators ketoconazole, diltiazem, rifampin, and quinidine were used as provided in the software (Supplementary Appendix), with the addition of the inhibition rate constants (Ki values) for P‐gp, using the lowest reported value for each drug in the interaction simulations. The Ki values of 0.5, 3.5, 84.5, and 0.43 μM were applied for ketoconazole, diltiazem, rifampin, and quinidine inhibition of P‐gp, respectively. The approach reported by Friedman et al.13 was applied to simulate the plasma concentration‐time profile of sustained‐released diltiazem. The models were developed using the same study design and dosing regimen as in each aforementioned clinical DDI trial (see Supplementary Table S1). Both minimal and full PBPK models were used to simulate the DDI between naloxegol and ketoconazole, diltiazem, and rifampin, and the full PBPK model was applied to predict the interaction between naloxegol and quinidine.
Predictive simulations
The final minimal PBPK model was utilized to simulate changes in the PK profile of naloxegol in the presence of various moderate (erythromycin, fluconazole, verapamil, and ciprofloxacin)14 and weak (alprazolam, fluoxetine, amlodipine, atorvastatin, and cimetidine)14 CYP3A inhibitors that were commonly coadministered with naloxegol in phase III trials.15 Compound files were used as provided in the Simcyp software for erythromycin (250 or 400 mg every 6 hours [Q6h] for 5 days), fluconazole (200 mg q.d. for 5 days), verapamil (120 mg 3 times daily [t.i.d.] for 5 days), fluoxetine (80 mg q.d. for 5 days), ciprofloxacin (500 mg twice daily [b.i.d.] for 5 days), and alprazolam (0.5 mg t.i.d. for 1 day). In a previous clinical study, ciprofloxacin was reported to result in a greater than twofold increase in sildenafil AUC.16 However, an in vitro study indicated that ciprofloxacin at 2 mM inhibited erythromycin‐mediated CYP3A activity by only 65%.17 Using the in vitro study findings,17 the model was adapted to include the Ki value of 538 μM for CYP3A inhibition. For alprazolam, the only in vitro CYP3A inhibition study showed alprazolam to have an IC50 value >50 μM against CYP3A.18 This IC50 value was used to predict the potential for DDI between naloxegol and alprazolam. Compound files for amlodipine (10 mg q.d. for 15 days), atorvastatin (80 mg q.d. for 5 days), and cimetidine (800 mg q.d. for 5 days) were developed using available in vitro and in vivo absorption, distribution, metabolism, and excretion data (Supplementary Appendix).
A compound file for efavirenz, a moderate CYP3A inducer,14 was also constructed, using a previously published model,19 with additional parameters for pKa (10.2),20 and unbound plasma protein binding (0.029).21 The minimal PBPK model was used to simulate potential DDI between efavirenz 400 mg q.d. for 14 days and naloxegol 25 mg as a single dose administered on day 14, with induction of intestinal CYP3A by efavirenz.
Predictions of plasma drug concentration‐time profiles, clearance, DDIs, distribution, and absorption were performed in the Simcyp simulator using a population of virtual individuals. The default Simcyp parameter values for creating a virtual healthy volunteer population (demographic, anatomic, and physiologic parameters, including liver volume and blood flow, and enzyme levels) were applied in the current analysis. The PK simulations were conducted using various clinical study conditions (see Supplementary Table S2), and the standard 10 trials of 10 subjects per trial was applied for each simulation). Mean and distribution of demographic covariates and drug parameters were generated using a Monte Carlo approach, under predefined study designs, within Simcyp.
RESULTS
Predicted and observed DDI with CYP3A/P‐gp inhibitors and inducer
The initial simulations of the kinetics of naloxegol (minimal PBPK model) alone and in the presence of ketoconazole were used to optimize the metabolism parameter inputs. Simulated profiles for the naloxegol and ketoconazole interaction were then compared to observed data (Figure 1 a). The predicted and observed AUC ratios were 13.1‐fold and 12.9‐fold, respectively, and the predicted and observed maximum plasma concentration (Cmax) ratios were 7.8‐fold and 9.6‐fold (Table 2 , Figure 2). The predicted AUC and Cmax ratios were close to observed values, indicating that the assigned fraction metabolized value of 1 for CYP3A4 was close to the actual value, based on the assumption that ketoconazole 400 mg q.d. specifically inhibits all CYP3A activity.
Figure 1.
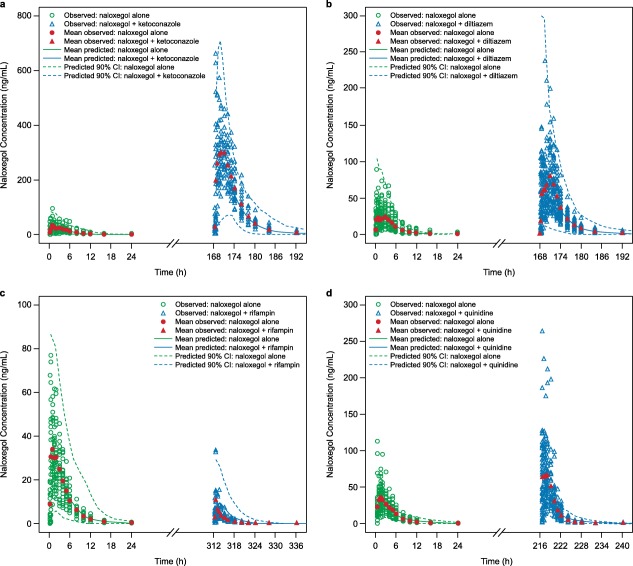
Simulated plasma drug concentration‐time profiles for 25 mg naloxegol alone and in the presence of the cytochrome P450 (CYP)3A4 modulators (a) ketoconazole 400 mg, minimal physiologically based pharmacokinetic (PBPK) model; (b) diltiazem 240 mg, minimal PBPK model; (c) rifampin 600 mg, minimal PBPK model; and (d) quinidine 600 mg, full PBPK model. Data for naloxegol alone are represented in green, and data for naloxegol in the presence of the respective CYP3A4 modulators are represented in blue. Observed values for naloxegol alone are represented by diamonds, and observed values for naloxegol + CYP3A4 modulators are represented by triangles. Red symbols represent mean observed values. Dashed lines represent the 90% confidence interval of the respective simulations.
Table 2.
Summary of predicted and observed AUC and Cmax ratios using minimal and full PBPK models for naloxegol 25 mg with coadministration of various CYP3A/P‐gp modulatorsa
| AUC | Cmax | ||||||
|---|---|---|---|---|---|---|---|
| Modulator | Dosing regimenb | Observed | Minimal PBPK model | Full PBPK model | Observed | Minimal PBPK model | Full PBPK model |
| Ketoconazole (CYP3A/P‐gp inhibitor) | 400 mg q.d. for 5 days (day 4) | 12.85 (11.3, 14.6) | 13.14 (11.9, 14.5) | 8.82 (7.17, 10.9) | 9.58 (8.1, 11.3) | 7.75 (6.9, 8.6) | 4.19 (3.38, 5.21) |
| Diltiazem (CYP3A/P‐gp inhibitor) | 240 mg q.d. for 5 days (day 4) | 3.41 (3.16, 3.68) | 2.80 (2.64, 2.98) | 2.45 (2.10, 2.86) | 2.85 (2.59, 3.14) | 2.28 (2.18, 2.39) | 1.70 (1.57, 1.83) |
| Rifampin (CYP3A/P‐gp inducer) | 600 mg q.d. for 10 days (day 10) | 0.11 (0.10, 0.13) | 0.24 (0.22, 0.26) | 0.37 (0.31, 0.43) | 0.25 (0.19, 0.31) | 0.27 (0.25, 0.30) | 0.35 (0.32, 0.39) |
| Quinidine (CYP3A/P‐gp inhibitor) | 600 mg single dose (day 1) | 1.38 (1.31, 1.46) | 1.13 (1.126, 1.14) | 1.23 (1.21, 1.25) | 2.44 (2.17, 2.75) | 1.19 (1.18, 1.21) | 1.87 (1.81, 1.93) |
AUC, area under the curve; Cmax, maximum concentration; CYP3A4, cytochrome P450 3A4; PBPK, physiologically based pharmacokinetic; P‐gp, p‐glycoprotein; q.d., once daily.
Data are geometric means with 90% confidence intervals.
Dosing regimen of modulator, with day of naloxegol 25 mg coadministration in parentheses.
Figure 2.
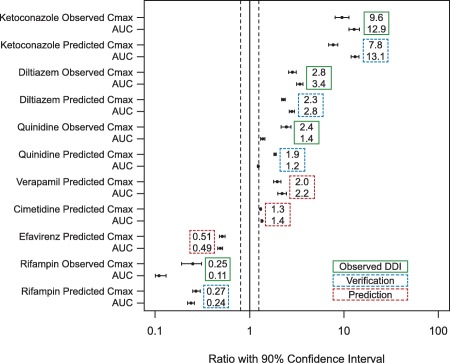
Simulated and observed naloxegol peak plasma concentration (Cmax) and area under the curve (AUC) ratios when coadministered with weak, moderate, and strong inhibitors and moderate and strong inducers of cytochrome P450 (CYP)3A, with 90% confidence intervals. Dashed lines represent the (80%, 125%) interval. Verification and prediction are both model‐predicted interaction results, but indicate cases in which the actual clinical drug‐drug interaction (DDI) studies were conducted (verification) or were not (prediction).
The minimal PBPK model of naloxegol was further evaluated by comparing predicted PK changes in naloxegol with coadministration of diltiazem 240 mg q.d. or rifampin 600 mg q.d. with observed changes. The predicted and observed change in AUC and Cmax ratio with concomitant diltiazem was 2.8‐fold and 3.4‐fold, respectively (AUC), and 2.3‐fold and 2.9‐fold (Cmax ratio, Table 2 , Figure 2). With coadministration of rifampin, the predicted and observed decrease in AUC ratio was 0.24‐fold and 0.11‐fold, respectively, and the predicted and observed decrease in Cmax ratio was 0.27‐fold and 0.25‐fold (Table 2 , Figure 2). Simulated concentration profiles for the naloxegol interaction with diltiazem and rifampin relative to observed data are presented in Figure 1 b,c. The reasonable predictions of naloxegol AUC and Cmax ratios with coadministered diltiazem and rifampin also demonstrate that the assigned parameters were appropriate to predict changes in the naloxegol plasma concentration‐time profile and potential DDIs.
The full PBPK model additionally produced adequate predictions for naloxegol PK changes with coadministration of a single dose of quinidine 600 mg (Table 2 , Figure 2). The predicted and observed increase in naloxegol AUC was 1.23‐fold and 1.38‐fold, respectively, and the predicted and observed increase in Cmax was 1.87‐fold and 2.44‐fold. The simulated PK profiles of naloxegol with and without coadministration of quinidine were consistent with the observed data (Figure 1 d).
The full PBPK model was also applied to predict changes in naloxegol exposure with coadministration of ketoconazole, diltiazem, and rifampin. Although the full PBPK model adequately predicted changes in naloxegol exposure in the presence of CYP3A/P‐gp modulators, there was a trend toward slight underprediction compared with observed values (Table 2 , Figure 2). Thus, the minimal PBPK model may be the preferred method for estimating changes in the PK of naloxegol in the presence of various moderate and weak CYP3A inhibitors or inducers by considering worst case scenarios.
Predicted DDI with moderate and weak CYP3A4 inhibitors and inducers
Simulations of naloxegol interaction with various moderate and weak CYP3A inhibitors were conducted using the minimal PBPK model (Table 3 , Figure 2). Fluconazole 200 mg q.d. was predicted to cause a 2.81‐fold increase in naloxegol AUC, and verapamil 120 mg t.i.d. was predicted to result in a 2.21‐fold increase in naloxegol AUC. Similarly, 250 and 400 mg of erythromycin Q6h were predicted to cause a 3.47‐fold and 4.63‐fold increase in naloxegol AUC, respectively. Ciprofloxacin 500 mg b.i.d. was predicted to have a negligible impact on naloxegol exposure, as was alprazolam 0.5 mg t.i.d. (based on the aforementioned in vitro IC50 value). The weak CYP3A inhibitors amlodipine 10 mg q.d., atorvastatin 80 mg q.d., fluoxetine 80 mg q.d., and cimetidine 800 mg q.d. were predicted to increase naloxegol AUC by 22%, 8%, 26%, and 35%, respectively, indicating a minimal impact on naloxegol exposure (Table 3).
Table 3.
Summary of predicted AUC and Cmax ratios using the minimal PBPK model for naloxegol 25 mg with coadministration of various moderate and weak CYP3A inhibitors or inducer
| Minimal PBPK modela | |||
|---|---|---|---|
| Modulator | Dosing regimensb | AUC ratio | Cmax ratio |
| Fluconazole (CYP3A inhibitor) | 200 mg q.d. for 5 days (day 4) | 2.81 (2.71, 2.92) | 2.40 (2.3, 2.51) |
| Verapamil (CYP3A/P‐gp inhibitor) | 120 mg t.i.d. for 5 days (day 4) | 2.21 (2.00, 2.46) | 1.97 (1.8, 2.15) |
| Erythromycin (CYP3A/P‐gp inhibitor) | 250 mg Q6h for 5 days (day 4) | 3.47 (3.16, 3.81) | 2.77 (2.55, 3.01) |
| 400 mg Q6h for 5 days (day 4) | 4.63 (4.18, 5.13) | 3.42 (3.12, 3.75) | |
| Ciprofloxacin (CYP3A inhibitor) | 500 mg b.i.d. for 5 days (day 4) | 1.01 | 1.02 |
| Alprazolam (CYP3A inhibitor) | 0.5 mg t.i.d. for 1 day (day 1) | 1.00 | 1.00 |
| Amlodipine (CYP3A inhibitor) | 10 mg q.d. for 15 days (day 14) | 1.22 (1.20, 1.24) | 1.20 (1.18, 1.21) |
| Atorvastatin (CYP3A inhibitor) | 80 mg q.d. for 5 days (day 4) | 1.08 (1.08, 1.09) | 1.13 (1.12, 1.14) |
| Fluoxetine (CYP3A inhibitor) | 80 mg q.d. for 5 days (day 4) | 1.26 (1.23, 1.29) | 1.22 (1.20, 1.24) |
| Cimetidine (CYP3A inhibitor) | 800 mg q.d. for 5 days (day 4) | 1.35 (1.33, 1.37) | 1.31 (1.30, 1.33) |
| Efavirenz (CYP3A inducer), with induction of intestinal CYP3A4 | 400 mg q.d. for 14 days (day 14)b | 0.49 (0.46, 0.52) | 0.51 (0.48, 0.55) |
AUC, area under the curve; b.i.d., twice daily; Cmax, maximum concentration; CYP3A, cytochrome P450 3A; CYP3A4, cytochrome P450 3A4; PBPK, physiologically based pharmacokinetic; q.d., once daily; Q6h, once every 6 hours; t.i.d., 3 times daily.
Data are geometric means with 95% confidence intervals.
Dosing regimen of modulator, with day of naloxegol 25 mg coadministration in parentheses.
The minimal PBPK model was also applied to predict DDI with naloxegol and the moderate CYP3A inducer, efavirenz 400 mg q.d. The model predicted that naloxegol AUC would decrease by 51% when the induction effect by efavirenz on both hepatic and intestine CYP3A was considered (Table 3 , Figure 2).
DISCUSSION
Minimal and full PBPK models of naloxegol were successfully developed by integrating physiochemical properties, in vitro metabolism data, and clinical PK results. The models were refined and qualified through calibration against one data set and adequately simulated outcomes with different comparators. Visual inspection of the model predictions compared with experimental values also contributed to the refinement and qualification of the models.
Results from clinical DDI studies showed that CYP3A enzymes predominate in the metabolism of naloxegol and that the impact of CYP3A inhibition on the PK of naloxegol is much greater than the impact of P‐gp inhibition.6, 7 For example, observed data showed that ketoconazole, a strong inhibitor of CYP3A and an inhibitor of P‐gp,14 increased naloxegol AUC by 1,185%, whereas quinidine, a weak inhibitor of CYP3A and a potent inhibitor of P‐gp,14 increased naloxegol AUC by only 38%. It is also established that, in contrast to CYP‐mediated DDIs, P‐gp‐mediated DDIs are generally not clinically significant and usually result in a less than twofold increase in exposure.22 The localization of P‐gp also tends to result in a greater impact on limiting uptake of drugs from blood circulation into the brain and from intestinal lumen into epithelial cells than on enhancing the excretion of drugs out of hepatocytes and renal tubules into the adjacent luminal space.23 With naloxegol treatment, it is hypothesized that P‐gp has a primary effect on absorption.23 However, because naloxegol is highly soluble and rapidly absorbed,3 the contribution of intestinal P‐gp to overall drug absorption is unlikely to be quantitatively important because P‐gp transport activity can be saturated by high concentrations of naloxegol in the intestinal lumen.23 In addition, the PK of naloxegol is linear or near linear with a dose of up to 1,000 mg,24 indicating that the P‐gp transporter was saturated in the dose range studied. Thus, the observed increase in the exposure of naloxegol when coadministered with quinidine is likely to be driven by the CYP3A inhibitory activity of quinidine.7
As CYP3A inhibition is evidenced to be the dominant factor affecting the PK of naloxegol, the minimal PBPK model was applied to predict changes in naloxegol exposure in the presence of other moderate and weak CYP3A inhibitors. Of the additional moderate CYP3A inhibitors included in this analysis, verapamil25 and erythromycin26 are also P‐gp inhibitors but may have less potency in this regard than ketoconazole and quinidine.27 The weak CYP3A inhibitors, alprazolam, amlodipine, atorvastatin, fluoxetine, and cimetidine, are not identified as P‐gp inhibitors. Therefore, including or excluding the P‐gp component in the PBPK model would have had minimal difference on the DDI predictions for these compounds.
Many of the inhibitors/inducers evaluated in the current analysis were standard in vivo CYP3A modulators that have been classified by the FDA. Molecules classified as moderate (fluconazole, verapamil, and erythromycin) and weak (alprazolam, fluoxetine, and cimetidine) CYP3A inhibitors by the FDA14 would result in twofold to fivefold and less than twofold, respectively, AUC increases of a CYP3A‐sensitive substrate. Similarly, efavirenz is classified by the FDA as a moderate CYP3A inducer14 and would cause a 50–80% decrease in the AUC of a sensitive CYP3A substrate. The minimal PBPK model successfully predicted these moderate/weak CYP3A inhibitors and moderate inducers could have similar effects on naloxegol as on other CYP3A‐sensitive substrates (Table 3), indicating that a dose adjustment might be necessary when naloxegol is coadministered with a moderate CYP3A inhibitor/inducer. Indeed, based on the current PBPK model and results from clinical DDI studies, naloxegol's package insert3 suggests the naloxegol dose should be reduced by one‐half when coadministered with moderate CYP3A inhibitors (e.g., diltiazem, erythromycin, and verapamil). It also recommends no dose adjustment when naloxegol is coadministered with weak CYP3A inhibitors (e.g., quinidine and cimetidine). The US package insert also includes PBPK modeling results for the interaction with efavirenz to guide naloxegol dosing.3
In conclusion, the developed PBPK models were successfully applied to simulate the PK profile of naloxegol and quantitatively predict the magnitude of interactions with various CYP3A/P‐gp modulators. Weak CYP3A inhibitors may be expected to have minimal impact on naloxegol exposure in routine clinical use, whereas moderate CYP3A inducers may reduce naloxegol exposure by 50%. Given the reasonable agreement found between model predictions and observed values, the PBPK model will be useful to guide dosing in clinical practice when naloxegol is coadministered with other CYP3A modulators.
Supporting information
Supporting Information
Acknowledgments
This study was funded by AstraZeneca Pharmaceuticals LP. Medical writing services were provided by Meg Shurak, MS, and Diane DeHaven–Hudkins, PhD, of Complete Healthcare Communications, Chadds Ford, PA, with funding from AstraZeneca Pharmaceuticals, Wilmington, DE.
Conflict of Interest
This study was funded by AstraZeneca Pharmaceuticals. D.Z., M.S., and N.A‐H. are employees of AstraZeneca Pharmaceuticals. K.B. was an employee of AstraZeneca Pharmaceuticals at the time this work was performed, and is currently employed as a contractor for AstraZeneca Pharmaceuticals. K.B., N.A‐H., and M.S. are shareholders of AstraZeneca.
Author Contributions
D.Z., K.B., M.S., and N.A‐H. wrote the manuscript. D.Z., K.B., and N.A‐H. designed the research. D.Z., K.B., M.S., and N.A‐H. performed the research. D.Z., K.B., and N.A‐H. analyzed the data.
References
- 1. Chey, W.D. , Webster, L. , Sostek, M. , Lappalainen, J. , Barker, P.N. & Tack, J. Naloxegol for opioid‐induced constipation in patients with noncancer pain. N. Engl. J. Med. 370, 2387–2396 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Webster, L. , Dhar, S. , Eldon, M. , Masuoka, L. , Lappalainen, J. & Sostek, M. A phase 2, double‐blind, randomized, placebo‐controlled, dose‐escalation study to evaluate the efficacy, safety, and tolerability of naloxegol in patients with opioid‐induced constipation. Pain 154, 1542–1550 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Movantik™ (naloxegol) [package insert] . Full prescribing information, AstraZeneca Pharmaceuticals, Wilmington, DE, 2015.
- 4. Odinecs, A. , Song, Y. , Harite, S. , Lee, M.G. , Kugler, A.R. & Eldon, M.A. NKTR‐118, an oral peripheral opioid antagonist, has low potential for drug‐drug interactions. J. Clin. Pharmacol. 49, 1091–1130 (2009). [Google Scholar]
- 5. Bui, K. , She, F. , Hutchison, M. , Brunnström, Å. & Sostek, M. Absorption, distribution, metabolism and excretion of [14C]‐labeled naloxegol in healthy subjects. Int. J. Clin. Pharmacol. Ther. 53, 838–846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bui, K. , Zhou, D. , Sostek, M. , She, F. & Al‐Huniti, N. Effects of CYP3A modulators on the pharmacokinetics of naloxegol. J. Clin. Pharmacol; e‐pub ahead of print 2015. [DOI] [PubMed] [Google Scholar]
- 7. Bui, K. , She, F. , Zhou, D. , Butler, K. , Al‐Huniti, N. & Sostek, M. The effect of quinidine, a strong P‐glycoprotein inhibitor, on the pharmacokinetics and central nervous system distribution of naloxegol. J. Clin. Pharmacol; e‐pub ahead of print 2015. [DOI] [PubMed] [Google Scholar]
- 8. Jamei, M. , Marciniak, S. , Feng, K. , Barnett, A. , Tucker, G. & Rostami–Hodjegan, A. The Simcyp population‐based ADME simulator. Expert Opin. Drug Metab. Toxicol. 5, 211–223 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Johnson, T.N. , Zhou, D. & Bui, K.H. Development of physiologically based pharmacokinetic model to evaluate the relative systemic exposure to quetiapine after administration of IR and XR formulations to adults, children and adolescents. Biopharm. Drug Dispos. 35, 341–352 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Rodgers, T. & Rowland, M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm. Res. 24, 918–933 (2007). [DOI] [PubMed] [Google Scholar]
- 11. Bui, K. , She, F. & Sostek, M. The effects of renal impairment on the pharmacokinetics, safety, and tolerability of naloxegol. J. Clin. Pharmacol. 54, 1375–1382 (2014). [DOI] [PubMed] [Google Scholar]
- 12. Troutman, M.D. & Thakker, D.R. Efflux ratio cannot assess P‐glycoprotein‐mediated attenuation of absorptive transport: asymmetric effect of P‐glycoprotein on absorptive and secretory transport across Caco‐2 cell monolayers. Pharm. Res. 20, 1200–1209 (2003). [DOI] [PubMed] [Google Scholar]
- 13. Friedman, E.J. et al Effect of different durations and formulations of diltiazem on the single‐dose pharmacokinetics of midazolam: how long do we go? J. Clin. Pharmacol. 51, 1561–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 14. Guidance for Industry . Drug Interaction Studies–Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Draft Guidance: US Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research, 2012. [Google Scholar]
- 15.Naloxegol, data on file. AstraZeneca, Wilmington, DE, 2013.
- 16. Hedaya, M.A. , El‐Afify, D.R. & El‐Maghraby, G.M. The effect of ciprofloxacin and clarithromycin on sildenafil oral bioavailability in human volunteers. Biopharm. Drug Dispos. 27, 103–110 (2006). [DOI] [PubMed] [Google Scholar]
- 17. McLellan, R.A. , Drobitch, R.K. , Monshouwer, M. & Renton, K.W. Fluoroquinolone antibiotics inhibit cytochrome P450‐mediated microsomal drug metabolism in rat and human. Drug Metab. Dispos. 24, 1134–1138 (1996). [PubMed] [Google Scholar]
- 18. Bohets, H. et al Identification of the cytochrome P450 enzymes involved in the metabolism of cisapride: in vitro studies of potential co‐medication interactions. Br. J. Pharmacol. 129, 1655–1667 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rekić, D. , Röshammar, D. , Mukonzo, J. & Ashton, M. In silico prediction of efavirenz and rifampicin drug‐drug interaction considering weight and CYP2B6 phenotype. Br. J. Clin. Pharmacol. 71, 536–543 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rabel, S.R. , Maurin, M.B. , Rowe, S.M. & Hussain, M. Determination of the pKa and pH‐solubility behavior of an ionizable cyclic carbamate, (S)‐6‐chloro‐4‐(cyclopropylethynyl)‐1,4‐dihydro‐4‐(trifluoromethyl)‐2H‐3,1‐benzoxazin‐2‐one (DMP 266). Pharm. Dev. Technol. 1, 91–95 (1996). [DOI] [PubMed] [Google Scholar]
- 21. Shou, M. et al Modeling, prediction, and in vitro in vivo correlation of CYP3A4 induction. Drug Metab. Dispos. 36, 2355–2370 (2008). [DOI] [PubMed] [Google Scholar]
- 22. Fenner, K.S. et al Drug‐drug interactions mediated through P‐glycoprotein: clinical relevance and in vitro‐in vivo correlation using digoxin as a probe drug. Clin. Pharmacol. Ther. 85, 173–181 (2009). [DOI] [PubMed] [Google Scholar]
- 23. Lin, J.H. & Yamazaki, M. Clinical relevance of P‐glycoprotein in drug therapy. Drug Metab. Rev. 35, 417–454 (2003). [DOI] [PubMed] [Google Scholar]
- 24. Eldon, M.A. , Kugler, A.R. , Medve, R.A. , Bui, K. , Butler, K. & Sostek, M. Safety, tolerability, pharmacokinetics, and pharmacodynamic effects of naloxegol at peripheral and central nervous system receptors in healthy male subjects: a single ascending‐dose study. Clin. Pharmacol. Drug Dev. 4, 434–441 (2015). [DOI] [PubMed] [Google Scholar]
- 25. Tang, F. , Horie, K. & Borchardt, R.T. Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa? Pharm. Res. 19, 765–772 (2002). [DOI] [PubMed] [Google Scholar]
- 26. Eberl, S. et al Role of p‐glycoprotein inhibition for drug interactions: evidence from in vitro and pharmacoepidemiological studies. Clin. Pharmacokinet. 46, 1039–1049 (2007). [DOI] [PubMed] [Google Scholar]
- 27. Schwarz, U.I. , Gramatté, T. , Krappweis, J. , Oertel, R. & Kirch, W. P‐glycoprotein inhibitor erythromycin increases oral bioavailability of talinolol in humans. Int. J. Clin. Pharmacol. Ther. 38, 161–167 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information