

Abstract
Introduction
Ascertainment of the pattern and temporal change of biomarkers in preclinical (asymptomatic) sporadic Alzheimer's disease (AD) will increase knowledge about early pathogenesis and facilitate interventional therapeutic trials.
Methods
In this prospective longitudinal study, repeated cerebrospinal fluid (CSF) collections and cognitive evaluations were performed in cognitively healthy elderly individuals during a 9-year period.
Results
Low CSF β-amyloid (Aβ)42 levels predicted subsequent development of clinical AD 9 years later. Noteworthy, one-third of individuals with pathologically low baseline Aβ42 levels remained cognitively intact during follow-up. No further decrease in Aβ42 was seen in those with low levels already at baseline.
Discussion
CSF Aβ42 predicts sporadic AD at least 9 years before dementia onset and has plateaued already at this time. However, many individuals can harbor brain amyloid accumulation over a decade without signs of cognitive deterioration, which could implicate how CSF biomarkers are used to identify preclinical AD in future interventional therapeutic trials.
Keywords: Dementia, Alzheimer's disease, Cognitive aging, Cerebrospinal fluid, Cohort studies, β-Amyloid1–42, Tau protein
1. Introduction
The slowly progressive nature of Alzheimer's disease (AD) implies a long preclinical phase before onset of cognitive symptoms. Increasing evidence suggests that cerebral accumulation of β-amyloid (Aβ) can be detected 5–20 years before dementia onset in AD, when using cerebrospinal fluid (CSF) Aβ42 or amyloid positron emission tomography (PET) imaging [1], [2], [3], [4]. Important evidence comes from studies evaluating asymptomatic individuals with autosomal dominant forms of AD [1], [5]. To determine the temporal evolution of AD biomarkers during the early phases of sporadic AD, we need longitudinal studies with repeated biomarker assessments over 5–15 years covering the preclinical phases of AD. A few studies with repeated longitudinal biomarker assessments in cognitively healthy individuals have been published [6], [7], [8], but studies over extended periods, of >4 years, are still lacking.
Several studies imply that CSF can identify cognitively healthy elderly individuals that are at increased risk of subsequent development of cognitive decline [9], [10], [11], [12], [13], [14]. However, the frequency of false positive cases is still unclear. To address this, we need long-term follow-up cognitively healthy individuals with deviant CSF biomarkers.
In this prospective and longitudinal study, we investigated CSF biomarkers repeatedly over 9–10 years in individuals, who all were cognitively healthy at baseline. The cognitive performance and development of dementia were determined during the clinical follow-up. Great care was taken to minimize drop-out during the study.
2. Methods
2.1. Study design
The objective of this study was to model within-person neurodegenerative biomarker trajectories in preclinical AD using repeated assessments of CSF biomarkers and cognitive performance as well as to investigate the predictive ability of CSF biomarkers to identify future development of clinical dementia. It is a prospective, longitudinal, observational study on initially cognitively healthy elderly volunteers recruited through advertisement in year 2002 in the city of Malmö, Sweden [13], for the purpose to constitute a healthy control group in dementia studies. Individuals who responded were included in the study unless they fulfilled any of the prospectively set exclusion criteria. Baseline exclusion criteria were (1) subjective cognitive decline, (2) presence of mild cognitive impairment (MCI) or dementia, (3) mini-mental state examination (MMSE) [15] score of <27, and (4) presence of other morbidities possibly affecting cognitive status such as major depressive episode, ongoing alcohol abuse, and severs disorders of the central nervous system. Treatable and reversible diseases that could affect cognition were treated and did not lead to exclusion. Included participants were then followed longitudinally in approximately every third year with focus on cognitive performance and CSF measurements.
2.2. Subjects
In total, 62 individuals could be recruited of which 54 performed baseline lumbar puncture and CSF collection. All participants also underwent comprehensive examination including physical, neurologic, and psychiatric evaluation, computed tomography (CT) of the brain, and cognitive testing at baseline. Cognitive follow-up was offered after 3, 5, and 9–10 years with renewed lumbar puncture after 5 and 9–10 years. Individuals with baseline CSF values and clinical cognitive follow-up after 9 years (n = 44) were included in the main analyses of the present study (Fig. 1). Only a handful of participants were evaluated after closer to 10 years at the last follow-up, whereas the overwhelming majority was evaluated after 9 years.
Fig. 1.
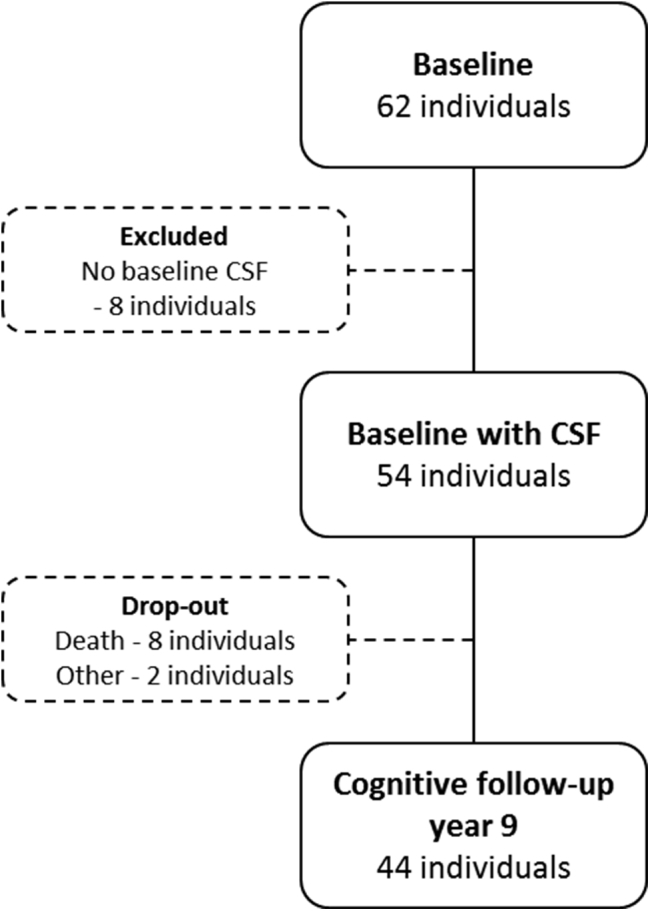
Flowchart of inclusion and drop-out in the study sample. Abbreviation: CSF, cerebrospinal fluid.
In the subgroup of participants who were not available for the 9-year follow-up visit (n = 10, Fig. 1), medical record was collected and antemortem cognitive follow-up performances in the study were evaluated in nine cases. In this subgroup, we found one individual who had developed MCI and the rest were cognitively normal at the last observation.
2.3. Cognitive evaluation
Cognitive testing included MMSE (all visits) [15], clock drawing test (all visits) [16], cube drawing (all visits) [17], delayed memory in Alzheimer's disease assessment scale cognitive subscale (ADAS-cog; baseline, follow-up years 5 and 9) [17], and a quick test (follow-up years 3, 5, and 9) [18]. At follow-up after 9 years, Stroop test [19], trail making test A and B [20], symbol digit modalities test [21], letter S fluency test (phonemic fluency) [22], animal fluency test (semantic fluency) [22], and month naming test (the task of naming the months backward as fast as possible starting with December) were also added. Delayed memory was scored as number of correctly recalled words, which gives higher scores when better delayed memory function.
Cognitive diagnosis was based on clinical evaluation by a physician experienced in dementia disorders and was later confirmed by a consensus group of experienced physicians. The consensus group was blinded to biomarker values and had only access to medical history, cognitive test results, and CT scan results. Diagnosis criteria used in regular clinical settings were applied, i.e. MCI [23], Alzheimer's dementia [24], vascular dementia [25], dementia with Lewy body (DLB) [26], and other dementia (OD) [27]. DLB and AD participants are studied together in this study because DLB patients often also have amyloid pathology next to their synucleinopathy [28].
2.4. Lumbar puncture and CSF analyses
Lumbar puncture was performed in a sitting position with CSF obtained from the L3/L4 or L4/L5 interspaces. All CSF were collected in plastic (polypropylene) tubes, gently mixed to avoid gradient effect. Samples were then centrifuged at 2000 × g at 4°C for 10 minutes to eliminate cells and other insoluble material. Pending biochemical analyses samples were immediately frozen and stored at −80°C without being thawed or refrozen.
Analysis of Aβ42, total tau (T-tau), and phosphorylated tau (P-tau) using xMAP technology (INNO-BIA AlzBio3 kit; Innogenetics, Ghent, Belgium) was performed using the same batch of reagents for each CSF acquisition. A large random sample of baseline and 5-year follow-up CSF was analyzed together with 9-year follow-up CSF to assure concordant assay values between all three analysis occasions. CSF values are given in nanograms per liter.
2.5. Ethics
The study was approved by the regional ethics committee at Lund University. All participants gave their written informed consent at baseline and at each follow-up.
2.6. Statistical analysis
IBM SPSS statistics version 22 was used for statistical analysis. Nonparametric tests were used because of the low number of cases and the nonnormal (Gaussian) distribution of CSF biomarker levels. Differences between diagnosis groups were calculated with Kruskal-Wallis test followed by Mann-Whitney U test when applicable. Mann-Whitney U test was used for dichotomized variables. Paired samples were analyzed using Wilcoxon signed-rank test. Cox regression models were set for prediction of AD and DLB as well as cognitive impairment. Baseline CSF levels and baseline delayed word recall score were added separately to the model with the following covariates: age, gender, and presence of apolipoprotein E (APOE) ε4 allele. Finally, a model including CSF levels, delayed word recall, and all covariates were created. Results are presented as hazard ratio (HR) with the 95% confidence interval (CI). For baseline CSF levels, standardized z-scores were used so that HR reflects change per one standard deviation (SD). Cox regression models were performed on all participants with baseline CSF measurements and at least one cognitive follow-up (n = 53), thus including the individuals that died during the follow-up period.
To estimate predictive ability, sensitivity, specificity, positive predictive value, and negative predictive value were calculated. Because of the bimodal distribution of Aβ42, this measure was dichotomized using 192 ng/L as a cutoff (Supplementary Fig. 1), which is the same as suggested by Shaw et al. [29]. In addition, Kaplan-Meier survival curves are used for temporal visualization of conversion to dementia diagnosis. Significance level is set to P < .05.
3. Results
3.1. Participants
Forty-four participants (n = 44) were included in the main analyses of the present study, as described in Fig. 1. Demographics, cognitive performance, and CSF biomarker levels are presented in Table 1. During the clinical follow-up period in total, 12 individuals (27%) developed cognitive impairment (Table 1).
Table 1.
Group characteristics at baseline and at follow-up year 5 and year 9, including follow-up diagnoses
| Characteristics | Baseline | Follow-up year 5 | Follow-up year 9 |
|---|---|---|---|
| Baseline demographics | |||
| Number (n) | 44 | ||
| Sex (F/M) | 29/15 | ||
| Age (y) | 72 (65–78) | ||
| Education (y) | 11 (9–15) | ||
| APOE ε4 (n) (homo-/heterozygous) | 1/14 | ||
| Follow-up time (mo) | 111 (110–112) | ||
| Cognitive tests | |||
| MMSE (points) | 30.0 (29.0–30.0) | 29.0 (27.0–29.0) | 28.0 (26.0–29.0) |
| AQT (s)∗ | 62.0 (52.5–69.0)† | 61.0 (53.5–74.5) | 67.0 (56.5–84.0) |
| Delayed memory (ADAS), correct words of 10 | 8.0 (7.0–9.0) | 8.0 (7.0–9.0) | 8.0 (6.0–9.0) |
| CSF biomarkers | n = 44 | n = 32 | n = 27‡ |
| Aβ42 | 252.0 (174.5–312.0) | 230.0 (180.5–292.5) | 238.0 (139.0–275.0) |
| T-tau | 67.5 (55.5–92.0) | 74.0 (58.0–99.0) | 79.0 (58.0–111.0) |
| P-tau | 27.0 (21.5–43.5) | 35.0 (21.0–50.0) | 29.0 (22.0–41.0) |
| Follow-up diagnosis, n | n = 44 (53)§ | ||
| Normal | 32 (39) | ||
| Mild cognitive impairment | 4 (5) | ||
| Alzheimer's dementia | 5 (5) | ||
| Lewy body dementia | 1 (1) | ||
| Vascular dementia | 1 (1) | ||
| Other dementia | 1 (1) | ||
Abbreviations: APOE, apolipoprotein E; MMSE, mini-mental state examination; AQT, a quick test; ADAS, Alzheimer's disease assessment scale; CSF, cerebrospinal fluid; Aβ42, β-amyloid 42; T-tau, total tau; P-tau, phosphorylated tau.
NOTE. Median values with 25th–75th interquartile range within brackets.
Color-form version of AQT is stated (reference value <70 s).
AQT was not performed at baseline and, therefore, values from follow-up year 3 are given.
Not all have CSF measurement from year 5, which results in 36 participants with two CSF assessments and 23 participants with all three CSF assessments. CSF levels are given in ng/L.
Numbers within brackets include premortem diagnosis for participants who deceased during follow-up and that were included in the Cox regression model.
3.2. Prediction of AD or DLB using baseline CSF biomarkers
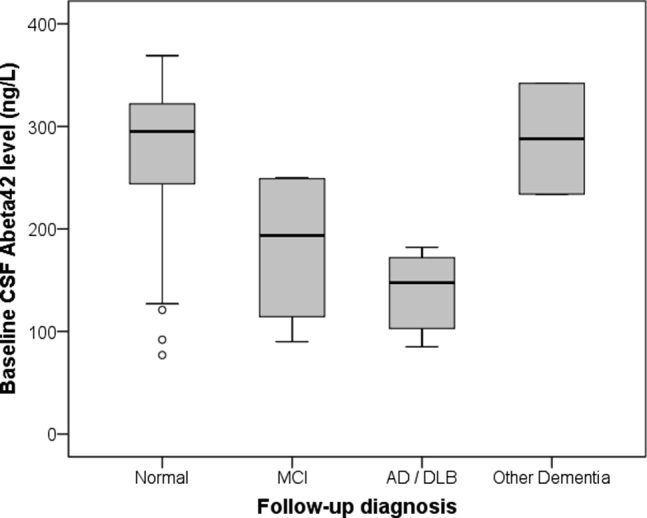
Individuals who developed AD or DLB during follow-up had lower CSF Aβ42 levels at baseline compared with the cognitive stable individuals and OD individuals, with MCI at intermediate levels (χ2, 12.2; degree of freedom [df], 3; P < .01; Supplementary Fig. 2). No differences in T-tau or P-tau levels were seen.
Twelve cognitively healthy individuals (27%) had CSF Aβ42 levels at baseline below the cutoff of <192 ng/L [29] and were, hence, interpreted as having pathologic levels. Six individuals in this group (50%) had developed AD (n = 5) or DLB (n = 1) after 9 years of follow-up. In contrast, no one with normal baseline CSF Aβ42 levels had developed AD or DLB. Thus, CSF Aβ42 predicted development of AD or DLB within 9 years with a sensitivity of 100%, specificity of 84%, positive predictive value of 50%, and negative predictive value of 100%.
Fig. 2A shows Kaplan-Meier survival curves of low baseline CSF Aβ42 levels compared with normal levels for prediction of subsequent development of AD or DLB. Cox regression model revealed that cognitively healthy individuals with low baseline Aβ42 levels had an increased risk of subsequent AD or DLB with an HR of 11.9 (95% CI, 1.5–94.2; P < .05) for each SD decrease of CSF Aβ42 levels when adjusted for age, gender, presence of APOE ε4 allele, and baseline delayed word recall. In contrast, CSF tau and P-tau did not predict development of AD/DLB over 9 years and the ratios of CSF Aβ42/tau or Aβ42/P-tau did not improve the predictive ability compared with CSF Aβ42 alone.
Fig. 2.

Kaplan-Meier curves for development of cognitive diagnosis depending on CSF Aβ42 status (A) Development of Alzheimer's dementia and dementia with Lewy bodies during follow-up in the group with low baseline CSF Aβ42 levels compared with the group with normal levels. (B) Development of cognitive impairment (AD, DLB, VaD, or MCI) during follow-up in the group with low baseline CSF Aβ42 levels compared with the group with normal levels. n = 44. Abbreviations: CSF, cerebrospinal fluid; Aβ42, β-amyloid 42; AD, Alzheimer's disease; DLB, dementia with Lewy bodies; MCI, mild cognitive impairment.
3.3. Prediction of MCI or dementia using baseline CSF biomarkers
Twelve individuals (27%) developed some form of cognitive impairment during the 9-year follow-up (Table 1). Of these, eight had low CSF Aβ42 at baseline (two MCI, five AD, and one DLB) and four had normal baseline levels (two MCI, one vascular dementia, and one other type of dementia). Thus, CSF Aβ42 predicted cognitive impairment in general (MCI or dementia) with a sensitivity of 67%, specificity of 88%, positive predictive value of 67%, and negative predictive value of 88%. Cox regression model revealed that cognitively healthy individuals with low baseline Aβ42 levels had an increased risk of subsequent cognitive impairment with an adjusted HR of 3.9 (95% CI, 1.8–8.4; P < .05) for each SD decrease of CSF Aβ42 (Fig. 2B).
3.4. Prediction of AD or DLB using baseline cognitive tests
No difference in baseline cognitive test results was seen between the individuals that developed AD/DLB at follow-up compared with those who remained cognitively stable (P > .05). However, the group of individuals that developed AD/DLB had significantly lower delayed memory scores already at the 5-year follow-up compared with those in the other groups (χ2, 8.46; df, 3; P < .05). When added to the Cox regression model, baseline delayed word recall score (ADAS-cog item 3) contributed to predict development of AD and DLB with HR of 2.8 (95% CI, 1.1–6.7; P < .05) for each not correctly given word. However, this predictive ability disappeared if CSF Aβ42 levels were removed from the model. This is in contrast to CSF Aβ42 that remained an independent predictive factor irrespective of delayed word recall. Hence, CSF Aβ42 levels is the driving predictive factor 9 years before diagnosis in this cohort, and episodic memory only contributes if amyloid status is taken into account.
3.5. Low baseline CSF Aβ42 levels and cognitive stability over 9 years
Individual demographics and cognitive follow-up data, for each of the participants with low baseline CSF Aβ42 level, are specified in Table 2. Note that 4 of 12 individuals (33%) exhibited no cognitive symptoms and performed well on cognitive testing even after 9 years of follow-up. On the other hand, these four individuals had higher CSF P-tau levels at both baseline (U = 96.0; P < .001) and at follow-up after 9 years (U = 41.5; P < .05) compared with the cognitively normal individuals with normal baseline CSF Aβ42 levels.
Table 2.
Individual follow-up cognitive diagnoses, performances, and baseline demographics for the subgroup of participants with low baseline CSF Aβ42 levels
| Participant | Follow-up year 9 |
Demography |
Baseline |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | Year of diagnosis | Age | MMSE (points) | Delayed word recall (correct of 10) | AQT (s)∗ | Clock test (correct) | Sex | Education (y) | APOE genotype | Aβ42 (ng/L) | T-tau (ng/L) | P-tau (ng/L) | MMSE (points) | Delayed word recall (correct of 10) | |
| A | AD | 2012 | 84 | 19 | 3 | 91 | Yes | Female | 7.0 | 3/4 | 130 | 97 | 49 | 30 | 7 |
| B | AD | 2012 | 93 | 21 | 2 | 83 | No | Female | 7.0 | 3/4 | 103 | 102 | 61 | 29 | 9 |
| C | AD | 2007† | 80 | 21 | 0 | 150 | No | Male | 9.0 | 3/4 | 172 | 45 | 26 | 29 | 8 |
| D | AD | 2010 | 92 | 21 | 0 | 176 | No | Female | 13.0 | 3/3 | 182 | 33 | 11 | 30 | 5 |
| E | AD | 2007† | 83 | 12 | 0 | 184 | No | Female | 9.0 | 3/4 | 85 | 157 | 55 | 30 | 6 |
| F | DLB | 2010† | 82 | 30 | 7 | 80 | No | Male | 18.0 | 3/3 | 165 | 94 | 45 | 30 | 8 |
| G | MCI | 2012 | 83 | 25 | 6 | 121 | No | Female | 8.0 | 3/3 | 90 | 62 | 25 | 30 | 10 |
| H | MCI | 2012 | 91 | 28 | 7 | 146 | Yes | Male | 12.5 | 3/4 | 139 | 82 | 34 | 28 | 8 |
| I | Normal | NA | 88 | 29 | 7 | 68 | Yes | Male | 16.5 | 3/3 | 77 | 70 | 62 | 30 | 8 |
| J | Normal | NA | 81 | 28 | 6 | 50 | Yes | Female | 15.0 | 3/3 | 127 | 145 | 74 | 28 | 7 |
| K | Normal | NA | 81 | 30 | 9 | 71 | Yes | Female | 7.0 | 3/4 | 121 | 139 | 69 | 30 | 10 |
| L | Normal | NA | 72 | 29 | 10 | 40 | Yes | Male | 10.0 | 3/3 | 92 | 63 | 45 | 27 | 9 |
Abbreviations: CSF, cerebrospinal fluid; Aβ42, β-amyloid 42; MMSE, mini-mental state examination; AQT, a quick test; APOE, apolipoprotein E; T-tau, total tau; P-tau, phosphorylated tau; AD, Alzheimer's disease; DLB, dementia with Lewy bodies; MCI, mild cognitive impairment; NA, not applicable.
NOTE. Year of diagnosis state when the participant was given its first cognitive diagnosis.
Color-form version of AQT is stated (reference value <70 s).
Year of preceding MCI diagnosis.
3.6. Longitudinal CSF biomarker levels over 9 years
Thirty-six of the individuals with CSF measurements at baseline had at least one repeated CSF acquisition during follow-up period (year 5 or year 9), of which 23 individuals had from all three occasions (Fig. 3 and Supplementary Figs. 3 and 4). On group level, there was a decrease in CSF Aβ42 levels (T = 85.0; P < .05) and an increase in CSF T-tau levels (T = 329.5; P < .01) between baseline and follow-up after 9 years, whereas CSF P-tau remained stable. Baseline CSF Aβ42 levels correlated negatively with baseline CSF P-tau levels (rs = −0.48, P < .001). Baseline CSF P-tau levels also correlated negatively with CSF Aβ42 levels at follow-up (rs = −0.61, P < .001). Moreover, baseline CSF P-tau levels were significantly higher in the individuals with low baseline CSF Aβ42 when compared with those with normal baseline CSF Aβ42 (U = 310.0; P < .01), but not at follow-up 9 years later.
Fig. 3.

Temporal development of CSF Aβ42 levels for each individual divided according to follow-up cognitive diagnoses n = 36 individuals. CSF: all occasions = 23 individuals, baseline + year 5 = 9 individuals, baseline + year 9 = 4 individuals. Cognitive groups: (A) Normal-Normal n = 26 individuals, (B) Normal-MCI n = 3 individuals, (C) Normal-AD/DLB n = 5 individuals, and (D) Normal-other dementia n = 2 individuals. Green dotted line represents change of mean value for each group with more than four participants. Red dotted line indicates cutoff 192 ng/L, suggested by Shaw et al [29]. Abbreviations: CSF, cerebrospinal fluid; Aβ42, β-amyloid 42; MCI, mild cognitive impairment; AD, Alzheimer's disease; DLB, dementia with Lewy bodies.
Low baseline CSF Aβ42 levels were observed in 10 of these individuals and no further decrease over time occurred (Fig. 3). Instead, the CSF Aβ42 levels were stably decreased in these 10 individuals during follow-up, of which 7 individuals had developed AD/DLB or MCI at the 9-year follow-up visit. In contrast, 5 of the remaining 26 individuals (19%) converted from normal CSF Aβ42 levels at baseline to pathologic low levels during follow-up. Compared with those with normal CSF Aβ42 levels also at follow-up, these individuals had higher CSF P-tau levels at baseline (U = 95.0, P < .05) as well as at follow-up after 9 years (U = 67.0, P < .01), whereas follow-up cognitive test results, age, sex, APOE genotype, and education did not differ.
Hence, significantly higher baseline and follow-up CSF P-tau levels were observed in all cognitively stable participants with low CSF Aβ42 levels (at baseline or converters during follow-up) compared with those cognitively stable with consistent normal CSF Aβ42 levels. Finally, we observe that all individuals with MCI and AD/DLB diagnoses tend to increase over time in CSF T-tau levels even though this was not statistically significant (Supplementary Figs. 3 and 4).
4. Discussion
To our knowledge, no previous study has measured CSF biomarkers repeatedly over an extended period during the preclinical phases of sporadic AD, which is needed to be able to determine the trajectories of biomarker changes during the preclinical phase of sporadic AD. In the present study, the data suggest that CSF Aβ42 is decreased up to 9 years before dementia onset and does not decrease further during this preclinical phase, i.e. CSF Aβ42 has already plateaued down to fully decreased level 9 years before AD dementia onset. Indeed, the current results do actually imply that the decrease in CSF Aβ42 occurs more than a decade before onset of sporadic AD/DLB because several cases with low CSF Aβ42 at baseline did not develop MCI or dementia during the 9-year follow-up and none of the individuals that converted from normal to low Aβ42 levels during follow-up did develop MCI or AD. These data are in agreement with data obtained from studies evaluating CSF Aβ42 levels in asymptomatic cases with autosomal dominant AD [1], [5]. The present data extend knowledge obtained from studies showing that CSF Aβ42 levels are quite stable during the dementia and MCI phases of AD [30], [31]. The annual incidence rate of CSF Aβ42 conversion was 2% in this study, which supports that CSF Aβ42 becomes reduced during adulthood rather than childhood/adolescence.
The changes in CSF tau and P-tau might be more subtle than the change in CSF Aβ42 during the preclinical stages of AD, and consequently more difficult to reliably detect in CSF [3], [32]. In the present study, development of AD/DLB is not associated with baseline CSF tau or P-tau levels. However, the individuals that developed AD/DLB had a quite slow disease progression rate with relatively low tau levels even at dementia onset. Instead, lower CSF Aβ42 levels are associated with higher CSF P-tau levels throughout follow-up. Together, these data imply that CSF P-tau might change as early as Aβ42 in preclinical AD, even though the changes are less pronounced compared with those of CSF Aβ42. Similar finding has recently been observed in a study examining cases with autosomal dominant AD [5].
Diagnostic methods are needed to accurately detect preclinical AD to be able to recruit individuals with yet only limited neurodegeneration for clinical trials. The increased risk of future development of AD or DLB in cognitively healthy elderly individuals with low CSF Aβ42 levels is in agreement with previous studies [9], [10], [11], [12], [14]. However, despite a high negative predictive value (100%), several individuals with low baseline CSF Aβ42 levels did not develop AD or DLB during a follow-up period of nearly a decade. In fact, a remarkable one-third (n = 4) of the individuals with low baseline CSF Aβ42 levels remained completely cognitive intact (Table 2). We later confirmed pathologic amyloid load in neocortex in all these four individuals at follow-up using [18F]flutemetamol PET (Supplementary Material). The study, therefore, highlights the possibility for elderly individuals to harbor amyloid pathology for a long period without development of cognitive dysfunction. Our findings, hence, demonstrate the difficulties of using amyloid markers, such as CSF Aβ42 or amyloid PET, alone in the diagnostic workup of preclinical AD. The combination with one or preferably several markers that reflects brain dysfunction or neurodegeneration has been suggested [33], [34], such as with regional brain atrophy [35], [36], regional cerebral hypoperfusion [37], [38], decreased regional glucose metabolism [36], [39], or altered cortical connectivity (through resting state functional magnetic resonance imaging) [40]. To optimize this diagnostic workup will in the future be very important when recruiting individuals with signs of brain amyloid accumulation, but no cognitive symptoms or impairment, for clinical trials evaluating new disease-modifying therapies that might cause significant side-effects. The tolerance for side-effects in such trials should be low if inclusion criteria are used that results in recruitment of population where up to 50% in the placebo group will be free of AD after more than a decade. Therefore, there is an urgent need for multimodal studies in cognitively healthy elderly individuals evaluating different diagnostic algorithms including amyloid biomarkers in combination with different biomarkers that reflect disease stage.
There are limitations to this study. First, the number of participants is too low for extended subgroup analyses. Second, cognitive impairment occurs late in the follow-up. This could indicate that the group at baseline did not represent the entire spectrum of noncognitively impaired individuals in the community. Third, only one participant developed DLB, which prevents statistical analysis of this disease group separately. However, DLB often presents amyloid pathology and presumably lies in the disease spectrum between Parkinson's disease (synucleinopathy) and AD (amyloidopathy) [28]. We, therefore, grouped AD and DLB, but the findings of this study remain also if the DLB participant is removed from this group. Fourth, CT was used for neuroimaging throughout the study, mainly owing to magnetic resonance imaging not being as accessible at the start of the study in 2002. The study was also planned primarily as a CSF biomarker study, in which CT is sufficient to exclude contraindications for lumbar puncture and to evaluate occurrence of clinically relevant cerebral lesions. Nevertheless, the study has invaluable strengths. We describe within-person changes in CSF biomarkers over a decade in healthy elderly individuals. Because a subgroup developed AD, we could also study these within-person changes during the preclinical (asymptomatic) stages of AD. All conversions to dementia occurred between follow-up year 5 and year 9 and would not have been identified with shorter follow-up. Hence, secondary end points are replaced by primary end points (i.e. dementia diagnosis). Finally, 44 of 54 individuals (81%) had clinical follow-up evaluation, which is very high in the light of 9 years of follow-up in an older population. With the medical record examination of the deceased, only two individuals (4%) of the included subjects lack 9-year follow-up. Hence, the results reflect the actual state of this entire study population.
5. Conclusions
Low CSF Aβ42 levels predict development of AD at least 9 years before dementia onset with a very high negative predictive value. Data obtained from the repeated CSF measurements imply that CSF Aβ42 levels have decreased and reached a plateau already a decade before clinical onset of dementia in sporadic AD. Surprisingly, many individuals can harbor brain amyloid accumulation over a decade without any signs of cognitive deterioration, which will clearly implicate how CSF biomarkers are used to identify preclinical AD in future interventional therapeutic trials.
Research in context.
-
1.
Systematic review: Traditional sources (such as PubMed) were reviewed with focus on predicting sporadic dementia in cognitively healthy individuals. A handful publications have investigated these elongated preclinical stages, although very few with follow-up exceeding 5 years. Instead, current evidence is mainly derived from evaluating autosomal dominant Alzheimer's disease (AD).
-
2.
Interpretation: Our findings improve the understanding of how cerebrospinal fluid AD-biomarkers can be interpreted in absence of cognitive symptoms. They align with current hypothesis that β-amyloid is an early marker of AD but also confirm difficulties of interpreting their predictive relevance on individual level. This could implicate their use for identifying preclinical AD in future therapeutic trials.
-
3.
Future directions: The article elucidates the need for long follow-up in preclinical AD studies. This to clarify (1) how long individuals can harbor brain amyloid accumulation without cognitive impact and (2) which biomarker combination that correctly identifies preclinical AD with impending risk of dementia conversion.
Acknowledgments
The authors acknowledge Åsa Wallin for participating in the diagnosis consensus group and Karina Simonsen for assistance at the 9-year follow-up visits. Both affiliated to the Memory Clinic at Skåne University Hospital, Malmö, Sweden.
The study was supported by the European Research Council, the Swedish Research Council, the Crafoord Foundation, the Swedish Brain Foundation, the Johan and Jakob Söderberg's Foundation, “Stiftelsen Gamla Tjänarinnor”, “Fredrik and Ingrid Thurings Stiftelse”, the Swedish Federal Government under the ALF agreement, and Swedish Brain Power. The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; or preparation, review, or approval of the article; or decision to submit the article for publication. No industry sponsorship/funding have taken place.
Footnotes
E.S., L.M., H.Z., and O.H. report no disclosures. K.B. is on the advisory board for Roche Diagnostics and for IBL International. All authors declare that they have no conflict of interest related to this study.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.dadm.2015.09.002.
Contributor Information
Erik Stomrud, Email: erik.stomrud@med.lu.se.
Oskar Hansson, Email: oskar.hansson@med.lu.se.
Supplementary data
Supplementary Fig. 1.

Supplementary Fig. 2.
References
- 1.Bateman R.J., Xiong C., Benzinger T.L., Fagan A.M., Goate A., Fox N.C. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buchhave P., Minthon L., Zetterberg H., Wallin A.K., Blennow K., Hansson O. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69:98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- 3.Jack C.R., Jr., Holtzman D.M. Biomarker modeling of Alzheimer's disease. Neuron. 2013;80:1347–1358. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villemagne V.L., Burnham S., Bourgeat P., Brown B., Ellis K.A., Salvado O. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: A prospective cohort study. Lancet Neurol. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 5.Fagan A.M., Xiong C., Jasielec M.S., Bateman R.J., Goate A.M., Benzinger T.L. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer's disease. Sci Transl Med. 2014;6:226ra30. doi: 10.1126/scitranslmed.3007901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouwman F.H., van der Flier W.M., Schoonenboom N.S., van Elk E.J., Kok A., Rijmen F. Longitudinal changes of CSF biomarkers in memory clinic patients. Neurology. 2007;69:1006–1011. doi: 10.1212/01.wnl.0000271375.37131.04. [DOI] [PubMed] [Google Scholar]
- 7.Toledo J.B., Xie S.X., Trojanowski J.Q., Shaw L.M. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013;126:659–670. doi: 10.1007/s00401-013-1151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertens D., Knol D.L., Scheltens P., Visser P.J., Alzheimer's Disease Neuroimaging Initiative Temporal evolution of biomarkers and cognitive markers in the asymptomatic, MCI, and dementia stage of Alzheimer's disease. Alzheimers Dement. 2015;11(5):511–522. doi: 10.1016/j.jalz.2014.05.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gustafson D.R., Skoog I., Rosengren L., Zetterberg H., Blennow K. Cerebrospinal fluid beta-amyloid 1-42 concentration may predict cognitive decline in older women. J Neurol Neurosurg Psychiatr. 2007;78:461–464. doi: 10.1136/jnnp.2006.100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fagan A.M., Roe C.M., Xiong C., Mintun M.A., Morris J.C., Holtzman D.M. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 11.Roe C.M., Fagan A.M., Grant E.A., Hassenstab J., Moulder K.L., Maue Dreyfus D. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology. 2013;80:1784–1791. doi: 10.1212/WNL.0b013e3182918ca6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vos S.J., Xiong C., Visser P.J., Jasielec M.S., Hassenstab J., Grant E.A. Preclinical Alzheimer's disease and its outcome: A longitudinal cohort study. Lancet Neurol. 2013;12:957–965. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stomrud E., Hansson O., Zetterberg H., Blennow K., Minthon L., Londos E. Correlation of longitudinal cerebrospinal fluid biomarkers with cognitive decline in healthy older adults. Arch Neurol. 2010;67:217–223. doi: 10.1001/archneurol.2009.316. [DOI] [PubMed] [Google Scholar]
- 14.Moghekar A., Li S., Lu Y., Li M., Wang M.C., Albert M. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology. 2013;81:1753–1758. doi: 10.1212/01.wnl.0000435558.98447.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folstein M.F., Folstein S.E., McHugh P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 16.Shulman K.I. Clock-drawing: is it the ideal cognitive screening test? Int J Geriatr Psychiatry. 2000;15:548–561. doi: 10.1002/1099-1166(200006)15:6<548::aid-gps242>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 17.Rosen W.G., Mohs R.C., Davis K.L. A new rating scale for Alzheimer's disease. Am J Psychiatry. 1984;141:1356–1364. doi: 10.1176/ajp.141.11.1356. [DOI] [PubMed] [Google Scholar]
- 18.Jacobson J.M., Nielsen N.P., Minthon L., Warkentin S., Wiig E.H. Multiple rapid automatic naming measures of cognition: Normal performance and effects of aging. Percept Mot Skills. 2004;98:739–753. doi: 10.2466/pms.98.3.739-753. [DOI] [PubMed] [Google Scholar]
- 19.Troyer A.K., Leach L., Strauss E. Aging and response inhibition: Normative data for the Victoria Stroop Test. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2006;13:20–35. doi: 10.1080/138255890968187. [DOI] [PubMed] [Google Scholar]
- 20.Reitan R.M. The relation of the trail making test to organic brain damage. J Consult Psychol. 1955;19:393–394. doi: 10.1037/h0044509. [DOI] [PubMed] [Google Scholar]
- 21.Smith A. Western Psychological Services; Los Angeles, CA: 1991. Symbol digit modalities test. [Google Scholar]
- 22.Strauss E., Sherman E.M.S., Spreen O. 3rd ed. Oxford University Press; New York, NY: 2006. A compendium of neuropsychological tests: Administration, norms, and commentary. [Google Scholar]
- 23.Petersen R.C. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 24.McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer's disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 25.Erkinjuntti T., Inzitari D., Pantoni L., Wallin A., Scheltens P., Rockwood K. Research criteria for subcortical vascular dementia in clinical trials. J Neural Transm Suppl. 2000;59:23–30. doi: 10.1007/978-3-7091-6781-6_4. [DOI] [PubMed] [Google Scholar]
- 26.McKeith I.G., Dickson D.W., Lowe J., Emre M., O'Brien J.T., Feldman H. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 27.American Psychiatric Association . American Psychiatric Association; Washington D.C.: 1994. Diagnostic and statistical manual of mental disorders. Fourth Edition (DSM-IV) [Google Scholar]
- 28.McKeith I., Mintzer J., Aarsland D., Burn D., Chiu H., Cohen-Mansfield J. Dementia with Lewy bodies. Lancet Neurol. 2004;3:19–28. doi: 10.1016/s1474-4422(03)00619-7. [DOI] [PubMed] [Google Scholar]
- 29.Shaw L.M., Vanderstichele H., Knapik-Czajka M., Clark C.M., Aisen P.S., Petersen R.C. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchhave P., Blennow K., Zetterberg H., Stomrud E., Londos E., Andreasen N. Longitudinal study of CSF biomarkers in patients with Alzheimer's disease. PLoS One. 2009;4:e6294. doi: 10.1371/journal.pone.0006294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Bastard N., Aerts L., Sleegers K., Martin J.J., Van Broeckhoven C., De Deyn P.P. Longitudinal stability of cerebrospinal fluid biomarker levels: fulfilled requirement for pharmacodynamic markers in Alzheimer's disease. J Alzheimers Dis. 2013;33:807–822. doi: 10.3233/JAD-2012-110029. [DOI] [PubMed] [Google Scholar]
- 32.Braak H., Zetterberg H., Del Tredici K., Blennow K. Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-beta changes occur before increases of tau in cerebrospinal fluid. Acta Neuropathol. 2013;126:631–641. doi: 10.1007/s00401-013-1139-0. [DOI] [PubMed] [Google Scholar]
- 33.Lista S., Garaci F.G., Ewers M., Teipel S., Zetterberg H., Blennow K. CSF Abeta1-42 combined with neuroimaging biomarkers in the early detection, diagnosis and prediction of Alzheimer's disease. Alzheimers Dement. 2014;10:381–392. doi: 10.1016/j.jalz.2013.04.506. [DOI] [PubMed] [Google Scholar]
- 34.Drago V., Babiloni C., Bartres-Faz D., Caroli A., Bosch B., Hensch T. Disease tracking markers for Alzheimer's disease at the prodromal (MCI) stage. J Alzheimers Dis. 2011;26(Suppl 3):159–199. doi: 10.3233/JAD-2011-0043. [DOI] [PubMed] [Google Scholar]
- 35.Kohannim O., Hua X., Hibar D.P., Lee S., Chou Y.Y., Toga A.W. Boosting power for clinical trials using classifiers based on multiple biomarkers. Neurobiol Aging. 2010;31:1429–1442. doi: 10.1016/j.neurobiolaging.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mormino E.C. The relevance of beta-amyloid on markers of Alzheimer's disease in clinically normal individuals and factors that influence these associations. Neuropsychol Rev. 2014;24:300–312. doi: 10.1007/s11065-014-9267-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansson O., Buchhave P., Zetterberg H., Blennow K., Minthon L., Warkentin S. Combined rCBF and CSF biomarkers predict progression from mild cognitive impairment to Alzheimer's disease. Neurobiol Aging. 2009;30:165–173. doi: 10.1016/j.neurobiolaging.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 38.Mattsson N., Tosun D., Insel P.S., Simonson A., Jack C.R., Jr., Beckett L.A. Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer's disease and mild cognitive impairment. Brain. 2014;137:1550–1561. doi: 10.1093/brain/awu043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prestia A., Caroli A., van der Flier W.M., Ossenkoppele R., Van Berckel B., Barkhof F. Prediction of dementia in MCI patients based on core diagnostic markers for Alzheimer disease. Neurology. 2013;80:1048–1056. doi: 10.1212/WNL.0b013e3182872830. [DOI] [PubMed] [Google Scholar]
- 40.Wang L., Brier M.R., Snyder A.Z., Thomas J.B., Fagan A.M., Xiong C. Cerebrospinal fluid Abeta42, phosphorylated Tau181, and resting-state functional connectivity. JAMA Neurology. 2013;70:1242–1248. doi: 10.1001/jamaneurol.2013.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.