

Summary
The role of platelets in hemostasis and thrombosis is dependent on a complex balance of activatory and inhibitory signaling pathways. Inhibitory signals released from the healthy vasculature suppress platelet activation in the absence of platelet receptor agonists. Activatory signals present at a site of injury initiate platelet activation and thrombus formation; subsequently, endogenous negative signaling regulators dampen activatory signals to control thrombus growth. Understanding the complex interplay between activatory and inhibitory signaling networks is an emerging challenge in the study of platelet biology, and necessitates a systematic approach to utilize experimental data effectively. In this review, we will explore the key points of platelet regulation and signaling that maintain platelets in a resting state, mediate activation to elicit thrombus formation, or provide negative feedback. Platelet signaling will be described in terms of key signaling molecules that are common to the pathways activated by platelet agonists and can be described as regulatory nodes for both positive and negative regulators.
Keywords: blood platelets, hemostasis, platelet activation, review, thrombosis
Introduction
The primary role of platelets lies in their ability to form aggregates when they encounter areas of blood vessel damage, allowing them to limit blood loss by forming a hemostatic clot. The capacity of platelets to aggregate also forms the basis of their role in disease, whereby platelet thrombus formation is triggered by an inappropriate stimulus such as the rupture of an atherosclerotic plaque. If the thrombus causes vessel occlusion in the heart or brain, this can result in a heart attack or stroke. The way in which platelets respond to their environment, often referred to as ‘platelet function’, defines their ability to fulfill their role in hemostasis and also their propensity to contribute towards disease. The environmental stimuli encountered by platelets are complex, as are the intracellular signaling pathways that regulate their responses to these stimuli.
To understand how platelets function within the vasculature, the three types of signals encountered by platelets and the signaling responses that they trigger must be considered simultaneously. Platelets encounter inhibitory signals that keep them in a quiescent state in the healthy vasculature, activatory signals at sites of vascular damage that rapidly trigger adhesion and aggregation, and negative regulatory signals that provide negative feedback once platelet activation is initiated and serve to limit thrombus formation (Fig. 1). There is a problem, however, with the conventional models used to understand platelet signaling. Platelets experience numerous extracellular signals at the same time and respond to complex combinations of primary and secondary activatory signals in addition to opposing inhibitory signals, yet we conceptualize such regulation as distinct linear pathways, each of which is triggered in response to a single agonist. This simplified approach is useful for designing experiments to delineate the order of events, but a more sophisticated approach is needed to incorporate multiple agonists and pathways, which themselves crosstalk, into a signaling network. Understanding these complex interacting signaling pathways as networks is a challenge that can only ultimately be met by systematic approaches that seek to model complex systems with many interacting parts. Systematic strategies that enable a holistic view of platelet function will be important to support the development of new antiplatelet therapies and new diagnostic methods, and successful prediction of hemostatic side effects for non‐platelet‐targeting drugs.
Figure 1.
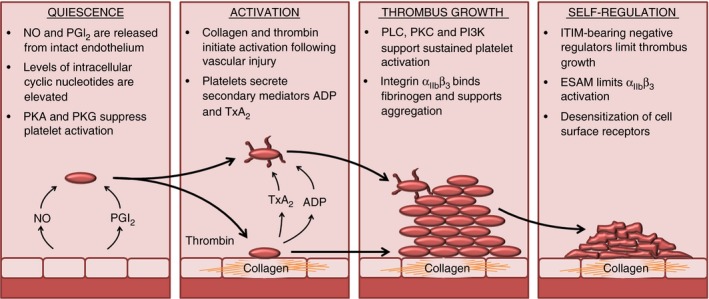
Stages of platelet activation and thrombus formation. Platelets in the circulation are kept in a quiescent state by nitric oxide (NO) and prostacyclin (prostaglandin I2; PGI 2), which are released by the vascular endothelium. In platelets, NO and PGI 2 increase the levels of cGMP and cAMP, and suppress platelet activity by the activation of protein kinase A (PKA) and protein kinase G (PKG). Following vessel injury, components of the subendothelial matrix are exposed, including collagen, which provides an adhesive surface for platelets to attach to and initiate signaling events and platelet activation. Local production of thrombin and secretion of secondary mediators also contribute to the initiation of platelet activation. Key components of platelet signaling pathways are activated, including phospholipase C (PLC), protein kinase C (PKC), and phosphatidylinositide‐3‐kinase (PI3K), supporting sustained platelet activation and thrombus formation through the initiation of cytoskeletal rearrangements, granule secretion, and activation of integrin αII bβ3. So as to limit thrombus growth and prevent the formation of occlusive thrombi, platelets contain self‐regulating negative feedback mechanisms that counteract positive signaling. These negative regulators include immunoreceptor tyrosine‐based inhibition motif (ITIM)‐containing receptors, endothelial cell‐selective adhesion molecule (ESAM), which negatively regulates integrin αII bβ3 activity, phosphatases that counteract phosphorylation‐dependent positive signaling, and receptor desensitization, which reduces the platelets’ response to secondary mediator signaling. TxA2, thromboxane A2.
Many of the complex positive and negative platelet signaling pathways converge on key regulators of platelet signaling. By understanding how platelets regulate important signaling molecules such as phospholipase C (PLC), protein kinase C (PKC), and phosphatidylinositide‐3‐kinase (PI3K), which underpin critical events in platelet activation such as granule secretion and integrin αIIbβ3 activation, we can understand the factors that define platelet function. In this review of platelet signaling pathways, we will refrain from describing signaling pathways as such, and instead focus on the impact of molecules that activate or inhibit platelet function on the core signaling processes that control platelet function, to begin to appreciate the interconnectedness of these systems.
Regulators of platelet inhibition
Within the vasculature, platelets exist in a complex, flowing environment where both rheologic and hemodynamic factors force platelets into close proximity to the vessel wall. The signals generated by healthy, undamaged blood vessels promote quiescence, and allow platelets to circulate in a resting state 1. Without inhibitory signaling mechanisms, platelets would become activated even in the absence of activating signals. Circumstances in which these inhibitory signals are defective or activatory factors are exposed can lead to inappropriate platelet activation and cause ischemic heart disease or stroke 2.
The inhibitory signaling pathways are relatively few in number, but suppress several key nodes in the platelet signaling network that support activation. The primary platelet‐inhibiting signals generated by healthy, intact vascular endothelial cells are nitric oxide (NO) and prostacyclin (prostaglandin I2 [PGI2]), which regulate levels of intracellular cyclic nucleotides that suppress platelet activity by activating protein kinase G (PKG) and protein kinase A (PKA). 2.
NO and PGI2
NO suppresses platelet activation by regulating intracellular levels of cGMP 3 via the activation of soluble guanyl cyclase, which regulates PKG activity. PGI2 binds to the prostaglandin receptor (IP receptor) on the platelet surface 4, and activates G‐protein α‐s‐coupled G‐protein‐coupled receptors (GPCRs). The active GTP‐bound form of Gs then binds to and activates adenylyl cyclase, and stimulates the production of cAMP from AMP 5, which in turn causes the activation of cAMP‐dependent PKA.
Many substrates of PKA and PKG are yet to be thoroughly characterized, but there is significant overlap in the well‐established targets. These targets include regulators of platelet activation such as Rap1b, which controls integrin αIIbβ3 affinity, and is phosphorylated at Ser179 by PKA and PKG, causing it to be relocated away from the cell membrane, and downstream effectors of platelet activation 6. Cyclic nucleotides are also strong inhibitors of the release of Ca2+ into the cytosol, which underpins many events in platelet activation, including regulation of the Ca2+‐dependent Rap1 guanine nucleotide exchange factor (GEF), CALDAG‐GEFI 7. Phosphorylation of the inositol 1,4,5‐trisphosphate (IP3) receptor by PKA and PKG inhibits its function as a Ca2+ channel, and prevents Ca2+ release into the cytosol from the dense tubular system (DTS) 2. Targets specific to PKA include proteins involved in cell adhesion and spreading, such as Gα13 and GPIbβ. cGMP regulates cyclic nucleotide levels by activating phosphodiesterase (PDE)2A and PDE5A, which degrade cAMP and cGMP, respectively, but also inhibits PDE3A activity, reducing constitutive cAMP degradation 2.
Platelets from humans deficient in PKG have enhanced Ca2+ responses to agonists, indicating a role for cGMP‐mediated inhibition of Ca2+ release pathways 8. Mice lacking PKG have a prothrombotic phenotype and increased intravascular adhesion and aggregation following ischemia 9. Mice deficient in the IP receptor show enhanced platelet activation, highlighting the importance of Gs‐coupled receptors that regulate cAMP in suppressing platelet activation 10. Additionally, deletion of the IP receptor in atherosclerosis‐prone apolipoprotein E‐deficient mice significantly accelerates the development of atherosclerosis, which may be attributable, in part, to increased platelet reactivity 11.
CD39
The endothelium and red blood cells can release several molecules, such as ADP and ATP, that are capable of stimulating platelets, and removal of these molecules is essential for the prevention of unwanted activation. CD39, an ectonucleotidase found at the platelet membrane and on endothelial cells, prevents platelet activation by hydrolyzing secreted ADP and ATP to AMP and adenosine 12. Adenosine activates the Gs‐coupled adenosine receptor, causing inhibition of platelets through elevation of cAMP 13.
Junctional adhesion molecule A (JAM‐A)
JAM‐A, a member of the immunoglobulin superfamily of surface membrane proteins, has been identified in human platelets, and is thought to prevent platelet aggregation and thrombus formation in resting platelets through inactivation of integrin αIIbβ3. In resting platelets, JAM‐A is phosphorylated and associates with integrin αIIbβ3 to allow the recruitment of C‐terminal src kinase (Csk). Csk regulates autoinhibition of c‐Src through phosphorylation of the inhibitory Tyr529 site of c‐Src, thus preventing c‐Src‐dependent phosphorylation and activation of integrin αIIbβ3 14, 15. Upon strong agonist stimulation, JAM‐A is dephosphorylated, and this removes the brake on integrin αIIbβ3 activation. As a consequence of this, mice deficient in JAM‐A show a hyper‐reactive platelet phenotype 15.
Key regulators of activation
Following damage to the vascular endothelium, endogenous inhibitory signals are overcome, enabling platelets to react instantly to limit blood loss at sites of vascular injury. Platelets located close to the vessel wall within the blood flow come into contact with exposed extracellular matrix proteins such as collagen, and this, in combination with vascular shear forces, enables them to adhere to the site of injury and aggregate to form a thrombus 1. Platelets bind to the collagen‐containing adhesive surface through glycoprotein (GP)Ib–V–XI via von Willebrand factor (VWF) in the presence of high shear forces. Activation of the platelet receptors for collagen, integrin α2β1 and GPVI enables stable adhesion and initiates platelet signaling and activation 16. The release of secondary mediators such as ADP and thromboxane A2 (TxA2) amplifies the platelet response and subsequent activation of the coagulation pathway at the platelet surface. Exposure of phosphatidylserine (PS) on the surface of highly activated platelets enables assembly of the prothrombinase complex and the generation of thrombin. Thrombin is a potent platelet activator that can stimulate and enhance platelet aggregation and thrombus formation through the protease‐activated receptors (PARs) 17. Thrombin also stabilizes the growing thrombus by cleaving fibrinogen to form a fibrin mesh.
These activating stimuli ultimately regulate a core set of signaling mediators that support activation. Three central mediator families of platelet activation are PLC, PKC, and PI3K, which are well characterized and underlie two of the critical events in platelet activation, i.e. secretion of secondary mediators and activation of integrin αIIbβ3. These are by no means the only critical regulators of platelet function, but they represent key nodes in the complex network of platelet signaling that regulate many other important signaling elements (Fig. 2) 18, 19, 20, 21.
Figure 2.
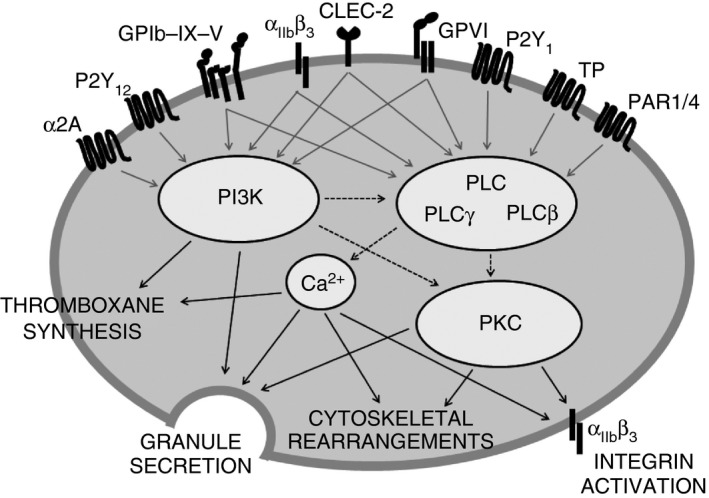
Phospholipase C (PLC), protein kinase C (PKC) and phosphatidylinositide‐3‐kinase (PI3K) are key mediators of platelet activation. All main activating platelet agonist receptors identified to date activate at least one of the following key activatory signaling mediators: PLC, PKC, or PI3K. These signaling nodes underlie several key processes required for platelet activation, including secretion of secondary mediators and activation of integrin αII bβ3, facilitating fibrinogen binding and platelet aggregation, and also cytoskeletal rearrangements, which enable platelet shape change and spreading. CLEC‐2, C‐type lectin receptor 2; GPVI, glycoprotein VI (collagen receptor); GPIb–IX–V, glycoprotein Ib–IX–V (von Willebrand factor receptor); PAR, protease‐activated receptor; P2Y1/P2Y12, ADP receptors; TP, thromboxane A2 receptor; αII bβ3, receptor for fibrinogen; α2A, adrenergic receptor.
PLC activation
Activation of PLC is a critical event in platelet activation, because it gives rise to two of the most important activatory signaling molecules in platelets. PLC family members catalyze the cleavage of phosphatidylinositol 4,5‐bisphosphate (PIP2) to generate diacylglycerol (DAG), which activates PKC, and IP3, which binds to IP3 receptors on the DTS of platelets to activate Ca2+ flux into the cytosol. Patients with low platelet PLC activity fail to respond normally to platelet agonists and even to Ca2+ ionophore, only achieving normal aggregation following addition of the direct PKC activator DiC8 22. This is because both Ca2+ release and PKC activation are critical for integrin αIIbβ3 activation and secretion. How and when the two main PLC isoforms in platelets, PLCγ2 and PLCβ, are activated therefore determines platelet activation.
PLCγ2 is activated downstream of immunoreceptor tyrosine‐based activation motif (ITAM)‐bearing (consensus sequence Yxx[L/I]x6–12Yxx[L/I]) adhesion receptors in platelets, i.e. GPVI‐FcRγ/FcγRIIA and C‐type lectin receptor 2 (CLEC‐2) (which bears a half‐ITAM motif [hemITAM]) 23. The pathway that activates PLCγ2 is complex, and involves assembly of a signaling protein complex, the linker of activated T cells (LAT) signalosome, around the scaffolding protein LAT, which is phosphorylated and activated by Syk downstream of Src family kinases 24. Activation of PLCγ2 requires binding of Slp76 to LAT via Gads, which enables Btk 25, 26 to phosphorylate PLCγ2 27. Clustering induced by collagen and fibrinogen binding to integrins α2β1 and αIIbβ3, respectively, also stimulates PLCγ2 signaling, although for this LAT is not required 28. Owing to its role in the signaling cascade of receptors that mediate interactions stimulated by matrix proteins, PLCγ2 plays a significant role in initial platelet adhesion and thrombus formation. Genetic deletion of PLCγ2 in mice results in severely diminished thrombus formation following minor laser‐induced vessel injury, whereas its role was less significant following major injury, when thrombin generation plays a greater role 29.
PLCβ is activated by Gq‐coupled GPCRs such as P2Y1 (ADP receptor), TP (TxA2 receptor), and PAR1 and PAR4 (activated by thrombin). PLCβ is an important mediator of signaling evoked by the secreted, soluble secondary mediators ADP and TxA2, and therefore mediates positive feedback signaling and recruitment of platelets to growing thrombi. The consequences of preventing PLCβ activation via gene deletion of Gq in mice is profound, because it is the major route to Ca2+ release and PKC activation in platelets that are not in direct contact with matrix proteins 29.
PKC activation
PKC constitutes a family of several different isoforms that play key regulatory roles in platelet intracellular signaling. Several isotypes/isoforms of PKC have been identified in platelets, and are divided into three different subtypes according to their structures and mechanisms of regulation: the conventional (classic) PKCs, i.e. PKCα, PKCβI/II, and PKCγ, which require both DAG and Ca2+ for full activation; the novel PKCs, i.e. PKCδ, PKCθ, PKCη, and PKCε (only in mice), which require DAG binding but are insensitive to Ca2+; and the atypical PKCs, i.e. PKCζ and PKCι/λ, which are insensitive to both DAG and Ca2+. Expression of some isoforms in platelets is controversial 19. Phosphorylation at key regulatory sites by phosphoinositide‐dependent kinase‐1 (PDK‐1) and autophosphorylation prime PKC for second messenger binding and activation 19. Studies that have utilized broad‐spectrum inhibitors and mice deficient in the individual isoforms of PKC have identified an overall positive role for the PKC family in the regulation of granule secretion, TxA2 synthesis, integrin activation, aggregation, and thrombus formation 19. Negative regulatory roles have also been identified, including roles in receptor desensitization and Ca2+ release, although some of the isoform‐specific roles remain controversial 19, 30, 31, 32. Substrates and targets of the PKC isoforms include components of the secretion machinery, such as SNAP23, syntaxin 4, and Munc18c, and cytoskeleton‐associated proteins, including pleckstrin, MARCKS, and vasodilator‐stimulated phosphoprotein (VASP), among other proteins that have been shown to be phosphorylated in a PKC‐dependent manner 19. These differences in function for the PKC family are attributed to different and distinct roles for the different isoforms in cell signaling and regulation, which could be attributable to different modes of regulation, different substrate specificities, and/or the association with distinct binding proteins and different subcellular localizations 33.
PI3K activation
Appreciation of the role of class I PI3K in platelets as a promoter of sustained activation and stable thrombus formation has developed as its different functions have become understood. All class I PI3K isoforms phosphorylate PIP2 to generate phosphatidylinositol 3,4,5‐trisphosphate (PIP3), enabling signaling to or recruitment of proteins containing PH domains to the plasma membrane. This is believed to localize signaling kinases within close proximity of their downstream effectors. One such protein is the Ser/Thr kinase protein kinase B (Akt), which has numerous targets but is believed to exert some of its effects via inhibition of glycogen synthase kinase 3 (GSK3). Inhibition of GSK3 appears to potentiate responses to thrombin but not to GPVI agonists such as collagen 34. Following stimulation with thrombin, PKCα mediates early phosphorylation of GSK3, whereas phosphorylation by Akt occurs at later time points 35. Mitogen‐activated protein kinases such as extracellular signal‐regulated kinase (ERK) are also downstream effectors of PI3K, with an apparently minor role downstream of GPVI 36, 37. PI3K has an important role in the regulation of Btk and its substrate PLCγ2 by causing them to colocalize at the plasma membrane 38.
Mice deficient in each type I PI3K expressed in platelets have been generated and characterized 39. PI3Kδ is expressed at low levels, and genetic deletion or knock‐in of a catalytically non‐functional form had little measurable impact on platelet function. PI3Kγ is predominantly involved in signal transduction downstream of P2Y12, but plays a semi‐redundant role with PI3Kβ downstream of GPCRs. PI3Kβ is the dominant form downstream of GPVI, where it has a role in PLCγ2 activation and Ca2+ elevation 40. PI3Kα is believed to function cooperatively with PI3Kβ downstream of GPVI, where it also has a role in PLCγ2 activation 40, 41, possibly through the regulation of Btk. PI3K‐dependent activation of PDK‐1 may also contribute to the regulation of PKC activation.
Key events during platelet activation
Cytoskeletal rearrangement
Platelet activation is associated with major changes in the actin cytoskeleton that initially support shape change and later allow platelets to spread once they come into contact with adhesive surfaces such as exposed collagen or other platelets. Shape change occurs rapidly when platelets are stimulated; it results in the formation of pseudopodia that increase platelet surface area, and is dependent on Ca2+ elevation mediated by Gq or activation of G13, which couples to the small GTPase Rho. Rho mediates cytoskeletal reorganization through activation of myosin light chain kinase. Cytoskeletal reorganization enables relocalization of platelet granules and organelles to the center of the platelet, the transient formation of filopodia, and the sustained formation of lamellipodia, which enables secretion and spreading over the area of vessel damage 42. For spreading to occur, activation of either PLCβ or PLCγ, mobilization of intracellular Ca2+, the generation of phosphoinositides and activation of integrin αIIbβ3 are required 43. Rapid reorganization of the actin cytoskeleton is characterized by a combination of uncapping, severing and nucleation of the actin filaments, and interaction with and activation of myosin II 44. Several regulators of these processes have been identified, including the Rho GTPases Rac, Cdc42 and RhoA, VASP, and PKC 19, 45, 46, 47.
Secretion of secondary mediators
Secretion or release of secondary mediators is critical for the positive feedback that supports clot consolidation, but can also serve as the primary activating signal for platelets encountering an established and growing thrombus. The most prominent secondary mediators are ADP and TxA2. Like integrin αIIbβ3 activation, the secretion pathways are dependent on several convergent signaling pathways activated by multiple receptors.
Granule secretion
Platelets contain different types of granule, including the dense granules, which contain high concentrations of the secondary mediators ADP and 5‐hydroxytryptamine, and the α‐granules, which contain many coagulation and adhesive proteins. The mechanisms governing the secretion of both types of platelet granule appears to be largely shared, and involve assembly of a secretion complex containing soluble NSF‐associated attachment protein receptor (SNARE) proteins such as the vesicle‐associated membrane proteins (VAMPs) 48, and SNARE regulators such as Munc13‐4 49. Ca2+ has a critical role in granule secretion in all cell types, but the precise mechanism of Ca2+‐mediated granule secretion in platelets is unclear. It is believed that the Ca2+‐sensing protein calmodulin mediates phosphorylation of myosin light chain 50, leading to interaction with VAMP and the exocytotic cell machinery, causing granule secretion 51. Slp1 inhibits dense granule secretion until the inhibition is relieved by binding of Rap1GAP2, although the role of Ca2+ in the process is unclear 52.
The DAG mimetic and PKC activator phorbol 12‐myristate 13‐acetate can induce granule secretion in the absence of intracellular Ca2+ elevation 53. Although it has long been understood that PKCs have a net positive regulatory role in granule secretion, the precise roles of the different isoforms are difficult to dissect. Negative roles for both PKCδ and PKCθ following stimulation by GPVI agonists have, however, been described 19. Many key regulators of granule secretion, such as Munc18c, syntaxin‐4, and SNAP23, are phosphorylated, and therefore potentially regulated, by PKCs 54, 55, 56.
As a consequence of the complex crosstalk between pathways, the secreted secondary mediators have a role in potentiating secretion, creating an unintuitive, cyclical relationship. For example, following GPVI activation, P2Y12 responds to secreted ADP by potentiating granule secretion via activation of Rac1, leading to an increase in ADP secretion and further activation of P2Y12 and P2Y1 57.
TxA2 synthesis
TxA2 is synthesized de novo upon platelet activation. Synthesis is mediated by a cascade of enzymes, including cyclooxygenase‐1, the target of aspirin, but it is regulated at the level of liberation of arachidonic acid from membranes by phospholipase A2. This enzyme is activated by elevated Ca2+, which induces translocation to the plasma membrane and phosphorylation by the stress kinase P38 58 and ERK1/2 59. As is the case in granule secretion, P2Y12 has a regulatory role in TxA2 synthesis, whereby stimulation of platelets with ADP causes PI3K‐dependent TxA2 generation, which potentiates Ca2+ signaling 60.
Novel secretion pathways
A novel platelet secretion mechanism has recently been reported whereby ATP is secreted from pannexin hemichannels to activate P2X1, the only ligand‐gated Ca2+ channel in platelets, causing influx of Ca2+. This enhances activation stimulated by low concentrations of GPVI agonists 61.
Integrin activation
In many ways, integrin αIIbβ3 activation represents the central point of platelet signaling pathways, as the majority of inhibitory and activatory signaling pathways contribute towards its regulation in some way. The activation of integrin αIIbβ3 is the culmination of a chain of signaling events that induce a conformational change enabling high‐affinity binding of fibrinogen and VWF. Without the capacity to bind fibrinogen, platelets are unable to adhere to each other and form aggregates. The binding partners that are assembled around integrin αIIbβ3 and support activation and association with the actin cytoskeleton include talin 62 and kindlin 63. The small GTPase Rap1 is an important regulator of integrin αIIbβ3 activation 57, 64, and appears to be the integrating point of many platelet‐activating signals. Elevation of Ca2+ modulates Rap1 activation via the highly expressed platelet GEF CalDAG‐GEFI 65, which binds Ca2+ via EF‐hand domains to become activated 66. Activation of PI3K is another central event in sustained platelet activation, and this too modulates Rap1 67. PI3K has recently been shown to act by inhibiting the GTPase‐activating protein RASA3, which negatively regulates Rap1 activation 68. This mechanism might explain why activators of PI3K, such as the ADP receptor P2Y12, are unable to elicit integrin αIIbβ3 activation unless positive regulation via Ca2+ elevation or PKC occurs simultaneously. A study using genetic reconstitution of the integrin αIIbβ3 activation pathway in cell lines, RIAM, indicated that the Rap1b effector was a critical mediator of integrin activation in platelets, linking the GTPase to integrin αIIbβ3 69. However, a recent study has demonstrated that the adaptor molecule might not have a critical role in platelets, as platelet‐specific RIAM deficiency in mice was not accompanied by a platelet functional defect 70.
Advanced microscopy techniques are now revealing the spatial and temporal regulation of integrin activation, and have demonstrated that knowledge of the signaling cascades that support activation of key platelet receptors such as integrin αIIbβ3 is only part of the story. Coordination of the cytoskeleton with clustering of adhesive receptors and mediators of activation underlies the ability of platelets to form stable aggregates under shear stress 71.
Negative feedback and self‐regulation
Following initiation of platelet activation and thrombus formation, negative signaling mechanisms restrain activation to ensure that platelet aggregation does not progress out of control, thus preventing excessive thrombus formation.
The pathways that contribute to negative regulation have been studied less extensively than those that mediate platelet activation, and consequently have not been fully characterized. Negative regulators, such as immunoreceptor tyrosine‐based inhibition motif (ITIM)‐containing receptors, are thought to reduce the activation of critical players such as PLC, PI3K, and integrin αIIbβ3 72. Other endogenous inhibitory mechanisms include endothelial cell‐selective adhesion molecule (ESAM), Wnt–β‐catenin, and semaphorin 3A (Sema3A), which negatively regulate integrin αIIbβ3 activity, phosphatases that limit phosphorylation‐dependent mechanisms, receptor desensitization, which limits the response to secondary mediator signaling, and intracellular nuclear receptors with different mechanisms of action 73, 74.
ITIM signaling
ITIM‐containing proteins are predominantly associated with negative regulation of platelet signaling and activation. The ITIM consensus sequence (L/I/V/S‐x‐Y‐x‐x‐L/V) in the cytoplasmic tail has been identified in several proteins that are expressed in platelets, including platelet endothelial cell adhesion molecule‐1 (PECAM‐1) 75, 76, carcinoembryonic antigen cell adhesion molecule‐1 (CEACAM‐1) 77, carcinoembryonic antigen cell adhesion molecule‐2 (CEACAM‐2) 78, G6b‐B 79, 80, and LILRB2/paired immunoglobulin‐like receptor B (PIRB) 81, all of which negatively regulate platelet activation. PIRB is activated by binding of its endogenous ligand, ANGPTL2, which is secreted by platelets 81. In contrast, PECAM‐1 appears to be activated by receptor clustering that is stimulated by homophilic interactions 82. For other ITIM receptors, it is not clear which type of interaction might trigger activation, but their function can be inferred from the phenotypes of mice with genetically induced deficiencies for different ITIM‐containing receptors. Activation of ITIM receptors evokes src family kinase‐dependent phosphorylation of the ITIM tyrosine residues, which enables the recruitment of negative regulators of platelet activation, including phosphatases such as SHP1/SHP2 83 and SHIP1/SHIP2, to the receptor 84. Binding to the ITIM brings these proteins in close proximity to their substrates. The resulting inactivation of tyrosine kinases such as Syk, inactivation of the LAT signalosome and PLCγ2 and inhibition of PIP3 leads to inactivation of PI3K and Akt, and inhibition of downstream signaling pathways 85. Recruitment to the ITIM can also result in the relocalization of molecules away from other signaling partners, preventing further transmission of positive signals such as that observed with PECAM‐1, which associates with PI3K and sequesters it away from LAT, preventing its activation and downstream signaling 85.
ITIM‐containing proteins were generally considered to constitute the off ‘switch’ that counteracts the positive signaling initiated by ITAM‐bearing receptors, such as GPVI‐FcRγ/FcγRIIA, and CLEC‐2 (which contains a hemITAM). However, studies on PECAM‐1 75, 76, 86, G6b‐B 79, 80 and LILRB2/PIRB 81 have identified inhibitory regulatory roles following stimulation by GPCR agonists and in the regulation of integrin αIIbβ3 function, suggesting that their role is not solely limited to inhibition of ITAM signaling. In support of an inhibitory role in the regulation and limitation of platelet activation, studies of thrombus formation in PECAM‐1 87, CEACAM‐1 77, CEACAM‐2 78 or LILRB2/PIRB 81 receptor‐deficient mice have shown that the mice have hyper‐reactive platelets, increased thrombus size and increased stability of thrombi as compared with wild‐type controls.
Interestingly, despite the negative roles of PECAM‐1 and CEACAM‐1 in the regulation of platelet activity, studies using either PECAM‐1−/− or CEACAM‐1−/− mouse platelets show impaired outside‐in signaling, as spreading and adhesion on fibrinogen, clot retraction and phosphorylation of focal adhesion kinase are reduced in comparison with wild‐type platelets 88, 89. This indicates positive roles for both PECAM‐1 and CEACAM‐1 in the regulation of integrin αIIbβ3 signaling following fibrinogen binding.
Human studies have revealed that the expression levels of PECAM‐1 on the surface of human platelets correlates negatively with platelet reactivity to CRP‐XL and ADP. The contribution of PECAM‐1 expression levels as a determinant of platelet reactivity is around half that of GPVI levels, demonstrating the importance of both activatory and inhibitory pathways in controlling responsiveness. 76.
Other negative regulators
ESAM, which is usually contained in the α‐granules, is translocated to the cell surface following platelet activation 90, and is suggested to be involved in the negative regulation of integrin αIIbβ3 outside‐in signaling, as ESAM‐deficient mouse platelets show increased aggregation in response to GPCR agonists, inhibition of clot retraction, increased thrombus formation in vivo, and reduced tail bleeding 91. The mechanism by which ESAM functions is currently unknown, although interaction with the scaffold protein NHERF‐1 highlights the possible regulation of several proteins, including GPCRs, G‐proteins, PLCβ, and components of the cytoskeleton.
The neurophillin‐1–plexin A receptor complex, which has been identified in platelets 92, is activated by Sema3A, which is secreted from endothelial cells 93 and negatively regulates platelet function through regulation of integrin αIIbβ3. Integrin activation, aggregation, adhesion and spreading are all impaired following Sema3A binding. The exact mechanisms by which Sema3A binding inhibits platelet function have yet to be fully elucidated; however, inhibition of the GTPase Rac1 could play a role 92.
Wnt3a is a GP that has been found to be released from TRAP‐stimulated platelets, enabling platelets to self‐regulate and limit activation 94. In platelets, Wnt3a is thought to activate the canonical Wnt–β‐catenin signaling pathway, as several components of this pathway have been identified in platelets 94. Wnt binding leads to the inhibition of platelet adhesion and shape change, dense granule secretion, integrin αIIbβ3 activation, and aggregation, possibly through regulation of the activity of small GTPases, including Rap1, Cdc42, Rac1, and RhoA 95.
Intracellular nuclear receptors have recently been identified in platelets and found to have a role in platelet inhibition. Usually associated with genomic regulation of transcription, these receptors have also been shown to have non‐genomic regulatory roles. Several intracellular nuclear receptors have been identified and characterized in human platelets, including peroxisome proliferator‐activating receptor (PPAR)α, PPARβ/δ, PPARγ, retinoid X receptor (RXR), liver X receptor (LXR), and glucocorticoid receptor 96, 97, 98, 99, 100. Treatment of platelets with ligands for these receptors results in inhibition of platelet aggregation in response to several platelet agonists, supporting previously published cardioprotective effects of these treatments 96, 97, 98, 99, 100, 101, 102. The different nuclear receptors can interact with each other to form heterodimers, although, to date, little is known about the interactions between these receptors in platelets. There is some overlap in the mechanisms by which they inhibit platelet activation, highlighting possible points of receptor interaction. Ligands for PPARα and PPARβ/δ appear to elicit their inhibitory effects through regulation of cellular cAMP levels 103, whereas PPARγ and LXR ligands alter association of the receptor with components of GPVI and integrin signaling pathways such as Syk, LAT, and PLCγ2 96, 98.
Protein modification by phosphorylation of tyrosine or serine and threonine residues is a key reversible mechanism of signal transduction. Dephosphorylation by phosphatases enables bidirectional regulation of signal transduction and platelet function. Phosphatases, including SHP1/SHP2, SHIP1/SHIP2, protein phosphatase (PP)2, phosphatase and tensin homolog (PTEN), and T‐cell ubiquitin ligand‐2 (TULA2), are integral mediators of several negative signaling mechanisms in platelets preventing key platelet processes such as mobilization of intracellular Ca2+, granule secretion, and integrin activation. SHP2 has well‐established roles in the negative regulation of platelet signaling downstream of both GPCRs and GPVI, and is a key mediator of ITIM receptor signaling downstream of PECAM‐1 and CEACAM‐1, negatively regulating proximal signaling events, leading to reduced PLC activation and therefore reduced mobilization of Ca2+ 73. TULA2 dephosphorylates and inactivates Syk, preventing signaling that has been shown in mathematical modeling studies to be critical in determining the rate of GPVI‐mediated platelet activation 104. The inositol phosphatases PTEN and SHIP1 play key roles in altering the phosphoinositide cycle and antagonizing PI3K function 105. The role of the serine/threonine kinase phosphatase PP2A in the negative regulation of integrin αIIbβ3 function and signaling has been attributed to the dephosphorylation of PKCζ, and protein‐tyrosine phosphatase 1B, which subsequently reduces Src phosphorylation and activation 106, 107, 108. Although several phosphatases are associated with negative regulation of platelet function, it is important to note that many positive regulatory functions for phosphatases, e.g. CD148, have also been identified in platelets 73.
Receptor desensitization enables platelets to regulate their responsiveness to platelet agonists. The process of receptor desensitization includes inactivation via phosphorylation events, and internalization, which removes receptors from the plasma membrane. As previously described, ADP is a critical secondary mediator of platelet activation, but the receptors for ADP are rapidly desensitized following exposure to ADP, resulting in a subsequently reduced agonist response 30, 74. This diminished response is attributed to desensitization of both the P2Y1 and P2Y12 receptors via agonist‐mediated internalization through two different protein kinase‐mediated pathways, with P2Y1 desensitization being mediated by both classic and novel isoforms of PKC, and P2Y12 desensitization being mediated by GPCR kinase and the novel PKC isoforms 30, 74. In contrast, the PARs have been shown, following platelet activation by thrombin or the PAR1 peptide agonist SFLLRN, to be internalized or shed into membrane microparticles, resulting in a decrease in the number of receptors detected at the platelet surface. This decrease is thought to underlie the inability of platelets to recover their responsiveness to PAR agonists 109.
Novel approaches to platelet signaling and future directions
The understanding of platelet signaling has increased exponentially in recent decades, and it is now clear that the signaling that underpins platelet function is best described as a heavily interlinked network rather than as linear pathways (Fig. 3). The vast amount of data describing the components within this network presents new challenges regarding the best way to interpret and transform this information into improved diagnostic and therapeutic strategies. Progressive approaches to platelet function screening have been developed that utilize standardized assay plate technology, bringing the high‐throughput methodologies of the drug discovery industry into the clinical setting 110. The ability to easily test many parameters simultaneously may allow positive and negative pathways to be investigated simultaneously and enable more of the platelet signaling network to be considered during the diagnosis of platelet disorders. Systematic measurement and analysis of platelet function under flow is also a promising avenue of development, and provides a sensitive diagnostic approach under more physiologic conditions and incorporates many more aspects of the platelet signaling network into the diagnosis of platelet disorders 111. The use of both of these strategies in combination with genetic mutant models, patient studies and inhibitor screening would give us a thorough insight into the roles of particular molecules in the regulation of platelet activity as a whole.
Figure 3.
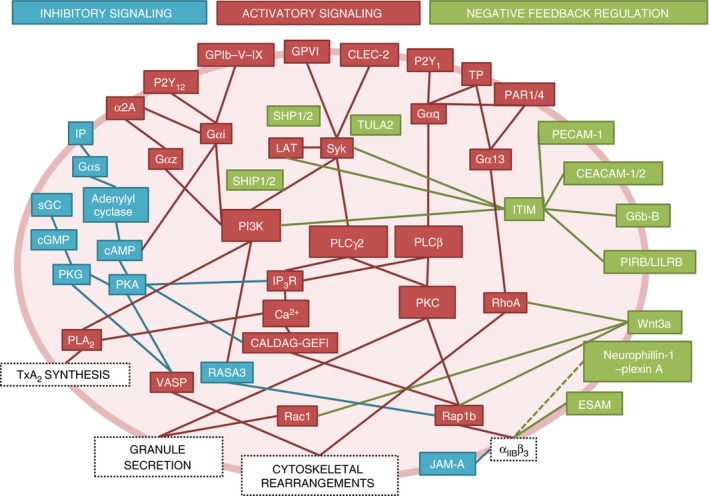
Complexity of platelet signaling networks. Platelet signaling models frequently describe signaling pathways activated by individual agonists; however, platelet signaling in vivo is highly complex, and involves simultaneous activation by multiple agonists and negative regulators, which form a complex signaling network. Several key signaling molecules, i.e. phospholipase C (PLC), protein kinase C (PKC), and phosphatidylinositide‐3‐kinase (PI3K), are common between the different pathways and form key nodes of platelet regulation. Blue boxes and lines represent mediators of inhibitory signaling that act to suppress platelet function in the absence of platelet activators in the healthy endothelium. Red boxes and lines represent mediators of activatory signaling following platelet activation by platelet agonists. Green boxes and lines represent mediators of negative feedback and inhibitory signaling that act to limit platelet activation following stimulation by platelet agonists. CALDAG‐GEFI, Ca2+‐dependent Rap1 guanine nucleotide exchange factor; CEACAM‐1/2, carcinoembryonic antigen cell adhesion molecule‐1/2; CLEC‐2, C‐type lectin receptor 2; ESAM, endothelial cell‐selective adhesion molecule; GP, glycoprotein; IP, prostaglandin receptor; IP 3 R, inositol trisphosphate receptor; ITIM, immunoreceptor tyrosine‐based inhibition motif; JAM‐A, junctional adhesion molecule A; LAT, linker of activated T cells; PAR, protease‐activated receptor; PECAM‐1, platelet endothelial cell adhesion molecule‐1; PIRB, paired immunoglobulin‐like receptor B; PKA, protein kinase A; PKG, protein kinase G; PLA 2, phospholipase A2; sGC, soluble guanyl cyclase; TULA2, T‐cell ubiquitin ligand‐2; TxA2, thromboxane A2; VASP, vasodilator‐stimulated phosphoprotein; α2A, adrenergic receptor.
Systems approaches that utilize mathematical modeling are increasingly being applied to help in our understanding of platelet biology. These can take two distinct forms: those using a top‐down approach, whereby the properties of relatively intact and complex systems are modeled; and those using a reductionist approach, whereby relatively few aspects of platelet signaling or function are modeled, but in greater detail. For example, the top‐down approach has revealed important details of how platelet thrombi are organized into a highly activated, dense ‘core’ of PS‐exposing platelets surrounded by a loosely packed and partially activated ‘shell’ 112, 113, 114. Reductionist models, in contrast, often focus on Ca2+ signaling, as it is easy to measure with high temporal resolution and to quantify, and is a point of signal integration downstream of many platelet pathways. Ca2+ models have proved to be useful in understanding how platelets organize important functional events, such as PS exposure, in subpopulations 115. Modeling of the processes that regulate cytosolic Ca2+ levels has provided insights into the mechanism of store‐operated Ca2+ entry and release from the DTS via IP3 receptors 116. Mathematical modeling has aided the understanding of kinase regulation downstream of GPVI and in particular the important role that negative regulation by phosphatases, such as TULA2, play in modulating signal transduction 104.
Future innovations will be required to integrate pathways into networks and to develop top‐down and reductionist models until they are able to meet in the middle. Such complex models will need to be validated by the use of data from many different platelet donors, in order to reflect the variability in platelet responsiveness that is present within the human population. Such modeling of platelet signaling networks will prove to be an important future direction for the field, as it will be a requirement for understanding how combinations of regulatory factors contribute collectively to platelet function and predicting how changes within specific parts of the network alter platelet reactivity or levels of activation. This knowledge will aid in the selection of proteins (individually or in combination) representing promising new antithrombotic targets, and will help in understanding the defects associated with an increased risk of thrombosis.
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
Acknowledgements
The authors would like to thank N. Kriek for assistance during the preparation of this manuscript. This work was supported by the British Heart Foundation (RG/09/011/28094 and RG/15/2/31224).
Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost 2016; 14: 918–30.
Manuscript handled by: P. H. Reitsma
Final decision: P. H. Reitsma, 11 February 2016
References
- 1. Aarts PA, van den Broek SA, Prins GW, Kuiken GD, Sixma JJ, Heethaar RM. Blood platelets are concentrated near the wall and red blood cells, in the center in flowing blood. Arteriosclerosis 1988; 8: 819–24. [DOI] [PubMed] [Google Scholar]
- 2. Smolenski A. Novel roles of cAMP/cGMP‐dependent signaling in platelets. J Thromb Haemost 2012; 10: 167–76. [DOI] [PubMed] [Google Scholar]
- 3. Mellion BT, Ignarro LJ, Ohlstein EH, Pontecorvo EG, Hyman AL, Kadowitz PJ. Evidence for the inhibitory role of guanosine 3′,5′‐monophosphate in ADP‐induced human platelet aggregation in the presence of nitric oxide and related vasodilators. Blood 1981; 57: 946–55. [PubMed] [Google Scholar]
- 4. Dutta‐Roy AK, Sinha AK. Purification and properties of prostaglandin E1/prostacyclin receptor of human blood platelets. J Biol Chem 1987; 262: 12685–91. [PubMed] [Google Scholar]
- 5. Gorman RR, Bunting S, Miller OV. Modulation of human platelet adenylate cyclase by prostacyclin (PGX). Prostaglandins 1977; 13: 377–88. [DOI] [PubMed] [Google Scholar]
- 6. Altschuler D, Lapetina EG. Mutational analysis of the cAMP‐dependent protein kinase‐mediated phosphorylation site of Rap1b. J Biol Chem 1993; 268: 7527–31. [PubMed] [Google Scholar]
- 7. Tertyshnikova S, Yan X, Fein A. cGMP inhibits IP3‐induced Ca2+ release in intact rat megakaryocytes via cGMP‐ and cAMP‐dependent protein kinases. J Physiol 1998; 512(Pt 1): 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eigenthaler M, Ullrich H, Geiger J, Horstrup K, Honig‐Liedl P, Wiebecke D, Walter U. Defective nitrovasodilator‐stimulated protein phosphorylation and calcium regulation in cGMP‐dependent protein kinase‐deficient human platelets of chronic myelocytic leukemia. J Biol Chem 1993; 268: 13526–31. [PubMed] [Google Scholar]
- 9. Massberg S, Sausbier M, Klatt P, Bauer M, Pfeifer A, Siess W, Fassler R, Ruth P, Krombach F, Hofmann F. Increased adhesion and aggregation of platelets lacking cyclic guanosine 3′,5′‐monophosphate kinase I. J Exp Med 1999; 189: 1255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 2002; 296: 539–41. [DOI] [PubMed] [Google Scholar]
- 11. Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi‐Kobayashi T, Yamashita JK, Katagiri H, Majima M, Yokode M, Kita T, Narumiya S. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE‐deficient mice. J Clin Invest 2004; 114: 784–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marcus AJ, Broekman MJ, Drosopoulos JH, Islam N, Alyonycheva TN, Safier LB, Hajjar KA, Posnett DN, Schoenborn MA, Schooley KA, Gayle RB, Maliszewski CR. The endothelial cell ecto‐ADPase responsible for inhibition of platelet function is CD39. J Clin Invest 1997; 99: 1351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnston‐Cox HA, Ravid K. Adenosine and blood platelets. Purinergic Signalling 2011; 7: 357–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naik MU, Caplan JL, Naik UP. Junctional adhesion molecule‐A suppresses platelet integrin alphaIIbbeta3 signaling by recruiting Csk to the integrin–c‐Src complex. Blood 2014; 123: 1393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Naik MU, Stalker TJ, Brass LF, Naik UP. JAM‐A protects from thrombosis by suppressing integrin alphaIIbbeta3‐dependent outside‐in signaling in platelets. Blood 2012; 119: 3352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gibbins JM. Platelet adhesion signalling and the regulation of thrombus formation. J Cell Sci 2004; 117: 3415–25. [DOI] [PubMed] [Google Scholar]
- 17. Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost 2002; 88: 186–93. [PubMed] [Google Scholar]
- 18. Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost 2005; 3: 1752–62. [DOI] [PubMed] [Google Scholar]
- 19. Harper MT, Poole AW. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J Thromb Haemost 2010; 8: 454–62. [DOI] [PubMed] [Google Scholar]
- 20. Offermanns S. Activation of platelet function through G protein‐coupled receptors. Circ Res 2006; 99: 1293–304. [DOI] [PubMed] [Google Scholar]
- 21. Born GV. The functional physiology of blood platelets. Adv Exp Med Biol 1972; 34: 3–21. [DOI] [PubMed] [Google Scholar]
- 22. Yang X, Sun L, Ghosh S, Rao AK. Human platelet signaling defect characterized by impaired production of inositol‐1,4,5‐triphosphate and phosphatidic acid and diminished Pleckstrin phosphorylation: evidence for defective phospholipase C activation. Blood 1996; 88: 1676–83. [PubMed] [Google Scholar]
- 23. Moroi AJ, Watson SP. Impact of the PI3‐kinase/Akt pathway on ITAM and hemITAM receptors: haemostasis, platelet activation and antithrombotic therapy. Biochem Pharmacol 2015; 94: 186–94. [DOI] [PubMed] [Google Scholar]
- 24. Spalton JC, Mori J, Pollitt AY, Hughes CE, Eble JA, Watson SP. The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC‐2 signaling in platelets. J Thromb Haemost 2009; 7: 1192–9. [DOI] [PubMed] [Google Scholar]
- 25. Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood 2003; 102: 3592–9. [DOI] [PubMed] [Google Scholar]
- 26. Bye AP, Unsworth AJ, Vaiyapuri S, Stainer AR, Fry MJ, Gibbins JM. Ibrutinib inhibits platelet integrin alphaIIbbeta3 outside‐in signaling and thrombus stability but not adhesion to collagen. Arterioscler Thromb Vasc Biol 2015; 35: 2326–35. [DOI] [PubMed] [Google Scholar]
- 27. Judd BA, Koretzky GA. The role of the adapter molecule SLP‐76 in platelet function. Oncogene 2001; 20: 6291–9. [DOI] [PubMed] [Google Scholar]
- 28. Wonerow P, Obergfell A, Wilde JI, Bobe R, Asazuma N, Brdicka T, Leo A, Schraven B, Horejsi V, Shattil SJ, Watson SP. Differential role of glycolipid‐enriched membrane domains in glycoprotein VI‐ and integrin‐mediated phospholipase Cgamma2 regulation in platelets. Biochem J 2002; 364: 755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nonne C, Lenain N, Hechler B, Mangin P, Cazenave JP, Gachet C, Lanza F. Importance of platelet phospholipase Cgamma2 signaling in arterial thrombosis as a function of lesion severity. Arterioscler Thromb Vasc Biol 2005; 25: 1293–8. [DOI] [PubMed] [Google Scholar]
- 30. Mundell SJ, Jones ML, Hardy AR, Barton JF, Beaucourt SM, Conley PB, Poole AW. Distinct roles for protein kinase C isoforms in regulating platelet purinergic receptor function. Mol Pharmacol 2006; 70: 1132–42. [DOI] [PubMed] [Google Scholar]
- 31. Strehl A, Munnix IC, Kuijpers MJ, van der Meijden PE, Cosemans JM, Feijge MA, Nieswandt B, Heemskerk JW. Dual role of platelet protein kinase C in thrombus formation: stimulation of pro‐aggregatory and suppression of procoagulant activity in platelets. J Biol Chem 2007; 282: 7046–55. [DOI] [PubMed] [Google Scholar]
- 32. Unsworth AJ, Smith H, Gissen P, Watson SP, Pears CJ. Submaximal inhibition of protein kinase C restores ADP‐induced dense granule secretion in platelets in the presence of Ca2+ . J Biol Chem 2011; 286: 21073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poole AW, Pula G, Hers I, Crosby D, Jones ML. PKC‐interacting proteins: from function to pharmacology. Trends Pharmacol Sci 2004; 25: 528–35. [DOI] [PubMed] [Google Scholar]
- 34. O'Brien KA, Stojanovic‐Terpo A, Hay N, Du X. An important role for Akt3 in platelet activation and thrombosis. Blood 2011; 118: 4215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moore SF, van den Bosch MT, Hunter RW, Sakamoto K, Poole AW, Hers I. Dual regulation of glycogen synthase kinase 3 (GSK3)alpha/beta by protein kinase C (PKC)alpha and Akt promotes thrombin‐mediated integrin alphaIIbbeta3 activation and granule secretion in platelets. J Biol Chem 2013; 288: 3918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Toth‐Zsamboki E, Oury C, Cornelissen H, De Vos R, Vermylen J, Hoylaerts MF. P2X1‐mediated ERK2 activation amplifies the collagen‐induced platelet secretion by enhancing myosin light chain kinase activation. J Biol Chem 2003; 278: 46661–7. [DOI] [PubMed] [Google Scholar]
- 37. Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X. Two distinct roles of mitogen‐activated protein kinases in platelets and a novel Rac1‐MAPK‐dependent integrin outside‐in retractile signaling pathway. Blood 2009; 113: 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bobe R, Wilde JI, Maschberger P, Venkateswarlu K, Cullen PJ, Siess W, Watson SP. Phosphatidylinositol 3‐kinase‐dependent translocation of phospholipase Cgamma2 in mouse megakaryocytes is independent of Bruton tyrosine kinase translocation. Blood 2001; 97: 678–84. [DOI] [PubMed] [Google Scholar]
- 39. Guidetti GF, Canobbio I, Torti M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv Biol Regul 2015; 59: 36–52. [DOI] [PubMed] [Google Scholar]
- 40. Gilio K, Munnix IC, Mangin P, Cosemans JM, Feijge MA, van der Meijden PE, Olieslagers S, Chrzanowska‐Wodnicka MB, Lillian R, Schoenwaelder S, Koyasu S, Sage SO, Jackson SP, Heemskerk JW. Non‐redundant roles of phosphoinositide 3‐kinase isoforms alpha and beta in glycoprotein VI‐induced platelet signaling and thrombus formation. J Biol Chem 2009; 284: 33750–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim S, Mangin P, Dangelmaier C, Lillian R, Jackson SP, Daniel JL, Kunapuli SP. Role of phosphoinositide 3‐kinase beta in glycoprotein VI‐mediated Akt activation in platelets. J Biol Chem 2009; 284: 33763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hartwig JH. The platelet cytoskeleton In: Michelson A, ed. Platelets, 3rd edn Cambridge, MA: Academic Press, 2013: 145–168. [Google Scholar]
- 43. Janmey PA. Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annu Rev Physiol 1994; 56: 169–91. [DOI] [PubMed] [Google Scholar]
- 44. Adelstein RS, Conti MA, Daniel JL, Anderson W Jr. The interaction of platelet actin, myosin and myosin light chain kinase. Ciba Found Symp 1975; 35: 101–9. [DOI] [PubMed] [Google Scholar]
- 45. Machesky LM, Insall RH. Signaling to actin dynamics. J Cell Biol 1999; 146: 267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost 2013; 11: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wentworth JK, Pula G, Poole AW. Vasodilator‐stimulated phosphoprotein (VASP) is phosphorylated on Ser157 by protein kinase C‐dependent and ‐independent mechanisms in thrombin‐stimulated human platelets. Biochem J 2006; 393: 555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Polgar J, Chung SH, Reed GL. Vesicle‐associated membrane protein 3 (VAMP‐3) and VAMP‐8 are present in human platelets and are required for granule secretion. Blood 2002; 100: 1081–3. [DOI] [PubMed] [Google Scholar]
- 49. Ren Q, Wimmer C, Chicka MC, Ye S, Ren Y, Hughson FM, Whiteheart SW. Munc13‐4 is a limiting factor in the pathway required for platelet granule release and hemostasis. Blood 2010; 116: 869–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nishikawa M, Tanaka T, Hidaka H. Ca2+‐calmodulin‐dependent phosphorylation and platelet secretion. Nature 1980; 287: 863–5. [DOI] [PubMed] [Google Scholar]
- 51. Quetglas S, Iborra C, Sasakawa N, De Haro L, Kumakura K, Sato K, Leveque C, Seagar M. Calmodulin and lipid binding to synaptobrevin regulates calcium‐dependent exocytosis. EMBO J 2002; 21: 3970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Neumuller O, Hoffmeister M, Babica J, Prelle C, Gegenbauer K, Smolenski AP. Synaptotagmin‐like protein 1 interacts with the GTPase‐activating protein Rap1GAP2 and regulates dense granule secretion in platelets. Blood 2009; 114: 1396–404. [DOI] [PubMed] [Google Scholar]
- 53. Rink TJ, Sanchez A, Hallam TJ. Diacylglycerol and phorbol ester stimulate secretion without raising cytoplasmic free calcium in human platelets. Nature 1983; 305: 317–19. [DOI] [PubMed] [Google Scholar]
- 54. Chung SH, Polgar J, Reed GL. Protein kinase C phosphorylation of syntaxin 4 in thrombin‐activated human platelets. J Biol Chem 2000; 275: 25286–91. [DOI] [PubMed] [Google Scholar]
- 55. Reed GL, Houng AK, Fitzgerald ML. Human platelets contain SNARE proteins and a Sec1p homologue that interacts with syntaxin 4 and is phosphorylated after thrombin activation: implications for platelet secretion. Blood 1999; 93: 2617–26. [PubMed] [Google Scholar]
- 56. Schraw TD, Lemons PP, Dean WL, Whiteheart SW. A role for Sec1/Munc18 proteins in platelet exocytosis. Biochem J 2003; 374: 207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stefanini L, Boulaftali Y, Ouellette TD, Holinstat M, Desire L, Leblond B, Andre P, Conley PB, Bergmeier W. Rap1–Rac1 circuits potentiate platelet activation. Arterioscler Thromb Vasc Biol 2012; 32: 434–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Borsch‐Haubold AG, Kramer RM, Watson SP. Phosphorylation and activation of cytosolic phospholipase A2 by 38‐kDa mitogen‐activated protein kinase in collagen‐stimulated human platelets. Eur J Biochem 1997; 245: 751–9. [DOI] [PubMed] [Google Scholar]
- 59. Garcia A, Shankar H, Murugappan S, Kim S, Kunapuli SP. Regulation and functional consequences of ADP receptor‐mediated ERK2 activation in platelets. Biochem J 2007; 404: 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garcia A, Kim S, Bhavaraju K, Schoenwaelder SM, Kunapuli SP. Role of phosphoinositide 3‐kinase beta in platelet aggregation and thromboxane A2 generation mediated by Gi signalling pathways. Biochem J 2010; 429: 369–77. [DOI] [PubMed] [Google Scholar]
- 61. Taylor KA, Wright JR, Vial C, Evans RJ, Mahaut‐Smith MP. Amplification of human platelet activation by surface pannexin‐1 channels. J Thromb Haemost 2014; 12: 987–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin beta tails: a final common step in integrin activation. Science 2003; 302: 103–6. [DOI] [PubMed] [Google Scholar]
- 63. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin‐3 is essential for integrin activation and platelet aggregation. Nat Med 2008; 14: 325–30. [DOI] [PubMed] [Google Scholar]
- 64. Chrzanowska‐Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC 2nd. Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest 2005; 115: 680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Stefanini L, Roden RC, Bergmeier W. CalDAG‐GEFI is at the nexus of calcium‐dependent platelet activation. Blood 2009; 114: 2506–14. [DOI] [PubMed] [Google Scholar]
- 66. Kawasaki H, Springett GM, Toki S, Canales JJ, Harlan P, Blumenstiel JP, Chen EJ, Bany IA, Mochizuki N, Ashbacher A, Matsuda M, Housman DE, Graybiel AM. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc Natl Acad Sci USA 1998; 95: 13278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Woulfe D, Jiang H, Mortensen R, Yang J, Brass LF. Activation of Rap1B by G(i) family members in platelets. J Biol Chem 2002; 277: 23382–90. [DOI] [PubMed] [Google Scholar]
- 68. Stefanini L, Paul DS, Robledo RF, Chan ER, Getz TM, Campbell RA, Kechele DO, Casari C, Piatt R, Caron KM, Mackman N, Weyrich AS, Parrott MC, Boulaftali Y, Adams MD, Peters LL, Bergmeier W. RASA3 is a critical inhibitor of RAP1‐dependent platelet activation. J Clin Invest 2015; 125: 1419–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, Puzon‐McLaughlin W, Lafuente EM, Boussiotis VA, Shattil SJ, Ginsberg MH. Reconstructing and deconstructing agonist‐induced activation of integrin alphaIIbbeta3. Curr Biol 2006; 16: 1796–806. [DOI] [PubMed] [Google Scholar]
- 70. Stritt S, Wolf K, Lorenz V, Vogtle T, Gupta S, Bosl MR, Nieswandt B. Rap1‐GTP‐interacting adaptor molecule (RIAM) is dispensable for platelet integrin activation and function in mice. Blood 2015; 125: 219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Poulter NS, Pollitt AY, Davies A, Malinova D, Nash GB, Hannon MJ, Pikramenou Z, Rappoport JZ, Hartwig JH, Owen DM, Thrasher AJ, Watson SP, Thomas SG. Platelet actin nodules are podosome‐like structures dependent on Wiskott–Aldrich syndrome protein and ARP2/3 complex. Nat Commun 2015; 6: 7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jones CI, Barrett NE, Moraes LA, Gibbins JM, Jackson DE. Endogenous inhibitory mechanisms and the regulation of platelet function. Methods Mol Biol 2012; 788: 341–66. [DOI] [PubMed] [Google Scholar]
- 73. Senis YA. Protein‐tyrosine phosphatases: a new frontier in platelet signal transduction. J Thromb Haemost 2013; 11: 1800–13. [DOI] [PubMed] [Google Scholar]
- 74. Hardy AR, Conley PB, Luo J, Benovic JL, Poole AW, Mundell SJ. P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase‐dependent mechanisms. Blood 2005; 105: 3552–60. [DOI] [PubMed] [Google Scholar]
- 75. Cicmil M, Thomas JM, Leduc M, Bon C, Gibbins JM. Platelet endothelial cell adhesion molecule‐1 signaling inhibits the activation of human platelets. Blood 2002; 99: 137–44. [DOI] [PubMed] [Google Scholar]
- 76. Jones CI, Garner SF, Moraes LA, Kaiser WJ, Rankin A, Bloodomics C, Ouwehand WH, Goodall AH, Gibbins JM. PECAM‐1 expression and activity negatively regulate multiple platelet signaling pathways. FEBS Lett 2009; 583: 3618–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wong C, Liu Y, Yip J, Chand R, Wee JL, Oates L, Nieswandt B, Reheman A, Ni H, Beauchemin N, Jackson DE. CEACAM1 negatively regulates platelet–collagen interactions and thrombus growth in vitro and in vivo . Blood 2009; 113: 1818–28. [DOI] [PubMed] [Google Scholar]
- 78. Alshahrani MM, Yang E, Yip J, Ghanem SS, Abdallah SL, deAngelis AM, O'Malley CJ, Moheimani F, Najjar SM, Jackson DE. CEACAM2 negatively regulates hemi (ITAM‐bearing) GPVI and CLEC‐2 pathways and thrombus growth in vitro and in vivo . Blood 2014; 124: 2431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Newland SA, Macaulay IC, Floto AR, de Vet EC, Ouwehand WH, Watkins NA, Lyons PA, Campbell DR. The novel inhibitory receptor G6B is expressed on the surface of platelets and attenuates platelet function in vitro . Blood 2007; 109: 4806–9. [DOI] [PubMed] [Google Scholar]
- 80. Mori J, Pearce AC, Spalton JC, Grygielska B, Eble JA, Tomlinson MG, Senis YA, Watson SP. G6b‐B inhibits constitutive and agonist‐induced signaling by glycoprotein VI and CLEC‐2. J Biol Chem 2008; 283: 35419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fan X, Shi P, Dai J, Lu Y, Chen X, Liu X, Zhang K, Wu X, Sun Y, Wang K, Zhu L, Zhang CC, Zhang J, Chen GQ, Zheng J, Liu J. Paired immunoglobulin‐like receptor B regulates platelet activation. Blood 2014; 124: 2421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sun QH, DeLisser HM, Zukowski MM, Paddock C, Albelda SM, Newman PJ. Individually distinct Ig homology domains in PECAM‐1 regulate homophilic binding and modulate receptor affinity. J Biol Chem 1996; 271: 11090–8. [DOI] [PubMed] [Google Scholar]
- 83. Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997; 386: 181–6. [DOI] [PubMed] [Google Scholar]
- 84. Bruhns P, Vely F, Malbec O, Fridman WH, Vivier E, Daeron M. Molecular basis of the recruitment of the SH2 domain‐containing inositol 5‐phosphatases SHIP1 and SHIP2 by fcgamma RIIB. J Biol Chem 2000; 275: 37357–64. [DOI] [PubMed] [Google Scholar]
- 85. Moraes LA, Barrett NE, Jones CI, Holbrook LM, Spyridon M, Sage T, Newman DK, Gibbins JM. Platelet endothelial cell adhesion molecule‐1 regulates collagen‐stimulated platelet function by modulating the association of phosphatidylinositol 3‐kinase with Grb‐2‐associated binding protein‐1 and linker for activation of T cells. J Thromb Haemost 2010; 8: 2530–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jones CI, Moraes LA, Gibbins JM. Regulation of platelet biology by platelet endothelial cell adhesion molecule‐1. Platelets 2012; 23: 331–5. [DOI] [PubMed] [Google Scholar]
- 87. Falati S, Patil S, Gross PL, Stapleton M, Merrill‐Skoloff G, Barrett NE, Pixton KL, Weiler H, Cooley B, Newman DK, Newman PJ, Furie BC, Furie B, Gibbins JM. Platelet PECAM‐1 inhibits thrombus formation in vivo . Blood 2006; 107: 535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wee JL, Jackson DE. The Ig‐ITIM superfamily member PECAM‐1 regulates the ‘outside‐in’ signaling properties of integrin alpha(IIb)beta3 in platelets. Blood 2005; 106: 3816–23. [DOI] [PubMed] [Google Scholar]
- 89. Yip J, Alshahrani M, Beauchemin N, Jackson DE. CEACAM1 regulates integrin alphaIIbbeta3‐mediated functions in platelets. Platelets 2015; 27: 1–10. [DOI] [PubMed] [Google Scholar]
- 90. Nasdala I, Wolburg‐Buchholz K, Wolburg H, Kuhn A, Ebnet K, Brachtendorf G, Samulowitz U, Kuster B, Engelhardt B, Vestweber D, Butz S. A transmembrane tight junction protein selectively expressed on endothelial cells and platelets. J Biol Chem 2002; 277: 16294–303. [DOI] [PubMed] [Google Scholar]
- 91. Stalker TJ, Wu J, Morgans A, Traxler EA, Wang L, Chatterjee MS, Lee D, Quertermous T, Hall RA, Hammer DA, Diamond SL, Brass LF. Endothelial cell specific adhesion molecule (ESAM) localizes to platelet–platelet contacts and regulates thrombus formation in vivo . J Thromb Haemost 2009; 7: 1886–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kashiwagi H, Shiraga M, Kato H, Kamae T, Yamamoto N, Tadokoro S, Kurata Y, Tomiyama Y, Kanakura Y. Negative regulation of platelet function by a secreted cell repulsive protein, semaphorin 3A. Blood 2005; 106: 913–21. [DOI] [PubMed] [Google Scholar]
- 93. Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M, Tessier‐Lavigne M, Taniguchi M, Puschel AW, Bussolino F. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 2003; 424: 391–7. [DOI] [PubMed] [Google Scholar]
- 94. Steele BM, Harper MT, Macaulay IC, Morrell CN, Perez‐Tamayo A, Foy M, Habas R, Poole AW, Fitzgerald DJ, Maguire PB. Canonical Wnt signaling negatively regulates platelet function. Proc Natl Acad Sci USA 2009; 106: 19836–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Steele BM, Harper MT, Smolenski AP, Alkazemi N, Poole AW, Fitzgerald DJ, Maguire PB. WNT‐3a modulates platelet function by regulating small GTPase activity. FEBS Lett 2012; 586: 2267–72. [DOI] [PubMed] [Google Scholar]
- 96. Moraes LA, Spyridon M, Kaiser WJ, Jones CI, Sage T, Atherton RE, Gibbins JM. Non‐genomic effects of PPARgamma ligands: inhibition of GPVI‐stimulated platelet activation. J Thromb Haemost 2010; 8: 577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Moraes LA, Swales KE, Wray JA, Damazo A, Gibbins JM, Warner TD, Bishop‐Bailey D. Nongenomic signaling of the retinoid X receptor through binding and inhibiting Gq in human platelets. Blood 2007; 109: 3741–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Spyridon M, Moraes LA, Jones CI, Sage T, Sasikumar P, Bucci G, Gibbins JM. LXR as a novel antithrombotic target. Blood 2011; 117: 5751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Moraes LA, Paul‐Clark MJ, Rickman A, Flower RJ, Goulding NJ, Perretti M. Ligand‐specific glucocorticoid receptor activation in human platelets. Blood 2005; 106: 4167–75. [DOI] [PubMed] [Google Scholar]
- 100. Ali FY, Davidson SJ, Moraes LA, Traves SL, Paul‐Clark M, Bishop‐Bailey D, Warner TD, Mitchell JA. Role of nuclear receptor signaling in platelets: antithrombotic effects of PPARbeta. FASEB J 2006; 20: 326–8. [DOI] [PubMed] [Google Scholar]
- 101. Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, Curtiss LK. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science 2003; 302: 453–7. [DOI] [PubMed] [Google Scholar]
- 102. Ali FY, Armstrong PC, Dhanji A‐RA, Tucker AT, Paul‐Clark MJ, Mitchell JA, Warner TD. Antiplatelet actions of statins and fibrates are mediated by PPARs. Arterioscler Thromb Vasc Biol 2009; 29: 706–11. [DOI] [PubMed] [Google Scholar]
- 103. Ali FY, Hall MG, Desvergne B, Warner TD, Mitchell JA. PPARβ/δ agonists modulate platelet function via a mechanism involving PPAR receptors and specific association/repression of PKCα – Brief Report. Arterioscler Thromb Vasc Biol 2009; 29: 1871–3. [DOI] [PubMed] [Google Scholar]
- 104. Dunster JL, Mazet F, Fry MJ, Gibbins JM, Tindall MJ. Regulation of early steps of GPVI signal transduction by phosphatases: a systems biology approach. PLoS Comput Biol 2015; 11: e1004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Laurent PA, Severin S, Gratacap MP, Payrastre B. Class I PI 3‐kinases signaling in platelet activation and thrombosis: PDK1/Akt/GSK3 axis and impact of PTEN and SHIP1. Adv Biol Regul 2014; 54: 162–74. [DOI] [PubMed] [Google Scholar]
- 106. Mayanglambam A, Bhavanasi D, Vijayan KV, Kunapuli SP. Differential dephosphorylation of the protein kinase C‐zeta (PKCzeta) in an integrin alphaIIbbeta3‐dependent manner in platelets. Biochem Pharmacol 2011; 82: 505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pradhan S, Alrehani N, Patel V, Khatlani T, Vijayan KV. Cross‐talk between serine/threonine protein phosphatase 2A and protein tyrosine phosphatase 1B regulates Src activation and adhesion of integrin alphaIIbbeta3 to fibrinogen. J Biol Chem 2010; 285: 29059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gushiken FC, Patel V, Liu Y, Pradhan S, Bergeron AL, Peng Y, Vijayan KV. Protein phosphatase 2A negatively regulates integrin alpha(IIb)beta(3) signaling. J Biol Chem 2008; 283: 12862–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Molino M, Bainton DF, Hoxie JA, Coughlin SR, Brass LF. Thrombin receptors on human platelets. Initial localization and subsequent redistribution during platelet activation. J Biol Chem 1997; 272: 6011–17. [DOI] [PubMed] [Google Scholar]
- 110. Lordkipanidze M, Lowe GC, Kirkby NS, Chan MV, Lundberg MH, Morgan NV, Bem D, Nisar SP, Leo VC, Jones ML, Mundell SJ, Daly ME, Mumford AD, Warner TD, Watson SP; UK Genotyping and Phenotyping of Platelets Study Group . Characterization of multiple platelet activation pathways in patients with bleeding as a high‐throughput screening option: use of 96‐well Optimul assay. Blood 2014; 123: e11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. de Witt SM, Swieringa F, Cavill R, Lamers MM, van Kruchten R, Mastenbroek T, Baaten C, Coort S, Pugh N, Schulz A, Scharrer I, Jurk K, Zieger B, Clemetson KJ, Farndale RW, Heemskerk JW, Cosemans JM. Identification of platelet function defects by multi‐parameter assessment of thrombus formation. Nat Commun 2014; 5: 4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Welsh JD, Stalker TJ, Voronov R, Muthard RW, Tomaiuolo M, Diamond SL, Brass LF. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood 2014; 124: 1808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Stalker TJ, Welsh JD, Tomaiuolo M, Wu J, Colace TV, Diamond SL, Brass LF. A systems approach to hemostasis: 3. Thrombus consolidation regulates intrathrombus solute transport and local thrombin activity. Blood 2014; 124: 1824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tomaiuolo M, Stalker TJ, Welsh JD, Diamond SL, Sinno T, Brass LF. A systems approach to hemostasis: 2. Computational analysis of molecular transport in the thrombus microenvironment. Blood 2014; 124: 1816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol BioSyst 2015; 11: 1052–60. [DOI] [PubMed] [Google Scholar]
- 116. Dolan AT, Diamond SL. Systems modeling of Ca(2+) homeostasis and mobilization in platelets mediated by IP3 and store‐operated Ca(2+) entry. Biophys J 2014; 106: 2049–60. [DOI] [PMC free article] [PubMed] [Google Scholar]