

Abstract
We report the first study of the biological effect of fulvestrant on ER positive clinical breast cancer using sequential biopsies through to progression. Thirty‐two locally/systemically advanced breast cancers treated with first‐line fulvestrant (250 mg/month) were biopsied at therapy initiation, 6 weeks, 6 months and progression and immunohistochemically‐analyzed for Ki67, ER, EGFR and HER2 expression/signaling activity. This series showed good fulvestrant responses (duration of response [DoR] = 25.8 months; clinical benefit = 81%). Ki67 fell (p < 0.001) in 79% of tumours by 6 months and lower Ki67 at all preprogression time‐points predicted for longer DoR. ER and PR significantly decreased in all tumours by 6 months (p < 0.001), with some declines in ER (serine 118) phosphorylation and Bcl‐2 (p = 0.007). There were modest HER2 increases (p = 0.034, 29% tumours) and loss of any detectable EGFR phosphorylation (p = 0.024, 50% tumours) and MAP kinase (ERK1/2) phosphorylation (p = 0.019, 65% tumours) by 6 months. While ER remained low, there was some recovery of Ki67, Bcl‐2 and (weakly) EGFR/MAPK activity in 45–67% patients at progression. Fulvestrant's anti‐proliferative impact is related to DoR, but while commonly downregulating ER and indicators of its signaling and depleting EGFR/MAPK signaling in some patients, additional elements must determine response duration. Residual ER at fulvestrant relapse explains reported sensitivity to further endocrine therapies. Occasional modest treatment‐induced HER2 and weakly detectable EGFR/HER2/MAPK signaling at relapse suggests targeting of such activity might have value alongside fulvestrant in some patients. However, unknown pathways must drive relapse in most. Ki67 has biomarker potential to predict fulvestrant outcome and as a quantitative measure of response.
Keywords: breast cancer, fulvestrant, biology, endocrine resistance
Short abstract
What's New?
The steroidal drug fulvestrant is a powerful antiestrogen, blocking the estrogen receptor‐alpha to a greater extent than tamoxifen. However, the development of drug resistance is a considerable problem with fulvestrant. For the first time, the authors of the present study examined the biological effects of fulvestrant therapy using sequential breast tumor biopsies from untreated, treated, and relapsed patients. While residual estrogen receptor activity and tumor cell proliferation were detected at relapse, no meaningful increases were found in EGFR/HER2/MAPK activity. Ki67 expression was associated with duration of response, indicating promise as a predictive biomarker for fulvestrant outcome.
Abbreviations
- ABC
advanced breast cancer
- AF‐1
activation function domain‐1
- AI
aromatase inhibitor
- AKT
protein kinase B
- Bcl‐2
apoptosis regulator B‐Cell CLL/Lymphoma 2
- CB
clinical benefit
- CI
confidence intervals
- CONR/LP
continuing responders/late progressors (CB tumours with responses exceeding median DoCB)
- DoCB
duration of response in patients with clinical benefit
- DoR
duration of response in All‐patients
- EGFR
epidermal growth factor receptor
- EP
early progressors (CB tumours progressing prior to median DoCB)
- ER
estrogen receptor‐alpha
- ERK1/2
extracellular signal regulated kinases 1 and 2
- GF
growth factor
- HER2
receptor tyrosine‐protein kinase erbB‐2
- HER3
receptor tyrosine‐protein kinase erbB‐3
- HER4
receptor tyrosine‐protein kinase erbB‐4
- IHC
immunohistochemistry
- Ki67
proliferation‐related KI‐67 antigen
- LAPC
locally advanced breast cancer
- MAPK
mitogen‐activated protein kinase
- mTOR
mammalian target of rapamycin
- PD
progressive disease
- PI3K
phosphatidylinositol 3‐kinase
- PR
progesterone receptor
- pS2
trefoil factor 1
- Src
tyrosine‐protein kinase Src
- TIMP‐1
tissue inhibitor of metalloproteinases 1
Fulvestrant (Faslodex™) is a pure anti‐estrogen with no known agonistic activity, contrasting tamoxifen. The steroidal agent fulvestrant prevents estradiol binding to estrogen receptor‐alpha (ER) to a stronger extent than tamoxifen. It also has a distinct mode of action that causes severe receptor conformational changes, promoting receptor degradation and downregulation of ER protein level and depletion of ER transcriptional activation.1 Clinically, therefore, fulvestrant retains activity in postmenopausal tamoxifen or nonsteroidal aromatase inhibitor (AI) resistant estrogen receptor positive (ER+) breast cancers.2, 3, 4, 5 Fulvestrant, at 250 mg, had similar time to progression, survival and response rate to use of tamoxifen/AIs in Phase III trials.2, 3, 4, 6 Following observations of dose‐dependent decline in ER after short‐term fulvestrant treatment of clinical breast cancer,7 additional studies including the CONFIRM trial5 provided evidence of further benefit with fulvestrant at 500 mg in ER+ disease following prior endocrine failure. Clinical significance of fulvestrant is set to increase further since this anti‐hormone also has potential in neoadjuvant and first‐line advanced ER+ postmenopausal disease settings, evidenced by trials including NEWEST8 and FIRST,9, 10 respectively. Further studies are also exploring fulvestrant alongside AIs, exemplified by the FACT11 and SWOG trials.12
Despite its increasing clinical value, de novo and acquired resistance remains a significant problem with fulvestrant. This disease state is largely unexplored in the clinical setting. The erbB receptor family members EGFR and HER2, as well as mitogen‐activated protein kinase signaling activity (MAPK), can be elevated and growth contributory to acquired fulvestrant resistance in vitro, 13, 14 although this is not a unifying feature of all fulvestrant resistant models.15 These can also be reliant on further erbB receptors,16 Src kinase and PI3K/AKT/mTOR signaling.17, 18 Moreover, growth factor (GF) signaling pathways have been heavily implicated in tamoxifen and estrogen deprivation resistance models, where they cross‐talk with ER. For example, EGFR/HER2 and MAPK signaling onto ER, via activation function domain 1 (AF‐1) residue phosphorylation (e.g., serine 118), can permit either agonistic behaviour of tamoxifen or hypersensitivity to residual estrogens in vitro.19, 20, 21, 22 Theoretically, depletion of ER and thereby cross‐talk's critical “hub” should occur with fulvestrant, so that the development of resistance would potentially be delayed and possibly also ER‐independent. Fulvestrant is certainly able to promote ER degradation, decrease ER‐regulated proteins (e.g., progesterone receptor [PR], pS2, cell survival protein Bcl‐2) and proliferation, and delay resistance in ER+ models.23 In short‐term studies (<16weeks) fulvestrant also decreased ER, PR and pS2, proliferation and (modestly) increased apoptosis in patient samples.7, 8, 24, 25 Nevertheless, some fulvestrant resistant patients retain sensitivity to further endocrine challenge.
Clearly, if we are to better understand response and acquired fulvestrant resistance in patients, it remains important to profile ER expression/function and GF signaling pathways during long‐term fulvestrant treatment. Indeed, it is our hypothesis that knowledge of such profiles through to fulvestrant resistance should aid interpretation of various breast cancer trials examining fulvestrant with anti‐GFs, could provide rationale for development of new strategies to delay or treat this resistant state, and may identify predictive biomarkers to maximize benefit from fulvestrant. Sequential breast cancer biopsies taken from locally advanced disease with or without metastases, prior to, during first‐line fulvestrant treatment and at subsequent relapse provide an important resource to help achieve this goal.
Here, for the first time, we profile the impact of initial (6 weeks) and prolonged (6 months and beyond) fulvestrant treatment (250 mg/month) on key elements of ER and GF signaling cross‐talk and proliferation in clinical ER+ samples. The immunohistochemical (IHC) methodology employed provides an immediate indication of potential value and feasibility of the various biomarker assays to predict fulvestrant clinical outcome.
Material and Methods
Sequential core biopsies were obtained from 32 ER+ locally advanced or systemically advanced breast cancer patients treated with first‐line fulvestrant (250 mg/month). Thirty patients were from Faslodex™ 003 (an open label first‐line study to enable exploratory biological investigation; Nottingham Research Ethics Committee EC00/191). Two patients (with all sequential biopsies on unblinding) were from Faslodex™ 0025 (a randomized, double blind Phase III trial comparing 250 mg fulvestrant with 20 mg tamoxifen as first‐line therapy; EC98/239). Table 1 details criteria defining quality and duration of fulvestrant clinical response. Supporting Information Table S1a summarizes the patient series including baseline disease characteristics and clinical response (provided on a per case basis in Supporting Information Table S1b). The patient series showed good fulvestrant responses with a median duration of response (DoR) of 25.8 (1.8–60.7) months. Twenty‐six patients (81.25%) had clinical benefit (CB), with a median DoR in CB patients (DoCB) of 29.3 (10.9–60.7) months. Responses to any other treatments following fulvestrant progression were not a component of these response data. The median duration of overall survival (reflecting impact of first‐line fulvestrant and any subsequent disease management) was 35.5 (2.1–71.9) months, with 11 breast cancer‐specific deaths at analysis.
Table 1.
Criteria defining quality and duration of response to fulvestrant
| QUALITY OF CLINICAL RESPONSE |
|
Patients were assessed clinically every 6 weeks for the first 6 months using bi‐dimensional calliper measurements of their tumour and then at 12 weekly intervals (as per UICC criteria): CR = Complete response to fulvestrant PR = Partial response to fulvestrant SD = Stable disease on fulvestrant CB = clinical benefit on fulvestrant, i.e., Complete or Partial response or Stable disease for >= 6 months PD = progressive disease, i.e., progression of disease on fulvestrant within 6 months |
| CLINICAL RESPONSE DURATION |
|
Median DoR = median duration of response between fulvestrant treatment commencement and disease progression on this agent for All‐patients Median DoCB= median duration of response to fulvestrant in patients with CB CB patients were also subdivided into EP (early progressors, i.e., CB tumours progressing prior to median DoCB on fulvestrant) or CONR/LP sub‐sets (continuing responders/late progressors, i.e., CB tumours with fulvestrant responses exceeding median DoCB, including those progressing on fulvestrant after this time). Two CB patients with follow‐up <median DoCB were excluded from EP versus CONR/LP analyses. |
Core biopsies were taken from each tumour using a 14‐gauge needle (AA, JFR, KLC, EG) before commencing fulvestrant (T1), at 6 weeks (T2) and 6 months (T3) on treatment, and at disease progression (T4) for routine formalin‐fixation and paraffin‐embedding. Adequate cellularity (>100 tumour cells) was first verified in each biopsy (n = 31, 28, 25, 15 samples at T1‐T4, respectively) and cellularity was recorded. IHC was then performed on 3 μm sections for proliferation (Ki67) and biomarkers indicative of ER function [ER, PR, Bcl‐2, pER (serine 118 phosphorylated ER)], GF signaling [EGFR and HER2 expression or activity (Tyr845 and Tyr1248 phosphorylated pEGFR and pHER2, respectively)] or MAPK activity [Thr202/Tyr204 pMAPK, detecting phosphorylated MAPK1/3 (ERK1/2)]. HercepTestTM and an additional assay with increased sensitivity for cytoplasmic staining monitored HER2 expression. All sequential samples for a patient were immunostained simultaneously. Semiquantitative assessment was performed by consensus of two observers blinded to patient details (percentage positivity for Ki67 and Bcl‐2; 0–3 HercepTestTM scoring; H Score on a 0–300 scale for all other markers). Staining was nuclear for ER, PR, pER, pMAPK and Ki67, cytoplasmic for Bcl‐2, or plasma membrane (EGFRm, HER2m) and cytoplasmic (EGFRc, HER2c). Any samples with insufficient cellularity or nonspecific staining after each assay were excluded. The IHC staining and assessment (using standardized and internally validated protocols) is further detailed in Supplementary Assay Information.
Focussing on the All‐patient (n = 32) and clinical benefit (CB, n = 26) groups, early progressors (EP, n = 11) and continuing responders/late progressors (CONR/LP, n = 13) as defined in Table 1, biomarker changes were analyzed between matched T1, T2, T3 and T4 biopsies in each patient using Friedman's ANOVA and Wilcoxon Paired Signed‐Rank test (significance p <= 0.05). At each biopsy time point, Kaplan–Meier analysis (Log rank test) determined biomarker relationship to DoR on fulvestrant using the respective median staining cut‐point (Supporting Information Table S2), with disease progression on fulvestrant as the event. Staining relationship to DoCB was determined by Mann Whitney analysis in EP versus CONR/LP. Patient numbers were insufficient for analysis (i) within de novo fulvestrant resistant disease (PD, n = 6) or within specific CB patient subgroups and (ii) with respect to DoR according to T4 biomarker expression.
Results
Figure 1 (with Supporting Information Table S2) shows the median staining obtained for the IHC biomarkers at each treatment time‐point in the All‐patient group. Staining data for individual patients are shown in Figure 2. Supporting Information Tables S3 and S4 provide median staining data for the CB (clinical benefit) and the EP (early progressors) and CONR/LP (continuing responders/late progressors) cohorts, respectively. Table 2 summarizes the key statistical findings for the biomarkers in these various patient cohorts during treatment.
Figure 1.
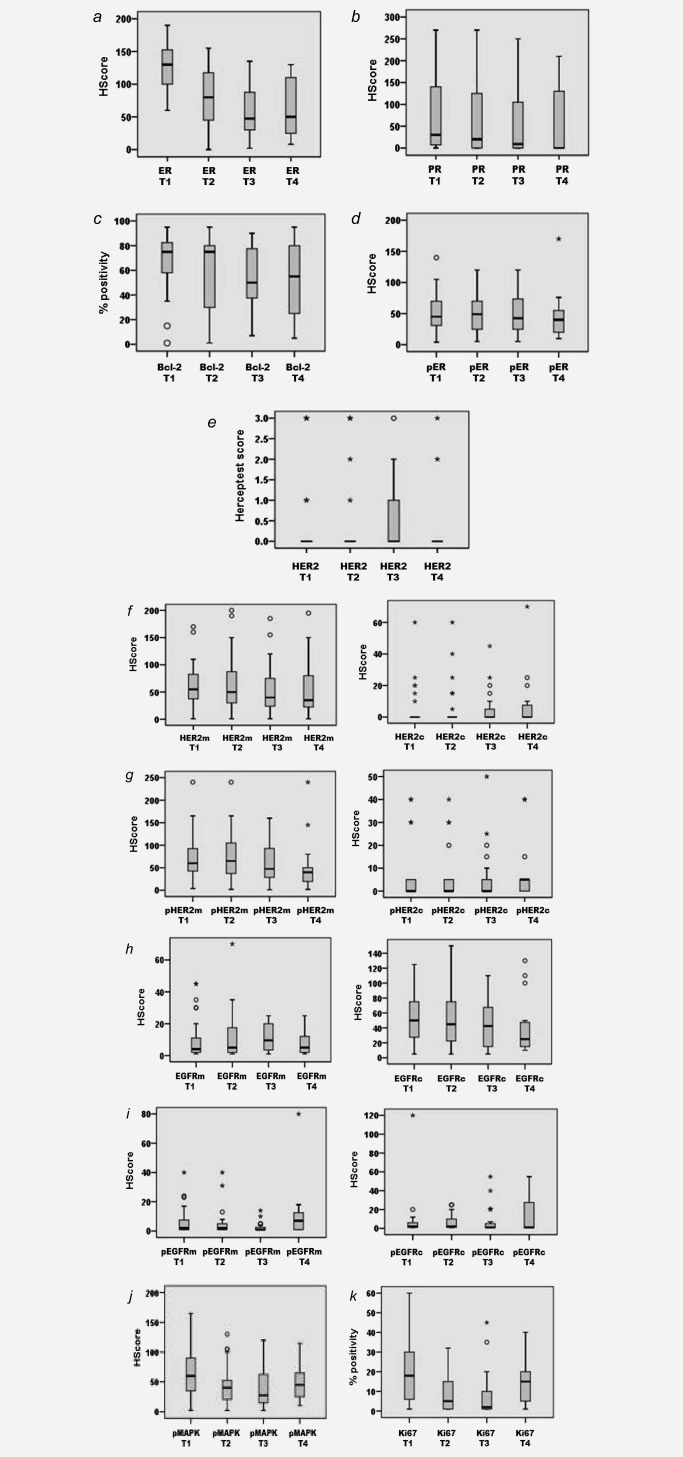
Box and whisker plots displaying marker profile (unmatched data) across T1 (pre‐treatment) and T2, T3 and T4 (6 week, 6 month and at progression on fulvestrant, respectively) for the All‐patient cohort. (a) ER H Score; (b) PR H Score; (c) % Bcl‐2; (d) pER (serine 118 phosphorylation) H Score; (e) HercepTest™ score; (f) HER2m (membrane) and HER2c (cytoplasmic) H Scores; (g) pHER2m (membrane) and pHER2c (cytoplasmic) H Scores; (h) EGFRm (membrane) and EGFRc (cytoplasmic) H Scores; (i) pEGFRm (membrane) and pEGFRc (cytoplasmic) H Scores; (j) pMAPK H Score and (k) % Ki67 staining (o = outliers and *= extreme outliers with values more than 3× the box height).
Figure 2.
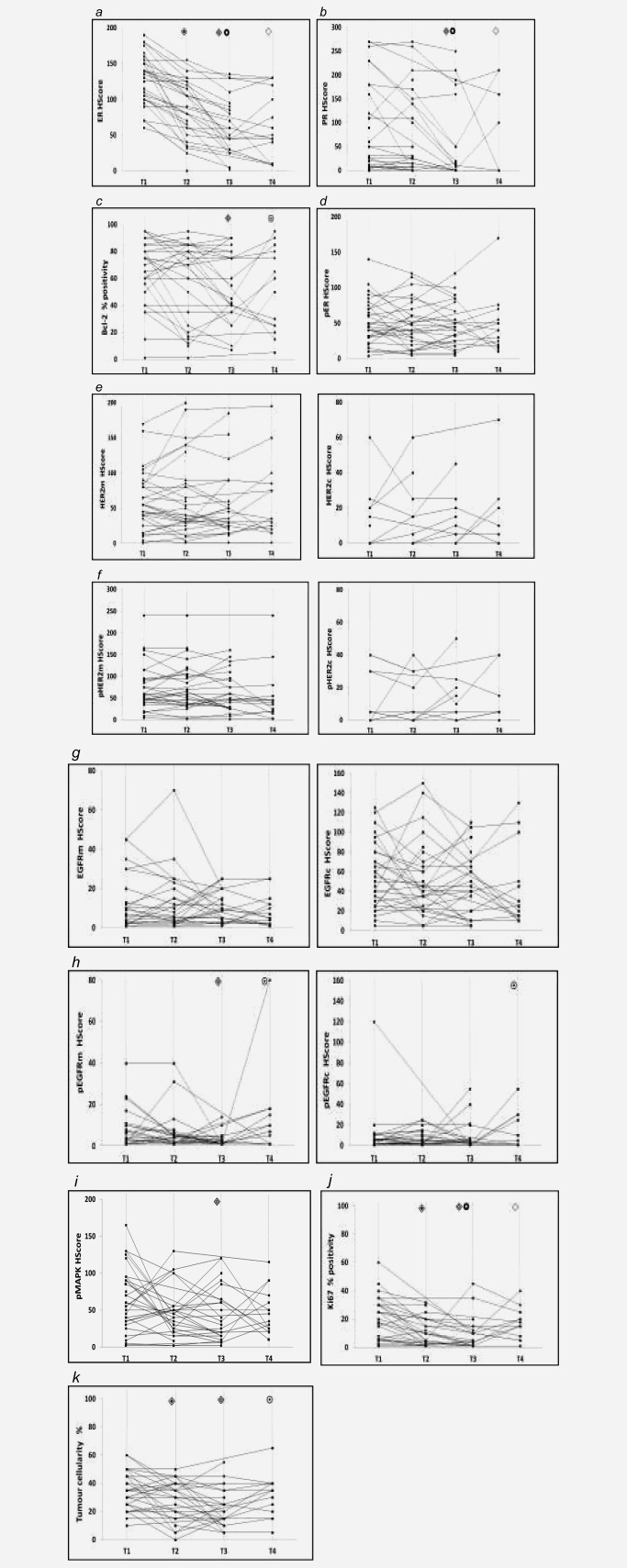
Line plots of individual changes in markers (matched data) across T1 (pretreatment) and T2, T3 and T4 (6 week, 6 month and at progression on fulvestrant respectively) for each patient. Wilcoxon Paired Signed‐Rank test compared staining between time‐points using paired sample data from each patient. Significance between T1‐T2, T1‐T3, T2‐T3, T1‐T4 or T3‐T4 is indicated by  ,
,  ,
,  ,
,  and
and  , respectively. To aid visualisation of profile over the multiple time‐points in these plots, marker data have been connected in the same patient if any sample was unavailable. (a) ER H Score (T1‐T2, T1‐T3, T2‐T3 falls p < 0.001; T1‐T4 fall p = 0.001); (b) PR H Score (T1‐T3 p < 0.001, T2‐T3 falls p = 0.006; T1‐T4 fall p = 0.012); (c) Bcl‐2% positivity (T1‐T3 fall p = 0.007; T3‐T4 increase p = 0.066); (d) pER (serine 118 phosphorylation) H Score; (e) HER2m (membrane) and HER2c (cytoplasmic) H Scores; (f) pHER2m (membrane) and pHER2c (cytoplasmic) H Scores (NB. no measurable staining was detected in any sample from 20 patients for HER2c and 16 patients for pHER2c); (g) EGFRm (membrane) and EGFRc (cytoplasmic) H Scores; (h) pEGFRm (membrane; T1‐T3 fall p = 0.024; T3‐T4 increase p = 0.012) and pEGFRc H Scores (cytoplasmic; T3‐T4 increase p = 0.041); (i) pMAPK H Score (T1‐T3 fall p = 0.019); (j) Ki67% positivity (T1‐T2 p = 0.001, T1‐T3 p = 0.012, T2‐T3 falls p = 0.048; T1‐T4 fall p = 0.028); (k) Tumour cellularity % (T1‐T2 fall p = 0.019; T1‐T3 fall p = 0.003; T3‐T4 increase p = 0.065).
, respectively. To aid visualisation of profile over the multiple time‐points in these plots, marker data have been connected in the same patient if any sample was unavailable. (a) ER H Score (T1‐T2, T1‐T3, T2‐T3 falls p < 0.001; T1‐T4 fall p = 0.001); (b) PR H Score (T1‐T3 p < 0.001, T2‐T3 falls p = 0.006; T1‐T4 fall p = 0.012); (c) Bcl‐2% positivity (T1‐T3 fall p = 0.007; T3‐T4 increase p = 0.066); (d) pER (serine 118 phosphorylation) H Score; (e) HER2m (membrane) and HER2c (cytoplasmic) H Scores; (f) pHER2m (membrane) and pHER2c (cytoplasmic) H Scores (NB. no measurable staining was detected in any sample from 20 patients for HER2c and 16 patients for pHER2c); (g) EGFRm (membrane) and EGFRc (cytoplasmic) H Scores; (h) pEGFRm (membrane; T1‐T3 fall p = 0.024; T3‐T4 increase p = 0.012) and pEGFRc H Scores (cytoplasmic; T3‐T4 increase p = 0.041); (i) pMAPK H Score (T1‐T3 fall p = 0.019); (j) Ki67% positivity (T1‐T2 p = 0.001, T1‐T3 p = 0.012, T2‐T3 falls p = 0.048; T1‐T4 fall p = 0.028); (k) Tumour cellularity % (T1‐T2 fall p = 0.019; T1‐T3 fall p = 0.003; T3‐T4 increase p = 0.065).
Table 2.
Summary of significant results from matched statistical analysis of biomarkers in the fulvestrant treated series for the All‐patients, CB (clinical benefit), EP (early progressors) and CONR/LP (continuing responders/late progressors) cohorts
| All‐patients | CB | EP and CONR/LP | |
|---|---|---|---|
| ER HScore |
T1‐T2‐T3: ER fall, p < 0.001 T1‐T2 or T1‐T3: ER fall, p < 0.001 T2‐T3: ER fall, p < 0.001 T1‐T4: ER fall, p = 0.001 |
T1‐T2‐T3: ER fall, p < 0.001 T1‐T2 or T1‐T3: ER fall, p < 0.001 T2‐T3: ER fall, p < 0.001 T1‐T4: ER fall, p = 0.004 |
EP → ER fall, T1‐T2: p = 0.018 T1‐T3: p = 0.003 T2‐T3: p = 0.028 CONR/LP → ER fall, T1‐T2: p = 0.003 T1‐T3: p = 0.005 T2‐T3: p = 0.008 |
| PR HScore |
T1‐T2‐T3: PR fall, p < 0.001 T1‐T2: PR unchanged in many patients T1‐T3: PR fall, p < 0.001 T2‐T3: PR fall, p = 0.006 T1‐T4: PR fall, p = 0.012 |
T1‐T2‐T3: PR fall, p < 0.001 T1‐T2: PR unchanged in many patients T1‐T3: PR fall, p = 0.008 T2‐T3: PR fall, p = 0.042 T1‐T4: PR fall, p = 0.018 |
EP → PR fall, T1‐T3: p = 0.008 T2‐T3: p = 0.042 CONR/LP → PR fall, T1‐T3: p = 0.093 T2‐T3: p = 0.063 |
| Bcl‐2% |
T1‐T2: Bcl‐2 unchanged in many patients T1‐T3: Bcl‐2 fall, p = 0.007 T3‐T4: Bcl‐2 rise in 64% patients, p = 0.066 |
T1‐T2: Bcl‐2 unchanged in many patients T1‐T3: Bcl‐2 fall, p = 0.012 T3‐T4: Bcl‐2 rise in 70% patients, p = 0.04 |
EP → Bcl‐2 fall, T1‐T2; p = 0.042 T1‐T3: p = 0.028 CONR/LP → T1‐T2: unchanged T1‐T3: p = 0.138 |
| pER HScore | 54% patients show decrease by T3, but no dominant change in pER during treatment | Some decreases in patients by T3, but no dominant change in pER during treatment | Some decreases in patients by T3, but no dominant change in pER during treatment |
|
HER2 expression: HercepTestTM or HER2m and HER2c H Score HER2 activity: pHER2m and HER2c H Score |
HercepTest™: T1‐T2‐T3: HER2 rise, p< 0.001 T1‐T3: HER2 rise, p = 0.034 |
HercepTest™: T1‐T3: HER2 rise, p = 0.034 HER2c assay: T2‐T3: HER2c rise, p = 0.041 |
pHER2m assay: EP → pHER2m rise, T2‐T3: p = 0.078 |
|
EGFR expression: EGFR activity: pEGFRm andpEGFRc H Score |
EGFR: no dominant change T1‐T3: pEGFRm fall, p = 0.024 T3‐T4: pEGFRm rise in 67% patients, p = 0.012 T3‐T4: pEGFRc rise in 50% patients, p = 0.041 |
EGFR: no dominant change T1‐T3: pEGFRm fall, p = 0.028 T3‐T4: pEGFRm rise in 72% patients, p = 0.011 T3‐T4: pEGFRc rise in 55% patients, p = 0.041 |
EGFR: No dominant change Some falls but no dominant change in pEGFR |
| pMAPK Hscore | T1‐T3: pMAPK fall, p = 0.019 | T1‐T3: pMAPK fall, p = 0.028 | Some falls but no dominant change in pMAPK |
| Ki67% |
T1‐T2‐T3: Ki67 fall, p < 0.001 T1‐T2: Ki67 fall, p = 0.001 T1‐T3: Ki67 fall, p = 0.012 T2‐T3: Ki67 fall, p = 0.048 T1‐T4: Ki67 fall, p = 0.028 T1‐T2‐T3‐T4: Ki67 rise in 45% patients, p = 0.077 |
T1‐T2‐T3: Ki67 fall, p < 0.001 T1‐T2: Ki67 fall, p = 0.001 T1‐T3: Ki67 fall, p = 0.012 T2‐T3: Ki67 fall, p = 0.048 T1‐T4: Ki67 fall, p = 0.074 |
EP → Ki67 fall T1‐T2‐T3: p = 0.032 T1‐T2: p = 0.027 T1‐T3: p = 0.033 T2‐T3: unchanged CONR/LP → Ki67 fall T1‐T2‐T3: p = 0.003 T1‐T2: p = 0.021 T1‐T3: p = 0.007 T2‐T3: p = 0.021 |
|
Tumour
cellularity % |
T1‐T2‐T3: cellularity fall, p = 0.007 T1‐T2: cellularity fall, p = 0.019 T1‐T3: cellularity fall, p = 0.003 T3‐T4: cellularity rise in 50% patients, p = 0.065 |
T1‐T2‐T3: cellularity fall, p = 0.007 T1‐T2: cellularity fall, p = 0.01 T1‐T3: cellularity fall, p = 0.004 T3‐T4: cellularity rise in 46% patients, p = 0.088 |
EP → cellularity fall T1‐T2‐T3: p = 0.018 T1‐T2: p = 0.045 T1‐T3: p = 0.028 CONR/LP → cellularity fall T1‐T2‐T3: p = 0.067 T1‐T2: p = 0.057 T1‐T3: p = 0.027 |
Statistical analyses were performed in SPSSTM using Friedman's ANOVA for T1‐T2‐T3 and T1‐T2‐T3‐T4 and Wilcoxon Paired Signed‐Rank test for all paired analysis. T1= pre‐treatment; T2= 6 week fulvestrant treatment; T3= 6 month fulvestrant treatment; T4= fulvestrant progression; c= cytoplasmic; m= membrane; CB= clinical benefit patient group; EP= early progressor patient group; CONR/LP= continuing responder/late progressor patient group.
ER expression, activity and ER‐regulated proteins PR and Bcl‐2
ER declined significantly versus T1 at all fulvestrant treatment time‐points and in all cohorts reaching its lowest levels by T3 which were then generally maintained at T4 (Figs. 1 a and 2a; Table 2; Supporting Information Tables S2–S4). No patient had lost all ER at relapse even in the longest T1‐T4 interval of 60.7 months. An example of sequential biopsy ER immunostaining is provided in Supporting Information Fig. S1a.
PR was detectable in most patients at T1. Despite small numbers of progressive disease patients, it was noted that PR was significantly lower than in CB patients at T1 (p = 0.012), with three of the 4 PR‐negative tumours being progressive disease. Although falls were generally modest at T2, by T3 PR had declined in all of the patient cohorts versus T1 (Figs. 1 b and 2b; Table 2; Supporting Information Tables S2–S4). Supporting Information Fig. S1b shows an example of sequential biopsy PR immunostaining. Interestingly, PR fall at T2 was more substantial and by T3 reached significance in EP patients, contrasting a more modest decline in the longer‐responding CONR/LP patients (Table 2; Supporting Information Table S4). PR remained significantly lower at T4 than T1 in the All‐patient cohort (Figs. 1 b and 2b; Supporting Information Table S2) and CB cohort following matched analysis (Table 2), with complete PR loss in one third of tumours (as predominantly seen in Supporting Information Fig. S1b). However, three tumours maintained substantial PR (H‐Score >= 100) at T4 exceeding T3 (Fig. 2 b).
Bcl‐2 was detected in all T1 samples irrespective of response status. Bcl‐2 fell significantly in 58% patients by T3 (Figs. 1 c, 2c; Table 2; Supporting Information Tables S2, S3 and Supporting Information Fig. S1c). Significant Bcl‐2 decreases were apparent in early progressors at both T2 and T3, but there was no T2 fall in CONR/LP patients and a marginal decline by T3 (Table 2; Supporting Information Table S4). Some T3‐T4 Bcl‐2 recovery occurred in approximately 65% patients (Fig. 2 c), reaching significance in about 70% CB so there was no significant staining difference between T1 and T4 (Table 2; Supporting Information Table S3). Supporting Information Figure S1c shows such Bcl‐2 staining.
ER activity (phosphorylated serine 118), again detected in all T1 samples irrespective of response status, decreased very modestly in 54% patients by T3 on matched analysis (Fig. 2 d; Table 2). Supporting Information Figure S1d provides an example of such sequential biopsy pER staining. No patient was pER negative at T4 and there was no consistent pattern of pER change at this time‐point (Figs. 1 d, 2d; Supporting Information Tables S2 and S3). Spearman's analysis showed that pER weakly correlated with ER expression at all time‐points (T1 p = 0.018, r = 0.42; T2 p = 0.054, r = 0.38; T3 p = 0.025, r = 0.46; T4 p = 0.003, r = 0.74) and with Bcl‐2 at T1 (p = 0.005, r = 0.5) and T3 (p = 0.008, r = 0.53).
Kaplan–Meier analysis revealed no significant relationship between ER expression or activity and DoR on fulvestrant at any time‐point or at T1 or T3 for PR or Bcl‐2 (data not shown). However, DoR was significantly prolonged where PR or Bcl‐2 staining at T2 exceeded the median cut‐point [Figs. 3 a (p = 0.008) and 3b (p = 0.01), respectively], while Bcl‐2 level in CONR/LP patients also significantly exceeded early progressors at this time‐point (p = 0.01; Supporting Information Table S4).
Figure 3.
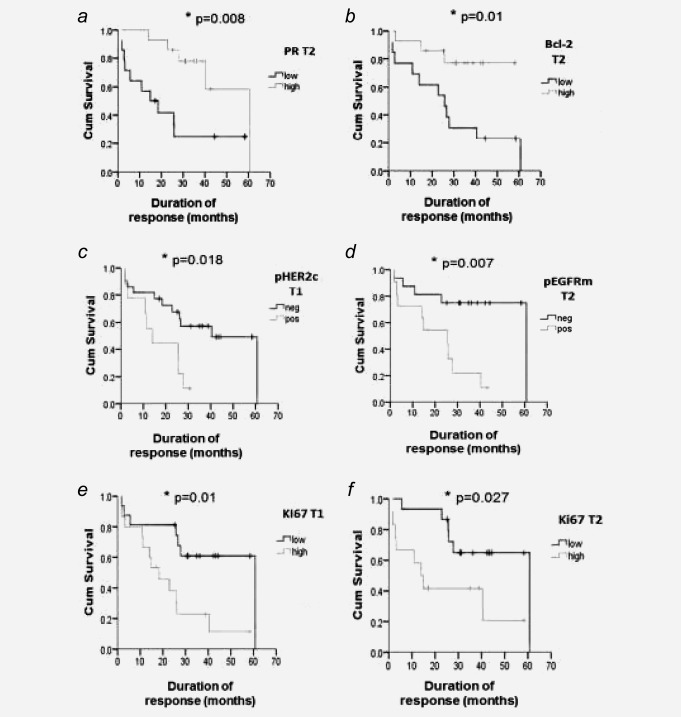
Kaplan–Meier association (using the median staining level from Supporting Information Table S2 at each time point as a cut‐point) between biomarkers and duration of response (DoR) to fulvestrant (*p < 0.05 using Log Rank test). Responses to any other treatments following fulvestrant progression were not a component of these response data. (a) PR H Score at T2; p = 0.008 for higher PR [mean DoR= 48.2 months (95% CI=36.9–59.5; n = 14)] vs. lower PR [mean DoR= 23.4 months (95% CI= 11.6–35.1; n = 14)]; (b) Bcl‐2% at T2; p = 0.01 for higher Bcl‐2% [mean DoR= 48.4 months (95% CI=38.5–58.4; n = 13)] vs. lower Bcl‐2% [mean DoR= 27.5 months (95% CI= 15.3–39.6; n = 14)]; (c) pHER2c (cytoplasmic) H Score at T1; p = 0.018 for any detectable pHER2c [mean DoR= 16.8 months (95% CI= 10.1–23.5); n = 9] versus no pHER2c [mean DoR= 39.1 months (95% CI= 28.5–49.7; n = 22)]; (d) pEGFRm (membrane) H Score at T2; p = 0.007 for any detectable pEGFRm [mean DoR= 21.1 months (95% CI= 12.5–29.7; n = 11)] vs. no pEGFRm [mean DoR= 48.1 months (95% CI= 36.0–60.3; n = 16)]; (e) Ki67% at T1; p = 0.01 for higher Ki67 [mean DoR= 22.2 months (95% CI= 13.2–31.3; n = 15)] vs. lower Ki67 [mean DoR= 43.1 months (95% CI= 30.6–55.5; n = 16)]; (f) Ki67% at T2; p = 0.027 for higher Ki67 [mean DoR= 24.7 months (95% CI= 11.6–37.9; n = 12)] vs. lower Ki67 [mean DoR= 47.1 months (95% CI= 36.2–58.0; n = 15)].
HER2, EGFR and their activated forms (pHER2, pEGFR)
Generally, very modest HER2 membrane (HER2m) expression and activity (pHER2m) were detectable in this series, with little or no HER2 cytoplasmic (HER2c, pHER2c) staining (Figs. 1 e‐g; Figs. 2e and 2f; Supporting Information Tables S2–S4). Staining with HercepTestTM was seen at T1 in 17% patients (5 of the 29 patients with adequate HercepTestTM data), including three 3+ samples comprising one CB and two PD. T1 samples with HercepTestTM score >0 also showed some positivity for HER2m/pHER2m. There were few significant changes in HER2 expression or activity with fulvestrant in this patient series (Figs. 1 e‐g; Figs. 2e and 2f; Supporting Information Tables S2–S4; Supporting Information Figs. S1e and S1g). HercepTestTM staining increased in 29% patients by T3 (Fig. 1 e). These increases reached significance by matched analysis in the whole series and in CB patients (Table 2) but were modest (HercepTestTM score 0‐to‐1: n = 2; 0/1‐to‐2: n = 3) with no 3+ gains. An additional HER2 expression assay also detected T2‐T3 HER2c increases in approximately 25% paired samples from the whole series (Figs. 1 f and 2e), although many remained HER2c negative. This increase reached significance in CB (Table 2), where there were additionally occasional modest T3 increases in HER2m and pHER2 on matched analysis in approximately 40% of patients (Figs. 2 e and 2 f; Supporting Information Figs. S1f and S1h). There was a trend for a T2‐T3 pHER2m increase in 60% EP samples (Table 2). HER2m expression (p = 0.028, r = 0.45) and activity at T3 (p = 0.016, r = 0.49) directly associated with pER by Spearman's analysis. Although occasional samples showed changes (including slight HER2c and pHER2c increases and one patient with a 0‐to‐2 HercepTestTM score increase), there was no dominant pattern in HER2 expression/activity at relapse with generally modest levels in T4 samples (Figs. 1 f and 1 g; Figs. 2e and 2f; Supporting Information Tables S2 and S3; Supporting Information Figs. S1e and S1g).
This series also had low EGFR membrane (EGFRm) and cytoplasmic (EGFRc) staining, excepting four patients at T1 (H Score>=100) of which three were CB. Some EGFR changes were seen in samples during treatment but membrane H Scores remained very low (median <=10) along with modest cytoplasmic signals (Figs. 1 h and 2g; Supporting Information Fig. S1i; Supporting Information Tables S2–S4). There was no dominant pattern of EGFR expression change on matched analysis at any time‐point (Fig. 2 g; Table 2). Similarly, pEGFR levels were generally very low (median <=10; Fig. 1i; Supporting Information Tables S2–S4). There was significant loss of detectable pEGFRm staining in approximately 50% patients by T3 including in CB (Figs. 1 i 2h; Table 2; Supporting Information Fig. S1j). While still generally at extremely low levels at T4 (median H Score <=10), small significant increases were subsequently detected for pEGFRc or pEGFRm in 50–67% T4 samples including 72% of CB (Figs. 1 i and 2h; Table 2; Supporting Information Tables S2 and S3; Supporting Information Fig. S1j). Tumours with weakly detectable T3‐T4 increases in pEGFRm also showed modest HER2m increases (p = 0.027) and Spearman's analysis revealed direct associations at T4 between EGFR activity and pER (p = 0.006), Bcl‐2 (p = 0.055) and Ki67 (p = 0.047).
Kaplan–Meier analysis was not meaningful for total EGFR or HER2 expression since this ER+ series contained very few highly HER2 or EGFR expressing tumours. Patients with any pHER2c positivity at T1 had a shortened DoR (p = 0.018; Fig. 3 c) but no relationship was seen for pHER2m. Although levels were extremely low, a shortened DoR was also observed for patients with any detectable pEGFRm at T2 (p = 0.007; Fig. 3 d). pHER2c (p = 0.051) and pEGFRm (p = 0.037) were also weakly increased in EP versus CONR/LP patient samples at these respective time‐points.
MAPK (ERK1/2) activity
Nuclear MAPK activity (pMAPK), detected at moderate levels in T1 samples irrespective of response status, significantly declined in approximately 65% of All‐patient and CB samples at T3 (Figs. 1 j, 2i; Table 2; Supporting Information Tables S2 and S3; Supporting Information Fig. S1k). Some falls were also apparent in EP and CONR/LP patients (Supporting Information Table S4). Such tumours with a pMAPK decrease by 6 months (n = 16) also showed significant decline in pEGFRm (p = 0.013), Bcl‐2 (p = 0.031) and Ki67 (p = 0.003). Subsequent non‐significant nuclear pMAPK increases occurred in approximately 50% T4 samples versus T3 (Figs. 1 j, 2i; Supporting Information Tables S2 and S3; Supporting Information Fig. S1k). Kaplan–Meier analysis revealed no relationship between pMAPK and DoR (data not shown).
Proliferative activity (Ki67)
Ki67 staining was modest for most samples, with significant decreases across T1‐T2‐T3 during fulvestrant treatment in all groups (Figs. 1 k and 2j; Table 2; Supporting Information Tables S2–S4; Supporting Information Fig. S1l). T2 and T3 Ki67 positivity was significantly lower than T1 in up to 79% patients for all groups (Fig. 2 j; Table 2). Tumour cellularity also decreased during fulvestrant response with T2 and T3 level significantly lower than T1 on matched analysis (Table 2; Fig. 2k; Supporting Information Tables S2–S4). There was a further very small but significant T2‐T3 Ki67 fall in approximately 60% of the All‐patient and CB cohorts (Figs. 1 k and 2j; Table 2). Interestingly, this continued T2‐T3 decline was only seen in the longer‐responding CONR/LP cohort and not in early progressors, where by T3 there was an indication of some modest recovery in proliferation (Table 2; Supporting Information Table S4). Although T4 Ki67 remained a little lower than T1 for approximately 70% patients (Figs. 1 k, 2j; Table 2) there was partial recovery in Ki67 in some T4 samples (Table 2: T1‐T2‐T3‐T4 p = 0.077; Fig. 2j; Supporting Information Tables S2 and S3; Supporting Information Fig. S1l) with T3‐T4 increases in 5/11 (about 45%) CB patients. Tumour cellularity also recovered in approximately 50% patients at T4 (Fig. 2 k; Table 2; Supporting Information Tables S2 and S3).
Kaplan–Meier analysis demonstrated a significantly shortened DoR at T1 (p = 0.01) for patients with Ki67 above the median cut‐point (>18% staining; Fig. 3 e). Concordantly, Ki67 was at a significantly higher level in early progressors versus the longer‐responding CONR/LP patient cohort at T1 (p = 0.037; Supporting Information Table S4). Multivariate analysis using Cox's proportional hazards model [considering univariate‐significant covariates baseline disease site and grade (Supporting Information Table S1), T1 pHER2c and T1 Ki67 (Figs. 3 c and 3 e)] showed T1 Ki67 was an independent predictor of fulvestrant DoR (p = 0.012), with high staining patients having a hazard 6.6‐fold that of low staining patients (Supporting Information Table S5). While levels were very low at T2 and T3 in CB patients, retention of any Ki67 at T2 was also adversely associated with DoR (p = 0.027; Fig. 1 f). With a trend at T2 (p = 0.071), early progressors had a significantly higher Ki67 level at T3 versus the longer‐responding CONR/LP cohort (p = 0.043; Supporting Information Table S4).
Discussion
This is the first clinical investigation of the biological impact of short (6 weeks), medium (6 months) and long‐term (>2 years) fulvestrant (250 mg/month) through to acquired resistance using sequential biopsies. In this ER+ breast cancer series, there was superior benefit for fulvestrant, with 81% CB and a median DoR of 25.8 (1.77–60.73) months, compared with previous reports of up to 60% CB and 4‐18 months response.3, 4, 6, 26, 27 Our series had substantial ER‐regulated proteins, modest EGFR/HER2 signaling and elevated Ki67 expression, a profile similarly equated to better outcome for other anti‐hormones.
The ER downregulation we observed with fulvestrant at 6 weeks is consistent with previous short‐term (<3 weeks) pre‐surgical primary breast cancer studies for short‐acting (6 or 18 mg/daily subcutaneously28) and long‐acting formulations (50–250 mg/month intramuscularly7). ER decline is also seen in the neoadjuvant (4 and 16 weeks treatment) setting.8 Such studies demonstrated that ER downregulation is fulvestrant dose‐dependent but we demonstrate here that treatment duration is a further influence since superior ER decline was achieved by 6 months (the timeframe for 250 mg fulvestrant to reach steady‐state2). Critically, we have found that ER level at all time‐points fails to relate to fulvestrant response. Indeed, by 6 weeks virtually every tumour had significant ER decline irrespective of patient response status or duration [occurring in CB (clinical benefit), PD (progressive disease), EP (early progressors) and CONR/LP patients (continuing responders/late progressors)]. The CONFIRM trial5 demonstrated a longer duration of CB while the NEWEST trial8 reported greater ER depletion for 500 mg versus 250 mg fulvestrant. However, our findings indicate that despite ER being the required target for fulvestrant and ER downregulation a hallmark of this agent's mechanism of action, parameters other than receptor level must determine extent of clinical fulvestrant response in ER+ tumours.
To determine whether ER activity was more informative, we monitored fulvestrant impact on ER phosphorylation and two ER‐regulated proteins. Although patient number precluded meaningful analysis of pER or Bcl‐2, PD patients were commonly PR negative at baseline suggesting classical estrogen/ER signaling is needed to achieve CB with fulvestrant. However, these PR findings remain clinically controversial.29, 30 Moreover, tissue inhibitor of metalloproteinases 1 (TIMP‐1) overexpression has been noted to promote PR loss and fulvestrant resistance in vitro potentially via modifying nonclassical ER activity.31 Fulvestrant promoted a time‐dependent fall in PR and Bcl‐2 (and modest pER decline in about 50% patients) in our series by 6 months. Previous shorter‐term studies using 250 mg fulvestrant have reported significant PR decline,7, 32 but our findings again equate better with the timeframe for 250 mg dose steady state2 and corroborate with NEWEST trial observations that 500 mg is required to significantly repress PR during short‐term treatment.8 We found that baseline ER activity, PR and Bcl‐2 did not relate to duration of CB, and in the longer‐responding (CONR/LP) patients PR and Bcl‐2 decline during treatment was at best small. This suggests that an extended DoR with fulvestrant does not equate with superior blockade of these particular ER‐regulated proteins.
We also examined whether fulvestrant influenced ER/GF pathway cross‐talk and if this determined response. Experimentally, endocrine agents can deplete ER‐regulated growth factor ligands for upstream receptors of MAPK,22 and after 6 months fulvestrant we noted that any membrane EGFR activity was lost and MAPK (ERK1/2) activity decreased. Such pMAPK depletion may contribute toward the small fall in phosphorylated serine 118 ER seen in some fulvestrant‐treated patients since pMAPK activates this AF‐1 residue.22 This pMAPK fall after 6 months fulvestrant was paralleled by Ki67 decline, so inhibition of ER/MAPK cross‐talk may contribute towards fulvestrant's anti‐proliferative effect, as reported in some ER+ models.33, 34 However, as T3 pMAPK and pEGFR decreases occurred in only about one‐half of CB patients, were extremely modest for pEGFR and were unrelated to outcome, further mechanisms must contribute to fulvestrant response in patients.
Importantly, low baseline Ki67 (<=median 18%) significantly associated with durable fulvestrant response in univariate and multivariate analysis. The subsequent fall in proliferation in many patients by 6 weeks was consistent with previous short‐term fulvestrant studies7, 8, 28 but we also determined that patients with the very lowest resultant proliferation (<=median 5%) had a longer DoR. In the IMPACT trial,35, 36 reduced proliferation at 2 weeks similarly predicted for extended disease‐free interval with tamoxifen or anastrozole presurgically. Proliferation suppression by fulvestrant was also reported in the neoadjuvant NEWEST trial8 with lowest Ki67 achieved using the clinically superior 500 mg dose.5 We also found that depletion of proliferation was apparent with longer‐term fulvestrant in many CB patients. By 6 months, there was a very small, continued Ki67 decline in the longer‐responding CONR/LP group contrasting partial recovery in early progressors. Thus, patients with the very lowest Ki67 after 6 months fulvestrant demonstrated superior response. Although further verification (including at 500 mg) is required, we propose that measuring this proliferation marker could have clinical predictive utility both to determine patients likely to substantially benefit from fulvestrant and as a quantitative measure of response.
We also report for the first time the tumour biomarker profile on acquisition of fulvestrant resistance in patients. Ki67 recovered in some clinical relapse samples compared with 6 month's treatment and this is likely to contribute towards tumour re‐growth (evidenced by increased cellularity). Frequent Bcl‐2 recovery suggests increased cell survival also plays a part. Low ER levels were retained at relapse, as seen in some acquired resistant models developed after 3–12 months fulvestrant treatment of ER+ cells.14 Several studies37, 38, 39 suggest that this residual ER may be functional in some patients. Cheung et al.39 reported CB following further endocrine treatment in 46% (13/28) and 12% (3/26) of patients who had initial CB or PD respectively on fulvestrant, including responses in seven patients that overlap with our series and retain ER at fulvestrant relapse. The persistent low levels of ER activity, PR and Bcl‐2 that we observed in fulvestrant relapse samples and the ER phosphorylation and Bcl‐2 detectable at pretreatment in PD patients provide further evidence for functional ER in some fulvestrant resistant tumours. This begs the question whether an increased dosage of fulvestrant might further deplete ER signaling and improve response, and provides rationale for development of more potent ER‐downregulators. In further support, superior ER depletion was seen with 500 mg fulvestrant both in NEWEST8 (vs. 250 mg examined up to 4 weeks) and Study 57.40 Treatment at 500 mg was also associated with improved DoCB and overall survival in CONFIRM5 and increased time to progression in the FIRST trial,9, 10 the latter also recently reporting better overall survival with fulvestrant versus anastrozole.41 Nevertheless, as Cheung et al.39 observed insensitivity to further endocrine agents in 54% of patients with initial CB on fulvestrant, a significant proportion of fulvestrant relapse patients may be
ER‐independent despite retention of ER. Moreover, some cell models reveal more prolonged (>2 years in vitro) fulvestrant can promote complete ER protein and mRNA loss.42 Theoretically, while ER positivity is a stable phenotype over the treatment window of the current study with 250 mg drug, further prolonging fulvestrant might ultimately promote an undesirable ER negative phenotype.
Increased EGFR/HER2 and MAPK signaling cross‐talks with ER AF‐1, promotes PR loss and drives tamoxifen or estrogen deprivation resistance in vitro.43 It is also detectable in some clinical samples on tamoxifen progression.44, 45 Here, very modest increases in EGFR activity occurred at relapse in approximately 65% fulvestrant treated patients versus 6 months, associating with HER2, pER, Bcl‐2 and proliferation. pMAPK also modestly recovered and PR was lost in about one‐third of relapse patients. Furthermore, although PD patients were few, two were HER2+ and one had high EGFR at pretreatment. Anti‐estrogen induced EGFR/HER2 signaling can begin to emerge during response in ER+ cells in vitro.14, 33, 34 Here, HercepTestTM score modestly increased in about one‐quarter of fulvestrant‐treated patients by 6 months and HER2 activity also weakly increased (particularly in early progressors) and correlated with pER. Along with preclinical studies,19, 33, 34, 46 our findings suggest such GF signaling and its cross‐talk with residual ER might modestly contribute towards limiting response in a small number of fulvestrant‐treated patients. However, HER2 induction with fulvestrant was infrequent, relapse was not paralleled by substantially increased EGFR/HER2/MAPK activity, and PD did not obviously associate with such signaling since one ER+ HercepTestTM positive patient and three patients with substantial baseline EGFR had clinical benefit on fulvestrant. These findings suggest lack of a central role for HER2/EGFR signaling in clinical fulvestrant resistance and explain recent trials showing no benefit of combining fulvestrant with lapatinib47 or gefitinib.48 Our findings also add to evidence that fulvestrant can be considered for HER2 or EGFR overexpressors.29, 49 For most patients, additional pathways clearly drive fulvestrant progression and pre‐clinical data are implicating potential players including erbB receptors,13, 16 PI3K/AKT/mTOR, Src kinase17, 18 and TIMP‐1.31 IHC in this sequential fulvestrant sample series is now continuing for further elements which should help interpret ongoing trials, including fulvestrant alongside PI3K or AKT inhibitors or with everolimus.50
Supporting information
Supporting Information Figure s1
Supporting Information Tables s1ab‐s5
Supporting Information LAB methods
Acknowledgements
Thanks go to P Finlay, SR Kyme, RA McClelland and HE Francies for biomarker immunohistochemistry, and to L Farrow for statistical analysis, under the direction of JMW Gee.
The study sponsor did not have any role in the collection, analysis and interpretation of data, in the writing of the report, and in the decision to submit the paper for publication.
Conflict of Interests: All clinical and biomarker studies were sponsored by AstraZeneca and the Tenovus Charity and Breast Cancer Now. A.A., E.G. and I.O.E. have no conflict. J.F.R.R., K.L.C., J.M.W.G. and R.I.N. have received honoraria and research funding from AstraZeneca. J.M.W.G. is in receipt of a research fellowship from Breast Cancer Now.
Previous Presentations: Preliminary results were presented at the San Antonio Breast Cancer Conference in 2004 by E. Gutteridge and at the European Breast Cancer Conference and the American Society of Clinical Oncology annual meeting in 2010 by A. Agrawal.
The copyright line for this article was changed on 24 May 2016 after original online publication.
References
- 1. Wakeling AE. Similarities and distinctions in the mode of action of different classes of antioestrogens. Endocr Relat Cancer 2000;7:17–28. [DOI] [PubMed] [Google Scholar]
- 2. Chia S, Gradishar W, Mauriac L, et al. Double‐blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor‐positive, advanced breast cancer: results from effect. J Clin Oncol 2008; 26:1664–70. [DOI] [PubMed] [Google Scholar]
- 3. Howell A, Robertson JFR, Quaresma Albano J, et al. Fulvestrant, Formerly ICI 182,780, Is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol 2002;20:3396–403. [DOI] [PubMed] [Google Scholar]
- 4. Osborne CK, Pippen J, Jones SE, et al. Double‐blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol 2002;20:3386–95. [DOI] [PubMed] [Google Scholar]
- 5. Di Leo A, Jerusalem G, Petruzelka L, et al. Final overall survival: fulvestrant 500 mg vs.250 mg in the randomized CONFIRM trial. J Natl Cancer Inst 2014;106:djt337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Howell A, Robertson JFR, Abram P, et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double‐blind, randomized trial. J Clin Oncol 2004;22:1605–13. [DOI] [PubMed] [Google Scholar]
- 7. Robertson JF, Nicholson RI, Bundred NJ, et al. Comparison of the short‐term biological effects of 7alpha‐[9‐(4,4,5,5,5‐pentafluoropentylsulfinyl)‐nonyl]estra‐1,3,5, (10)‐triene‐3,17beta‐diol (Faslodex) versus tamoxifen in postmenopausal women with primary breast cancer. Cancer Res 2001;61:6739–46. [PubMed] [Google Scholar]
- 8. Kuter I, Gee JM, Hegg R, et al. Dose‐dependent change in biomarkers during neoadjuvant endocrine therapy with fulvestrant: results from NEWEST, a randomized Phase II study. Breast Cancer Res Treat 2012;133:237–46. [DOI] [PubMed] [Google Scholar]
- 9. Robertson JF, Llombart‐Cussac A, Rolski J, et al. Activity of fulvestrant 500 mg versus anastrozole 1 mg as first‐line treatment for advanced breast cancer: results from the FIRST study. J Clin Oncol 2009;27:4530–5. [DOI] [PubMed] [Google Scholar]
- 10. Robertson JF, Lindemann JP, Llombart‐Cussac A, et al. Fulvestrant 500 mg versus anastrozole 1 mg for the first‐line treatment of advanced breast cancer: follow‐up analysis from the randomized 'FIRST' study. Breast Cancer Res Treat 2012;136:503–11. [DOI] [PubMed] [Google Scholar]
- 11. Bergh J, Jonsson PE, Lidbrink EK, et al. FACT: an open‐label randomized phase III study of fulvestrant and anastrozole in combination compared with anastrozole alone as first‐line therapy for patients with receptor‐positive postmenopausal breast cancer. J Clin Oncol 2012;30:1919–25. [DOI] [PubMed] [Google Scholar]
- 12. Mehta RS, Barlow WE, Albain KS, et al. Combination anastrozole and fulvestrant in metastatic breast cancer. N Engl J Med 2012;367:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Frogne T, Benjaminsen RV, Sonne‐Hansen K, et al. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat 2009;114:263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McClelland RA, Barrow D, Madden TA, et al. Enhanced epidermal growth factor receptor signaling in MCF7 breast cancer cells after long‐term culture in the presence of the pure antiestrogen ICI 182,780 (Faslodex). Endocrinology 2001;142:2776–88. [DOI] [PubMed] [Google Scholar]
- 15. Larsen SS, Egeblad M, Jaattela M, Lykkesfeldt AE. Acquired antiestrogen resistance in MCF‐7 human breast cancer sublines is not accomplished by altered expression of receptors in the ErbB‐family. Breast Cancer Res Treat 1999;58:41–56. [DOI] [PubMed] [Google Scholar]
- 16. Hutcheson IR, Goddard L, Barrow D, et al. Fulvestrant‐induced expression of ErbB3 and ErbB4 receptors sensitizes oestrogen receptor‐positive breast cancer cells to heregulin beta1. Breast Cancer Res 2011;13:R29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jordan NJ, Dutkowski CM, Barrow D, et al. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res 2014;16:R12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kirkegaard T, Hansen SK, Larsen SL, et al. T47D breast cancer cells switch from ER/HER to HER/c‐Src signaling upon acquiring resistance to the antiestrogen fulvestrant. Cancer Lett 2014;344:90–100. [DOI] [PubMed] [Google Scholar]
- 19. Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor‐HER2/neu cross‐talk in ER/HER2‐positive breast cancer. J Natl Cancer Inst 2004;96:926–35. [DOI] [PubMed] [Google Scholar]
- 20. Santen RJ, Song RX, Zhang Z, et al. Long‐term estradiol deprivation in breast cancer cells up‐regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer 2005;12 Suppl 1:S61–73. [DOI] [PubMed] [Google Scholar]
- 21. Martin LA, Pancholi S, Chan CM, et al. The anti‐oestrogen ICI 182,780, but not tamoxifen, inhibits the growth of MCF‐7 breast cancer cells refractory to long‐term oestrogen deprivation through down‐regulation of oestrogen receptor and IGF signalling. Endocr Relat Cancer 2005;12:1017–36. [DOI] [PubMed] [Google Scholar]
- 22. Britton DJ, Hutcheson IR, Knowlden JM, et al. Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen‐resistant growth. Breast Cancer Res Treat 2006;96:131–46. [DOI] [PubMed] [Google Scholar]
- 23. Osborne CK, Coronado‐Heinsohn EB, Hilsenbeck SG, et al. Comparison of the Effects of a Pure Steroidal antiestrogen With Those of Tamoxifen in a Model of Human Breast Cancer. J Natl Cancer Inst 1995;87:746–50. [DOI] [PubMed] [Google Scholar]
- 24. McClelland RA, Gee JM, Francis AB, et al. Short‐term effects of pure anti‐oestrogen ICI 182780 treatment on oestrogen receptor, epidermal growth factor receptor and transforming growth factor‐alpha protein expression in human breast cancer. Eur J Cancer 1996;32A:413–6. [DOI] [PubMed] [Google Scholar]
- 25. Ellis PA, Saccani‐Jotti G, Clarke R, et al. Induction of apoptosis by tamoxifen and ICI 182780 in primary breast cancer. Int J Cancer 1997;72:608–13. [DOI] [PubMed] [Google Scholar]
- 26. Gutteridge E, Agrawal A, Cheung K, et al. Early use of fulvestrant for the treatment of postmenopausal women with advanced breast cancer—the Nottingham experience. Eur J Cancer 2006;4:162 [Google Scholar]
- 27. Mauriac L, Pippen JE, Quaresma Albano J, et al. Fulvestrant (Faslodex) versus anastrozole for the second‐line treatment of advanced breast cancer in subgroups of postmenopausal women with visceral and non‐visceral metastases: combined results from two multicentre trials. Eur J Cancer 2003;39:1228–33. [DOI] [PubMed] [Google Scholar]
- 28. DeFriend DJ, Howell A, Nicholson RI, et al. Investigation of a new pure antiestrogen (ICI 182780) in women with primary breast cancer. Cancer Res 1994;54:408–14. [PubMed] [Google Scholar]
- 29. Bartsch R, Wenzel C, Altorjai G, et al. Her2 and Progesterone Receptor Status Are Not Predictive of Response to Fulvestrant Treatment. Clin Cancer Res 2007;13:4435–9. [DOI] [PubMed] [Google Scholar]
- 30. Mello CA, Chinen LT, da Silva SC, et al. Prolonged time to progression with fulvestrant for metastatic breast cancer. Med Oncol 2011;28:416–9. [DOI] [PubMed] [Google Scholar]
- 31. Bjerre C, Vinther L, Belling KC, et al. TIMP1 overexpression mediates resistance of MCF‐7 human breast cancer cells to fulvestrant and down‐regulates progesterone receptor expression. Tumour Biol 2013;34:3839–51. [DOI] [PubMed] [Google Scholar]
- 32. Robertson JF, Semiglazov V, Nemsadze G, et al. Effects of fulvestrant 250mg in premenopausal women with oestrogen receptor‐positive primary breast cancer. Eur J Cancer 2007;43:64–70. [DOI] [PubMed] [Google Scholar]
- 33. Massarweh S, Osborne CK, Jiang S, et al. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor‐positive, HER‐2/neu‐positive breast cancer. Cancer Res 2006;66:8266–73. [DOI] [PubMed] [Google Scholar]
- 34. Gee JM, Harper ME, Hutcheson IR, et al. The antiepidermal growth factor receptor agent gefitinib (ZD1839/Iressa) improves antihormone response and prevents development of resistance in breast cancer in vitro. Endocrinology 2003;144:5105–17. [DOI] [PubMed] [Google Scholar]
- 35. Dowsett M, Ebbs SR, Dixon JM, et al. Biomarker Changes During Neoadjuvant Anastrozole, Tamoxifen, or the Combination: Influence of Hormonal Status and HER‐2 in Breast Cancer–A Study from the IMPACT Trialists. J Clin Oncol 2005;23:2477–92. [DOI] [PubMed] [Google Scholar]
- 36. Smith IE, Dowsett M, Ebbs SR, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the immediate preoperative anastrozole, tamoxifen, or combined with tamoxifen (IMPACT) multicenter double‐blind randomized trial. J Clin Oncol 2005;23:5108–16. [DOI] [PubMed] [Google Scholar]
- 37. Robertson JF, Howell A, Gorbunova VA, et al. Sensitivity to further endocrine therapy is retained following progression on first‐line fulvestrant. Breast Cancer Res Treat 2005;92:169–74. [DOI] [PubMed] [Google Scholar]
- 38. Vergote I, Robertson JF, Kleeberg U, et al. Postmenopausal women who progress on fulvestrant ('Faslodex') remain sensitive to further endocrine therapy. Breast Cancer Res Treat 2003;79:207–11. [DOI] [PubMed] [Google Scholar]
- 39. Cheung KL, Owers R, Robertson JFR. Endocrine response after prior treatment with fulvestrant in postmenopausal women with advanced breast cancer: experience from a single centre. Endocr Relat Cancer 2006;13:251–5. [DOI] [PubMed] [Google Scholar]
- 40. Robertson JF, Dixon JM, Sibbering DM, et al. A randomized trial to assess the biological activity of short‐term (pre‐surgical) fulvestrant 500 mg plus anastrozole versus fulvestrant 500 mg alone or anastrozole alone on primary breast cancer. Breast Cancer Res 2013;15:R18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robertson J, Llombart‐Cussac A, Feltl D, et al. Fulvestrant 500 mg versus anastrozole as first‐line treatment for advanced breast cancer: overall survival from the phase II ‘first’ study. Abstract S6‐04. San Antonio Breast Cancer Conference. San Antonio 2014.
- 42. Giles M, Fiegl H, Widschwendter M, et al. Loss of estrogen receptor (ER) expression in MCF‐7 cells following long‐term exposure to fulvestrant. Breast Cancer Res Treat 2005;94:Abstract no. 5104. [Google Scholar]
- 43. Arpino G, Wiechmann L, Osborne CK, et al. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev 2008;29:217–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gee JM, Robertson JF, Gutteridge E, et al. Epidermal growth factor receptor/HER2/insulin‐like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr Relat Cancer 2005;12 Suppl 1:S99–S111. [DOI] [PubMed] [Google Scholar]
- 45. Gutierrez MC, Detre S, Johnston S, et al. Molecular changes in tamoxifen‐resistant breast cancer: relationship between estrogen receptor, HER‐2, and p38 mitogen‐activated protein kinase. J Clin Oncol 2005;23:2469–76. [DOI] [PubMed] [Google Scholar]
- 46. Osborne CK, Shou J, Massarweh S, et al. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 2005;11:865s–70s. [PubMed] [Google Scholar]
- 47. Burstein HJ, Cirrincione CT, Barry WT, et al. endocrine therapy with or without inhibition of epidermal growth factor receptor and human epidermal growth factor receptor 2: a randomized, double‐blind, placebo‐controlled phase III trial of fulvestrant with or without lapatinib for postmenopausal women with hormone receptor‐positive advanced breast cancer‐CALGB 40302 (Alliance). J Clin Oncol 2014;32:3959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carlson RW, O'Neill A, Vidaurre T, et al. A randomized trial of combination anastrozole plus gefitinib and of combination fulvestrant plus gefitinib in the treatment of postmenopausal women with hormone receptor positive metastatic breast cancer. Breast Cancer Res Treat 2012;133:1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robertson JF, Steger GG, Neven P, et al. Activity of fulvestrant in HER2‐overexpressing advanced breast cancer. Ann Oncol 2010;21:1246–53. [DOI] [PubMed] [Google Scholar]
- 50. Massarweh S, Romond E, Black EP, et al. A phase II study of combined fulvestrant and everolimus in patients with metastatic estrogen receptor (ER)‐positive breast cancer after aromatase inhibitor (AI) failure. Breast Cancer Res Treat 2014;143:325–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure s1
Supporting Information Tables s1ab‐s5
Supporting Information LAB methods