

Abstract
Affinity probes are useful tools for determining molecular targets and elucidating mechanism of action for novel, bioactive compounds. In the case of covalent inhibitors, activity based probes are particularly valuable for ensuring acceptable selectivity margins. However, there is a variety of bioorthogonalchemisty reactions available for modifying compounds of interest with clickable tags. Here, we describe a direct comparison of tetrazine ligation and strain promoted azide-alkyne cycloaddition using benzimidazole based probes to bind their known target, the gastric proton pump, ATP4A. This study validates the use of chemical probes for target identification and illustrates the superior efficiency of tetrazine ligation for copper-free click systems. In addition, we have identified several novel binding partners of benzimidazole probes: Isoform 2 of deleted in malignant brain tumors 1 protein (DMBT1) and three uncharacterized proteins.
Introduction
The development of covalent enzyme inhibitors is often avoided due to concerns of selectivity and possible immunogenicity of enzyme-inhibitors adducts. This is unfortunate, as covalent inhibitors often have increased efficiency and duration of action and also unnecessary as the development of clickable covalent probes can allow researchers to determine possible off-target effects1–3. Some of the most successful therapeutics in history are covalent inhibitors. For example, aspirin works as an NSAID by irreversibly acetylating a serine residue in the active site of cyclooxygenase COX-1 and COX-24, 5. Another example of successful covalent inhibitors are substituted benzimidazole compounds, which are widely used for the treatment of acid-related gastric diseases such as peptic ulcers and gastroesophageal reflux disease6, 7. Benzimidazole compounds are pro-drugs that must first undergo two protonations and a subsequent spontaneous rearrangement to become sufficiently reactive8, 9. Once activated, they form a disulfide bond with cysteine residues within the proton pump, the gastric H+/K+-ATPase, resulting in irreversible inhibition of enzymatic activity10–14. Because this reaction has been extensively studied and the target irrefutably identified as the catalytic subunit of the H+/K+-ATPase 14, 15, ATP4A, covalent probes derived from these compounds can serve as a useful model system for testing the efficiency of different bioorthogonal reactions in protein labeling. The copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction has widespread use in bioorthogonal labeling; however, this type of reaction is not always ideal because it requires a copper catalyst. In the case of benzimidazoles, reducing agents disrupt the disulfide bond between inhibitor and enzyme making CuAAC unsuitable for protein labeling with these compounds. By incorporating clickable moieties compatible with copper-free click chemistries, one can utilize covalent probes without being limited by toxicity or solubility issues16–18. Subsequently, the strain-promoted azide-alkyne cycloaddition (SPAAC)19, 20 and inverse electron demand diels-Alder (iEDDA)21–23 reaction for tetrazine ligation have become increasingly popular for protein labeling applications. We have synthesized a series of benzimidazole analogues containing different “clickable” moieties that can be used in a copper free setting to directly label the molecular target of these compounds: the H+/K+ exchanging ATPase or the gastric proton pump. We have directly compared tetrazine ligation and SPAAC and found that tetrazine ligation is far superior; not only in regard to reaction rate, but efficiency as well. Our results validate the development of chemical probes for discovery of unknown targets and also emphasize the ease of implementation when using copper-free clickable systems. In particular, this study highlights the utility and efficiency of tetrazine covalent probes.
Results and Discussion
Synthesis of Benzimidazole Probes
We synthesized two benzimidazole probes: Rabe-Tz and Rabe-N3 (Figure 1A). The compound 1 was treated with 2 in the presence of potassium hydroxide in DMSO under microwave condition to give compound 3 in 89% yield. Alcohol 3 was coupled with 4 in the presence of 1-ethyl-3-(3-dimetylaminopropyl) carbodiimide hydrochloride (EDC·HCl) and 4-dimethylaminopyridine (4-DMAP) in CH2Cl2, followed by oxidation with 3-chloroperoxybenzoic acid (mCPBA) to give the tetrazine-benzimidazole compound Rabe-Tz. Phenol 5 was coupled with alcohol 3 in the presence of diisopropyl azodicarboxylate (DIAD) and triphenylphosphine (polymer-bound), followed by oxidation with 3-chloroperoxybenzoic acid (mCPBA) yielding the azide-benzimidazole compound Rabe-N3.
Figure 1.

(A) Synthetic route for benzimidazole probes: Rabe-Tz and Rabe-N3. (B) Copper-free click chemistries available to synthesized probes.
Benzimidazole Probes are Potent Proton-Pump Inhibitors
In order to determine if the synthesized analogues retain inhibitory activity against the proton pump, we prepared gastric H+/K+-ATPase containing vesicles for use in a colorimetric activity assay. Differential centrifugation of gastric mucosa from pig stomachs, with slight modifications from procedures previously reported24–26, was used for vesicle isolation. To characterize the gastric H+/K+-ATPase in our sample, enzyme activity was detected by measuring the amount of inorganic phosphate released using the PiColorLock Gold Phosphate Detection System. Background phosphate release was measured using reactions carried out in the absence of MgCl2, in which gastric H+/K+-ATPase is inactive, as described previously by Shin et al27. Gastric H+/K+-ATPase activity was defined by the difference in signal in the absence and the presence of 100 μM Rabeprazole, a known proton-pump inhibitor (Figure 2A, B).
Figure 2.

(A) Structure of known proton-pump inhibitor, Rabeprazole (B) Gastric H+/K+-ATPase activity is inhibited by 100μM Rabeprazole. (C) Benzimidazole probes inhibit ATPase activity by approximately 50% at 10μM compound.
Each benzimidazole probe (10μM) was incubated with H+,K+-ATPase containing vesicles to determine their potency. Figure 2C shows the inhibitory activity of synthesized compounds in our assay. We tested both analogues in comparison to the known inhibitor, Rabeprazole, and found that all compounds tested inhibit the enzyme at similar levels. This suggests that the modifications to the core benzimidazole structure did not change potency of either compound.
Direct Labeling of Gastric H+/K+-ATPase, ATP4A, with Benzimidazole Analogues
Since the analogues completely retained inhibitory activity, we tested if they still directly bind and label their target. Gastric H+/K+-ATPase containing membranes were prepared using differential centrifugation, incubated with each probe, and copper-free click chemistry (Figure 1B) was utilized to attach a biotin tag. A streptavidin resin is then used to enrich for labeled proteins, which can be analyzed using western blot (Figure 3A). Because these compounds require low pH for activation, labeling should only occur under sufficiently acidic conditions. We performed the labeling experiment using Rabe-Tz at a pH of 2, 3, 4 and 7 and observed labeling of a protein with apparent molecular weight of 270kDa at pH=3 (Figure 3D). While a recent study proposes that the active form of the ATPase is the monomeric form (≈114kDa)26, extensive early research suggest the enzyme functions as a dimer or higher oligomer in conjunction with the β subunit28–33. Our finding suggests that active ATP4A is a dimer. Surprisingly, no labeled ATP4A was detected at pH 2, in which Rabe-Tz should be activated for covalently binding to the target. We reasoned that ATP4A is less soluble when it was treated at pH 2 and extracted from the membrane environment. For this reason, all subsequent experiments were run at pH=3. Specificity of labeling was shown by addition of a reducing agent, used to break the disulfide bond between the probe and its target. For both compounds, labeling is completely abolished by the addition of 50mM DTT (Figure 3C). Next, we directly compared the labeling efficiency of the two analogues by titrating the dose of probe used for labeling between 0.3 and 3.0 μM. As shown in Figure 3D, each compound labels ATP4A in a dose dependent manner, but Rabe-Tz is clearly more efficient, labeling with much stronger intensity at similar concentrations. There are two possibilities that explain the differences in labeling efficiency between Rabe-Tz and Rabe-N3. The first possibility is that Rabe-Tz binds to the proton pump with a higher affinity. The second option is that tetrazine ligation is more efficient than SPAAC, resulting in superior labeling. Based on the activity data showing that each analogue inhibits the proton pump with similar potency, it is plausible to assume that Rabe-Tz labels more strongly due to a more efficient click reaction34.
Figure 3.

Benzimidazole probes label the gastric H+/K+-ATPase, ATP4A. (A) Schematic drawing of labeling protocol. After incubation of probe and gastric membranes biotin is “clicked” onto the probe, the target enriched using streptavidin beads, separated using SDS-PAGE and analyzed by western blot. (B) Labeling is pH dependent, requiring a pH of 3.0 for acid activation of benzimidazole compounds. (C) Direct comparison of labeling effi ciency between probes at 1μM shows that labeling is blockable with DTT (50mM). (D) Labeling with 0.3, 1, and 3μM probe shows that labeling is dose dependent and Rabe-Tz labels more efficiently than Rabe-N3 at equivalent concentrations.
Fluorescent Imaging of Labeled Proteins
To take advantage of the efficient labeling of ATP4A with Rabe-Tz, we sought to identify Rabe-Tz labeled proteins using an unbiased approach: direct labeling with tetramethyl rhodamine (TAMRA) dye (Figure 4A). Following incubation of gastric vesicles with the tetrazine probe, we clicked a trans-cyclooctene-TAMRA dye to labeled proteins for visualization on SDS-PAGE gel. Figure 4B shows that Rabe-Tz directly labels two protein bands with apparent molecular weight of approximately 270 kDa and approximately 70kDa. Moreover, both bands are eliminated with the addition of DTT, and Coomassie blue staining confirms that equal amounts of protein were loaded to each lane.
Figure 4.

(A) Schematic representation of protocol used for unbiased protein labeling with TAMRA dye. (B) Rabe-Tz probe specifically labels protein at >250kDa and approximately 70kDa. Lane 1 and 2 are duplicate samples while Lane 3 demonstrates that labeling is blocked by 50mM DTT.
In order to identify these TAMRA labeled proteins, we performed a pull down experiment based on the protocol shown in Figure 3A, however, following SDS-PAGE, we stained the gel with Coomassie blue. No clear bands were visible, so gel slices at approximately 250kDa and ~70 kDa, where TAMRA labeling was observed, were excised and digested for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Peptide identifications were accepted if they could be established at greater than 95% probability to achieve an FDR less than 1.0% by the Scaffold Local FDR algorithm. Protein identifications were accepted if they contained at least 4 identified peptides. The band at 70kDa was found to correspond to serum albumin and is likely pulled down as a result of its extremely high abundance. For the higher molecular weight band, five proteins were identified exclusively in the sample not treated with DTT and can be seen in Table 1. As expected, the catalytic subunit of the gastric H+/K+-ATPase was a top hit, further confirming ATP4A as the target of Rabe-Tz. In addition, Isoform 2 of deleted in malignant brain tumors 1 protein (DMBT1) was also identified. This protein is a secreted scavenger receptor cysteine-rich protein that has been reported to be deleted in numerous human cancers including: brain35, lung36, and gastrointestinal cancers37. It is thought to play a role in both immune defense and epithelial differentiation38 and has also been reported to bind Helicobacter pylori39. Interestingly, Benzimidazole compounds are commonly used to treat H.pylori infection40 with an unknown cellular target and both DMBT1 and benzimidazole compounds have been reported to play a protective role for gastrointestinal mucosa41, 42. It would be interesting to carry out more studies to examine the physiological interaction between DMBT1 and benzimidazole compounds. Alternatively, the abundance of cysteine residues in this protein could account for benzimidazole binding under low pH. The remaining three proteins are currently uncharacterized in the literature, although, according to www.uniport.org, F1SKJ1 is inferred to have actin-dependent ATPase activity based on electronic annotation through Ensembl. Whether our probes bind to these proteins due to unrecognized ATPase activity or whether they only bind benzimidazoles under artificially low pH remains to be investigated.
Table 1.
Proteins identified by MS analysis of Rabe-Tz pull down.
| Protein | Accession Number | Molecular Weight |
|---|---|---|
| Gastric H+/K+ ATP-ase | ATP4A_PIG | 114 kDa |
| Isoform 2 of Deleted in malignant brain tumors 1 protein | Q4A3R3-2 | 148 kDa |
| Uncharacterized protein | F1SKJ1 | 225 kDa |
| Uncharacterized protein | F1SHC1 | 50 kDa |
| Uncharacterized protein | I3LT38 | 202 kDa |
Conclusion
We have synthesized two benzimidazole based probes and shown that both specifically label the catalytic component of the gastric H+/K+-ATPase, ATP4A. By directly comparing protein labeling using tetrazine ligation and SPAAC, we have demonstrated that tetrazine ligation is more useful; not only is tetrazine ligation faster, it is also more efficient. If tetrazine modification is tolerated on compounds of interest, we recommend this as the preferred approach for copper-free bio-orthogonal reactions.
Moreover, MS analysis of proteins targeted by benzimidazole probes confirm the use of covalent probes for target identification, as well as revealed a potential link between benzimidazole compounds and DMBT1 for H. pylori eradication and mucosal protection.
Materials and Methods
Compound Synthesis
Unless otherwise mentioned, all reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions. Dichloromethane (CH2Cl2), tetrahydrofuran (THF), acetonitrile, diethyl ether, were purchased from Fisher and purified by passage through two packed columns of neutral alumina under an argon atmosphere. All other reagents were purchased from Sigma-Aldrich and used without further purification, unless otherwise noted. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials. Reactions were monitored by thin-layer chromatography (TLC) carried out on silica gel plates (EMD Silica gel 60 F254) or LC-MS (Waters Autoprep HPLC-MS system). Visualization was achieved using UV light, phosphomolybdic acid in ethanol or potassium permanganate in water, each followed by heating. Sigma (high-purity grade, Merck Grade 9385, pore size 60 Å, 230–400 mesh particle size) silica gel was used for flash column chromatography. HPLC purification used Waters prep HPLC system (XBridge BEH C18 OBD Prep Column, 130Å, 5 μm, and 19 mm × 250 mm). NMR spectra were recorded on Bruker Avance III 600 (1H 600 MHz, 13C 150 MHz) spectrometers. Mass spectrometric data were obtained using Waters LCT Premiere XT. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad.
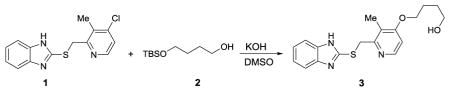
4-((2-(((1H-benzo[d]imidazol-2-yl)thio)methyl)-3-methylpyridin-4-yl)oxy)butan-1-ol (3)
A microwave reactor vail added compound 2 (2.19g, 10.7mmol), followed by added DMSO (10 mL), then KOH (600 mg, 10.7 mmol) added to the solution, the mixture heated to 70°C for 30min under argon atmosphere, after cool to room temperature, compound 143 (1.03g, 3.56 mmol) added, then microwave heating at 100°C for 5h. After cool to room temperature, remove the solvent by lyophilization, the residue was purified by flash chromatography (silica gel, CH2Cl2/MeOH = 100/1) to give compound S3 (1.09 g, 89%) as white solid. 1H NMR (DMSO-d6, 600 MHz): δ = 12.63 (br, 1 H), δ = 8.23 (d, J = 5.6 Hz, 1 H), 7.51 (br, 1 H), 7.40 (br, 1 H), 7.13–7.11 (m, 2 H), 6.95–6.94 (d, J = 5.8 Hz, 1 H), 4.69 (s, 2 H), 4.49–4.06 (t, J = 5.1 Hz 2 H), 4.08–4.06 (t, J = 6.5 Hz, 2 H), 3.47–3.44 (m, 1 H), 2.21 (s, 3 H),1.80–1.76 (m, 2 H), 1.60–1.55 (m, 2 H); 13C NMR (DMSO-d6, 150 MHz): δ = 162.8, 154.6, 150.3, 147.8, 143.7, 135.4, 121.5, 121.2, 119.7, 117.4, 110.4, 106.4, 67.9, 60.3, 36.3, 28.9, 25.2, 10.4; HRMS (ESI) Calcd. for C18H22N3O2S+ [M+H+] 344.1427, found 344.1447.
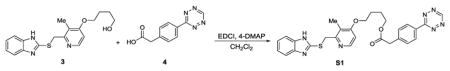
4-((2-(((1H-benzo[d]imidazol-2-yl)thio)methyl)-3-methylpyridin-4-yl)oxy)butyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (S1)
Compound 3 (146mg, 0.425 mmol) and compound 444 (184 mg, 0.851 mmol) in 10 mL dichloromethane, added 4-DMAP (5.2 mg, 0.0425 mmol), then cool to 0°C, follow by added N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI) (163 mg, 0.851 mmol), then allowed warm to room temperature. The reaction stirred for additional 48 hours. Remove the solvent under vacuum, the residue was purified by flash chromatography (silica gel, CH2Cl2/MeOH = 100/1) to give compound S1 (137mg, 59%) as red solid. 1H NMR (DMSO-d6, 600 MHz): δ = 12.63 (br, 1 H), 10.58 (s, 1 H), 8.46–8.44 (m, 2 H), 8.22–8.21 (d, J = 5.6 Hz,1 H), 7.58–7.56( m, 2 H), 7.52 (br, 1 H), 7.39 (br, 1 H), 7.13–7.11(m, 2 H), 6.93–6.92 (d, J = 5.7 Hz, 1 H), 4.68 (s, 2 H), 4.16–4.14 (t, J = 6.1 Hz, 2 H), 4.06–4.04 (t, J = 5.8 Hz, 2 H), 3.85 (s, 2 H), 2.19 (s, 3 H), 1.80–1.74 (m, 4 H); 13C NMR (DMSO-d6, 150 MHz): δ = 170.8, 165.4, 162.7, 158.2, 154.7, 150.3, 147.8, 143.7, 139.7, 135.4, 130.6, 130.5, 127.8, 121.7, 121.2, 119.7, 117.4, 110.4, 106.4, 67.6, 64.2, 40.2, 36.3, 25.1, 24.9, 10.4. HRMS (ESI) Calcd. for C28H28N7O3S+ [M+H+] 542.1969, found 542.1965.
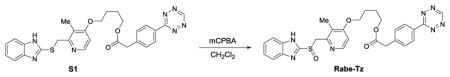
4-((2-(((1H-benzo[d]imidazol-2-yl)sulfinyl)methyl)-3-methylpyridin-4-yl)oxy)butyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (Rabe-Tz)
Compound S1 (50 mg, 0.0923 mmol) in CH2Cl2 (3 mL), cooled to −50°C, then added 3-Chloroperbenzoic acid (24 mg, 70%, 0.0969 mmol). The mixture was stirred for 1 hour at −50°C. Then added Et3N (18 μL, 0.129 mmol), and a 1:1 mixture of 6% aqueous Na2CO3 and 2% aqueous Na2S2O3 (8 mL) were successively added while allowing the mixture to warm to about room temperature over 1 hour. The aqueous layer were extracted with DCM, and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was purified by prepare HPLC to give compound Rabe-Tz (23 mg, 44%) as red solid. 1H NMR (DMSO-d6, 600 MHz): δ = 13.58 (br, 1 H), 10.58 (s, 1 H), 8.46–8.45 (m, 2 H), 8.21–8.20 (d, J = 5.6 Hz,1 H), 7.71 ( br, 1 H), 7.58–7.57 ( m, 3 H), 7.30 (br, 2 H), 6.94–6.93 (d, J = 5.7 Hz, 1 H), 4.79–4.68 (m, 2 H), 4.16–4.14 (t, J = 6.1 Hz, 2 H), 4.07–4.05 (t, J = 5.8 Hz, 2 H), 3.85 (s, 2 H), 2.12 (s, 3 H), 1.80–1.75 (m, 4 H); 13C NMR (DMSO-d6, 150 MHz): δ = 170.8, 165.4, 162.8, 158.2, 154.3, 150.1, 148.3, 143.1, 139.7, 134.7, 130.6, 130.5, 127.8, 123.9, 122.6,121.8, 119.8, 112.4, 106.5, 67.6, 64.2, 60.2, 40.2, 25.0, 24.9, 10.7; HRMS (ESI) Calcd. for C28H28N7O4S+ [M+H+] 558.1918, found 558.1905.
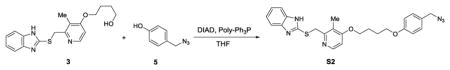
2-(((4-(4-(4-(azidomethyl)phenoxy)butoxy)-3-methylpyridin-2-yl)methyl)thio)-1H-benzo[d] imidazole (S2)
Compound 3 (343 mg, 1 mmol) and polymer-bound triphenyl phosphine (300 mg, 3 mol/g, 1.33 mmol) in THF (10 mL) at 0°C added DIAD (217 μL, 1.1 mmol), then compound 545 (164 mg, 1.1 mmol) added. The mixture was stirred for 2 days at room temperature. Filter and wash with MeOH, remove the solvent under reduced pressure, the resulting residue was purified by flash chromatography (silica gel, CH2Cl2/MeOH= 100/1) to give compound S2 (271 mg, 57%) as white solid. 1H NMR (DMSO-d6, 600 MHz): δ = 9.44 (s, 1 H), δ = 8.24–8.23 (d, J = 5.6 Hz, 1 H), 7.59–7.57 (m, 1 H), 7.50–7.48 (m, 1 H),7.18–7.14 ( m, 2 H), 7.06–7.05 ( m, 2 H), 6.97–6.96 ( d, J = 5.8 Hz, 1 H), 6.69–6.67 (m, 2 H), 5.25 (s, 2 H), 4.76 (s, 2H), 4.10–4.08 (t, J = 6.2 Hz, 2 H), 3.43–3.41 (t, J = 6.8 Hz, 2 H), 2.21 (s, 3 H), 1.83–1.79 (m, 2 H), 1.73–1.68 (m, 2 H); 13C NMR (DMSO-d6, 150 MHz): δ = 162.6, 156.9, 154.3, 151.5, 147.8, 143.0, 136.1, 128.6, 126.6, 121.7, 121.6, 119.7, 117.7, 115.4, 109.9, 106.4, 67.5, 50.3, 46.4, 37.3, 25.7, 25.0, 10.4. HRMS (ESI) Calcd. for C25H27N6O2S+ [M+H+] 475.1911, found 475.1907

2-(((4-(4-(4-(azidomethyl)phenoxy)butoxy)-3-methylpyridin-2-yl)methyl)sulfinyl)-1H-benzo[d]imidazole (Rabe-N3)
A solution of S2 (60 mg, 0.126 mmol) in CH2Cl2 (10 mL) cooled to −40°C. At this temperature, 3-Chloroperbenzoic acid (65 mg, 70%, 0.265 mmol) was added. Then reaction stirred for 2 hours at −25°C before quenched with Et3N (88 μL, 0.63 mmol), and a 1:1 mixture of 6% aqueous Na2CO3 and 2% aqueous Na2S2O3 (14 mL) were successively added while allowing the mixture to warm to about 0 °C. Stirring was continued for 1 hour at ambient temperature. The phases were separated and aqueous layers was extracted with CH2Cl2 (20mLx2), the combined the organic layer was washed with brine (10 mL each) before being evaporated to dryness. The resulting residue was purified by flash chromatography (silica gel, CH2Cl2/MeOH = 100/1) to give compound Rabe-N3 (28 mg, 45%) as white solid. 1H NMR (CD3CN, 600 MHz): δ = 8.11–8.10 (d, J = 5.6 Hz, 1 H), 7.74–7.73 (m, 1 H), 7.46–7.45 (m, 1 H),7.34–7.30 ( m, 2 H), 7.11–7.09 ( m, 3 H), 6.77–6.76 ( d, J = 5.7 Hz, 1 H), 6.74–6.72 (m, 2 H), 5.61–5.54 (m, 2 H), 4.96–4.83 (m, 2 H), 4.06–4.00 (m, 2 H), 3.37–3.35 (t, J = 6.8 Hz, 2 H), 2.14 (s, 3 H), 1.85–1.81 (m, 2 H), 1.75–1.70 (m, 2 H); 13C NMR (DMSO-d6, 150 MHz): δ = 164.2, 157.6, 154.5, 151.7, 148.8, 143.1, 136.6, 129.6, 128.3, 125.4, 124.2, 122.7, 121.4, 118.3, 112.3, 107.1, 68.6, 59.7, 51.7, 48.0, 26.8, 26.0, 11.0; HRMS (ESI) Calcd. for C25H27N6O3S+ [M+H+] 491.1860, found 491.1843.
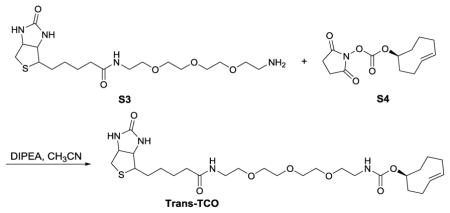
(S, E)-cyclooct-4-en-1-yl (13-oxo-17-(2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-3,6,9-trioxa-12-azaheptadecyl)carbamate (Trans-TCO)
To a solution of S3 (16 mg, 0.037 mmol) and S4 (10 mg, 0.037 mmol) in CH3CN (1 mL) was added DIPEA (13 μL, 0.074 mmol). The reaction mixture was stirred at room temperature for 3 minutes. The mixture was poured over EtOAc and washed with brine. The organic extract was dried with anhydrous NaSO4, filtered and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (CH2Cl2/MeOH) to afford Trans-TCO (15 mg, 71% yield) as white solid. HRMS (ESI) Calcd. for C27H46N4NaO7S+ [M+Na+] 593.2985, found 593.2968.
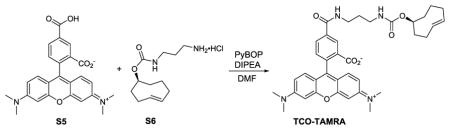
(S, E)-5-((3-(((cyclooct-4-en-1yloxy)carbonyl)amino)propyl) carbamoyl)-2-(6-(dimethylamino)-3-(dimethyliminio)-3H-xanthen-9-yl)benzoate (TCO-TAMRA)
To a stirred DMF (3 mL) solution of S6 (29.3 mg, 0.111 mmol), DIPEA (97 μl, 0.557 mmol), and 5-carboxytetramethylrhodamine (48 mg, 0.111 mmol) was added PyBOP (116 mg, 0.223 mmol). The mixture was stirred at room temperature for overnight. The solvent was evaporated. The residue was purified by prepare HPLC to give compound TCO-TAMRA (32 mg, 45%) as red solid. HRMS (ESI) Calcd. for C37H42N4NaO6+ [M+Na+] 639.3183, found 639.3201.
Preparation of Porcine Gastric H+/K+-ATPase Enzyme
Gastric H+/K+-ATPase was isolated using a protocol adapted from Dach et al. 201226 and Skrabanka et al 198425. Fresh porcine stomachs were obtained from Innovative Research Inc (Novi, MI) and kept on ice during each step. Gastric mucosa was scraped from the stomachs with a cell scraper and homogenized 1:1 v/v in homogenization buffer (0.25M Sucrose, 20mM TRIS-HCl, pH=7.4) using an immersion blender. The mucosa was further homogenized with 8 passes of piston-based motorized homogenizer. The sample was centrifuged at 27,000g (17,235rpm) for 20 minutes in a Sorval T-1250 rotor. The supernatant was transferred to a clean tube and re-centrifuged at 100,000g (33,000rpm in Sorval T-1250) for 1 hour. The pellet was re-suspended in homogenization buffer, homogenized with 5 passes of the motorized homogenizer and layered on a two-step gradient consisting of 37% sucrose (w/v) and 20 mM Tris-HCl (pH 7.4) at the bottom with 7% Ficoll at the top (GE Healthcare), 0.25 M sucrose, and 20 mM Tris-HCl (pH 7.4). The sample was centrifuged at 217,000g (35,000rpm in Thermo Scientific TH-641 rotor) for 1 hour H+/K+-ATPase containing vesicles were collected from the top of the ficoll layer and diluted with 20mM Tris buffer (pH=7.4) and centrifuged at 217,000g (49,000rpm in Sorval T-1250) for 30minutes. The pellet containing the vesicles was re-suspended in 20mM Tris (pH=7.4) and kept frozen in small aliquots at −20°C for short term and −80°C for long term storage.
Gastric H+/K+-ATPase Activity Assay
Isolated vesicles (15ug/mL) were pre-incubated for 15 minutes at 37°C with compound or vehicle in assay buffer (20mM TRIS-HCl, (pH=7), 10mM KCl, 2mM MgCl2) in the presence of 1ug/mL nigericin. The gastric H+/K+-ATPase was activated by the addition of 2mM NaATP (Sigma) and incubated at 37°C for 45 minutes. Activity was determined by measuring inorganic phosphate release using the PiColorLock Gold Phosphate Detection System (Innova Biosciences, Cambridge, UK). Assay background was measured by carrying out reactions in the absence of vesicles; while background phosphate release was measured using reactions carried out in the absence of MgCl2 Reactions without compounds were run with vehicle, DMSO, as a positive control. The following equations were used to calculate potency of inhibition:
Preparation of Gastric Mucosa Membranes
Fresh porcine stomachs were obtained from Innovative Research Inc (Novi, MI) and kept on ice during each step. Gastric mucosa was scraped from the stomachs with a cell scraper and homogenized 1:1 v/v in buffer A (50 mM MES, pH 6.0, 5 mM MgCl2, 5 mM CaCl2, 150 mM KCl) using an immersion blender. The mucosa was further homogenized with 10 passes of a piston-based motorized homogenizer. The homogenate was centrifuged at 800g for 10 minutes to remove cell debris and nuclei. The supernatant was centrifuged at 100,000g for 1 hour at 4°C. The pellet was resuspended in buffer A and the centrifugation at 100,000g repeated. The final membrane pellet was resuspended and homogenized in buffer A to a final concentration of approximately 18mg/mL and stored at −80°C.
Labeling of Mucosa Membrane with Benzimidazole Probes and Western Blot Analysis
Membrane fractions containing endogenous H+/K+-ATPase were isolated from porcine gastric mucosa using ultracentrifugation methods described above. Isolated membrane (800ug) was incubated with each benzimidazole analogue (1uM) in a glycine buffer at pH=3 for 1 hour at 37°C while shaking. After incubation, copper free click reactions were initiated by adding Dibenzocyclooctene-PEG4-biotin (Sigma) (100uM) to reactions with Rabe-N3 and trans-cyclooctene-biotin, (16uM) to reactions with Rabe-Tz. Reaction with either handle allows for conjugation of biotin to labeled proteins for pull down. Click reactions were allowed to proceed for three hours at room temperature. Reactions were centrifuged at 90,000g for 40 min at 4°C. Supernatants were aspirated, thus removing excess click reagents. The pellets were resuspended by homogenization with a TissueLyser (Qiagen) at 25 rps for 2 min and solubilized in RIPA buffer (50 mM Tris (pH= 8), 150 mM NaCl, 0.1% SDS, 1% NP40, 0.5% deoxycholate) then centrifuged at 12,000 rpm to remove particulate matter. To show blocking of labeling, 50mM DTT was added to select samples and incubated for 5 minutes at RT. Streptavidin ultralink resin slurry (Pierce) was added to all supernatants and incubated overnight at 4C. Streptavidin resins were washed 4 times by centrifugation at 0.5g followed by aspiration of supernatant and addition of 500μL RIPA buffer. Labeled proteins were eluted by incubation with excess biotin (2mM) in 2X Lamelli sample buffer (Biorad) at 70°C for 10min. Eluates were loaded onto a precast 4–15% Criterion™ Tris-HCl gel (BioRad) for protein separation, transferred to a PVDF membrane and blotted for the H,K-ATPase with Anti-ATP4A antibody (ab174293) (AbCam).
Fluorescent Labeling
Benzimidazole probe, Rabe-Tz (2uM), was incubated with 150ug of gastric vesicles (preparation described above) in Glycine buffer (pH=3) for 1h at 37°C in the presence or absence of 50mM DTT or TCEP. Targeted proteins were labeled with tetramethyl rhodamine via spontaneous reaction of trans-cyclooctene-TAMRA (16uM) and the tetrazine present on Rabe-Tz for 1 hr at room temperature in the dark. Labeled proteins were then precipitated with 1mL of cold acetone at −20°C for 2 hours, washed once with 500μL ammonium bicarbonate (100mM), then again with 500μL cold acetone. Precipitated proteins were centrifuged at 15,000g for 10 min, the acetone was removed, and the protein pellet was air-dried for 10 min. Protein pellets were resolubilized in 45μL PBS + 15μL 4x Lamelli Sample buffer (BioRad) while rotating at RT for 2 h. Lowry assay was used for total protein quantification and 25ug of each sample was loaded onto an SDS-PAGE gel for band separation and then scanned for fluorescent bands. The same gel was then immediately stained with Coomassie blue (BioRad) to compare the total amount of protein in each sample.
Pull down with Rabe-Tz for LC-MS/MS Analysis
The protocol described for “Labeling of Mucosa Membrane with Benzimidazole Probes” was followed using Rabe-Tz (1uM) and trans-cyclooctene-biotin (16uM)with the exception that the click reaction was only allowed to proceed for 1 hour. Following protein separation by SDS-PAGE protein bands of interest were excised, destained and SDS removed by washes with 50% methanol (v/v). These gel pieces were then dried using a speed-vac. To reduce the disulfide bonds, 10 mM DTT in 25 mM ammonium bicarbonate was added until the pieces were covered with 10 mM DTT in 25 mM ammonium bicarbonate for 1 hour at 56°C. For alkylation, the reducing agent was removed and the gel pieces were covered with freshly made 55 mM iodoacetamide in 25 mM ammonium bicarbonate at room temperature in the dark. After 45 minutes, the supernatant was removed and gel pieces were rinsed with 100 mM ammonium bicarbonate three times. Gel pieces were again dried using a speed-vac. To dried, gel-bound protein, 200 ng of trypsin (modified, sequencing grade, Promega) in 10 μl of 100 mM ammonium bicarbonate was added to swell the gel piece. Then an additional 90 μl of 100 mM ammonium bicarbonate was added or until the gel piece was covered with the digestion solution. Tryptic digestion was allowed to proceed at 37°C overnight. The supernatant containing the generated peptides was removed and desalted using Poros R2 beads as described in Erdjument-Bromage et al (1998). Peptides were eluted with 3 μl of 40% (v/v) acetonitrile containing 0.1% (v/v) formic acid and stored at −20°C if not analyzed immediately.
The purified peptides were diluted to 0.1% formic acid and each gel section was analyzed separately by microcapillary liquid chromatography with tandem mass spectrometry using the NanoAcquity (Waters) with a 100-μm-inner-diameter ×310-cm-length C18 column (1.7 um BEH130, Waters) configured with a 180-um × 2-cm trap column coupled to an OrbiElite mass spectrometer(Thermo Fisher Scientific) scanning 300–1650 m/z at 120000 resolution with AGC set at 1 × 10(6). Peptides were eluted with a linear gradient of 0–50% acetonitrile (0.1% formic acid) in water (0.1% formic acid) over 90 mins with a flow rate of 300nL/min. Key parameters for the data dependent MS were top 10 DDA, AGC 1e4, and CID ms/ms collected in the linear ion trap.
Database searching
Tandem mass spectra were extracted by ProteoWizard version 3.0.7529 from the raw files. Charge state deconvolution and deisotoping were not performed. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.3.02) and X! Tandem (The GPM, thegpm.org; version CYCLONE (2010.12.01.1)). Mascot was set up to search the uniprot_pig_20150721 database (selected for Mammalia, unknown version, 26153 entries) assuming the digestion enzyme trypsin. X! Tandem was set up to search a subset of the uniprot_sprot_20150223 database also assuming trypsin. Mascot and X! Tandem were searched with a fragment ion mass tolerance of 0.80 Da and a parent ion tolerance of 10.0 PPM. Carbamidomethyl of cysteine was specified in Mascot and X! Tandem as a fixed modification. Deamidation of asparagine and glutamine, oxidation of methionine, acetyl of the n-terminus and phospho of serine, threonine and tyrosine were specified in Mascot as variable modifications. Glu->pyro-Glu of the n-terminus, ammonia-loss of the n-terminus, gln->pyro-Glu of the n-terminus, deamidated of asparagine and glutamine, oxidation of methionine, acetyl of the n-terminus, carbamidomethyl of cysteine and phospho of serine, threonine and tyrosine were specified in X! Tandem as variable modifications.
Criteria for Protein Identification
Scaffold (version Scaffold_4.4.3, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95% probability to achieve an FDR less than 1.0% by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at greater than 99.9% probability to achieve an FDR less than 1.0% and contained at least 4 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii, Al et al Anal. Chem. 2003;75(17):4646-58). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. Proteins were annotated with GO terms from NCBI (downloaded Jul 22, 2015). (Ashburner, M et al Nat. Genet. 2000;25(1):25-9).
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health R01AG026660 (Y.M.L.) R01NS076117 (Y. M.L.), and training grant T32 GM073546-07 (CP). Authors also acknowledge the MSK Cancer Center Support Grant/Core Grant (Grant P30 CA008748) to Microchemistry and Proteomics Core Laboratory, Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, and the William Randolph Hearst Fund in Experimental Therapeutics. We are grateful to Elizabeth Chang and Ronald Hendrickson at the MSKCC Proteomics core for LC-MS/MS analysis.
Notes and references
- 1.Johnson DS, Weerapana E, Cravatt BF. Future medicinal chemistry. 2010;2:949–964. doi: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman LA, Zaro BW, Miller SM, Pratt MR. Journal of the American Chemical Society. 2013;135:14568–14573. doi: 10.1021/ja408322b. [DOI] [PubMed] [Google Scholar]
- 3.Cohen MS, Hadjivassiliou H, Taunton J. Nature chemical biology. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roth GJ, Stanford N, Majerus PW. Proceedings of the National Academy of Sciences of the United States of America. 1975;72:3073–3076. doi: 10.1073/pnas.72.8.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Der Ouderaa FJ, Buytenhek M, Nugteren DH, Van Dorp DA. European journal of biochemistry / FEBS. 1980;109:1–8. doi: 10.1111/j.1432-1033.1980.tb04760.x. [DOI] [PubMed] [Google Scholar]
- 6.Fellenius E, Berglindh T, Sachs G, Olbe L, Elander B, Sjostrand SE, Wallmark B. Nature. 1981;290:159–161. doi: 10.1038/290159a0. [DOI] [PubMed] [Google Scholar]
- 7.Robinson M. International journal of clinical practice. 2005;59:709–715. doi: 10.1111/j.1368-5031.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- 8.Lindberg P, Nordberg P, Alminger T, Brandstrom A, Wallmark B. Journal of medicinal chemistry. 1986;29:1327–1329. doi: 10.1021/jm00158a001. [DOI] [PubMed] [Google Scholar]
- 9.Shin JM, Cho YM, Sachs G. Journal of the American Chemical Society. 2004;126:7800–7811. doi: 10.1021/ja049607w. [DOI] [PubMed] [Google Scholar]
- 10.Shin JM, Besancon M, Simon A, Sachs G. Biochimica et biophysica acta. 1993;1148:223–233. doi: 10.1016/0005-2736(93)90133-k. [DOI] [PubMed] [Google Scholar]
- 11.Shin JM, Homerin M, Domagala F, Ficheux H, Sachs G. Biochemical pharmacology. 2006;71:837–849. doi: 10.1016/j.bcp.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 12.Shin JM, Besancon M, Prinz C, Simon A, Sachs G. Alimentary pharmacology & therapeutics. 1994;8(Suppl 1):11–23. doi: 10.1111/j.1365-2036.1994.tb00211.x. [DOI] [PubMed] [Google Scholar]
- 13.Shin JM, Sachs G. Biochemical pharmacology. 2004;68:2117–2127. doi: 10.1016/j.bcp.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 14.Besancon M, Shin JM, Mercier F, Munson K, Miller M, Hersey S, Sachs G. Biochemistry. 1993;32:2345–2355. doi: 10.1021/bi00060a028. [DOI] [PubMed] [Google Scholar]
- 15.Besancon M, Simon A, Sachs G, Shin JM. The Journal of biological chemistry. 1997;272:22438–22446. doi: 10.1074/jbc.272.36.22438. [DOI] [PubMed] [Google Scholar]
- 16.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su Y, Pan S, Li Z, Li L, Wu X, Hao P, Sze SK, Yao SQ. Scientific reports. 2015;5:7724. doi: 10.1038/srep07724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jang S, Sachin K, Lee HJ, Kim DW, Lee HS. Bioconjugate chemistry. 2012;23:2256–2261. doi: 10.1021/bc300364z. [DOI] [PubMed] [Google Scholar]
- 19.Debets MF, van Berkel SS, Dommerholt J, Dirks AT, Rutjes FP, van Delft FL. Accounts of chemical research. 2011;44:805–815. doi: 10.1021/ar200059z. [DOI] [PubMed] [Google Scholar]
- 20.Sletten EM, Bertozzi CR. Accounts of chemical research. 2011;44:666–676. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selvaraj R, Fox JM. Current opinion in chemical biology. 2013;17:753–760. doi: 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devaraj NK, Weissleder R. Accounts of chemical research. 2011;44:816–827. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seckute J, Devaraj NK. Current opinion in chemical biology. 2013;17:761–767. doi: 10.1016/j.cbpa.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saccomani G, Stewart HB, Shaw D, Lewin M, Sachs G. Biochimica et biophysica acta. 1977;465:311–330. doi: 10.1016/0005-2736(77)90081-5. [DOI] [PubMed] [Google Scholar]
- 25.Skrabanja AT, De Pont JJ, Bonting SL. Biochimica et biophysica acta. 1984;774:91–95. doi: 10.1016/0005-2736(84)90278-5. [DOI] [PubMed] [Google Scholar]
- 26.Dach I, Olesen C, Signor L, Nissen P, le Maire M, Moller JV, Ebel C. The Journal of biological chemistry. 2012;287:41963–41978. doi: 10.1074/jbc.M112.398768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin JM, Inatomi N, Munson K, Strugatsky D, Tokhtaeva E, Vagin O, Sachs G. The Journal of pharmacology and experimental therapeutics. 2011;339:412–420. doi: 10.1124/jpet.111.185314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soumarmon A, Grelac F, Lewin MJ. Biochimica et biophysica acta. 1983;732:579–585. doi: 10.1016/0005-2736(83)90234-1. [DOI] [PubMed] [Google Scholar]
- 29.Rabon EC, Gunther RD, Bassilian S, Kempner ES. The Journal of biological chemistry. 1988;263:16189–16194. [PubMed] [Google Scholar]
- 30.Shin JM, Sachs G. The Journal of biological chemistry. 1996;271:1904–1908. doi: 10.1074/jbc.271.4.1904. [DOI] [PubMed] [Google Scholar]
- 31.Abe K, Kaya S, Hayashi Y, Imagawa T, Kikumoto M, Oiwa K, Katoh T, Yazawa M, Taniguchi K. Biochemistry. 2003;42:15132–15138. doi: 10.1021/bi035686x. [DOI] [PubMed] [Google Scholar]
- 32.Abe K, Kaya S, Taniguchi K, Hayashi Y, Imagawa T, Kikumoto M, Oiwa K, Sakaguchi K. Journal of biochemistry. 2005;138:293–301. doi: 10.1093/jb/mvi127. [DOI] [PubMed] [Google Scholar]
- 33.Shin JM, Grundler G, Senn-Bilfinger J, Simon WA, Sachs G. Biochemistry. 2005;44:16321–16332. doi: 10.1021/bi051342q. [DOI] [PubMed] [Google Scholar]
- 34.Murrey HE, Judkins JC, Am Ende CW, Ballard TE, Fang Y, Riccardi K, Di L, Guilmette ER, Schwartz JW, Fox JM, Johnson DS. Journal of the American Chemical Society. 2015;137:11461–11475. doi: 10.1021/jacs.5b06847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mollenhauer J, Wiemann S, Scheurlen W, Korn B, Hayashi Y, Wilgenbus KK, von Deimling A, Poustka A. Nature genetics. 1997;17:32–39. doi: 10.1038/ng0997-32. [DOI] [PubMed] [Google Scholar]
- 36.Takeshita H, Sato M, Shiwaku HO, Semba S, Sakurada A, Hoshi M, Hayashi Y, Tagawa Y, Ayabe H, Horii A. Japanese journal of cancer research : Gann. 1999;90:903–908. doi: 10.1111/j.1349-7006.1999.tb00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mori M, Shiraishi T, Tanaka S, Yamagata M, Mafune K, Tanaka Y, Ueo H, Barnard GF, Sugimachi K. British journal of cancer. 1999;79:211–213. doi: 10.1038/sj.bjc.6690035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mollenhauer J, Herbertz S, Holmskov U, Tolnay M, Krebs I, Merlo A, Schroder HD, Maier D, Breitling F, Wiemann S, Grone HJ, Poustka A. Cancer research. 2000;60:1704–1710. [PubMed] [Google Scholar]
- 39.Prakobphol A, Xu F, Hoang VM, Larsson T, Bergstrom J, Johansson I, Frangsmyr L, Holmskov U, Leffler H, Nilsson C, Boren T, Wright JR, Stromberg N, Fisher SJ. The Journal of biological chemistry. 2000;275:39860–39866. doi: 10.1074/jbc.M006928200. [DOI] [PubMed] [Google Scholar]
- 40.Fiorini G, Zullo A, Gatta L, Castelli V, Ricci C, Cassol F, Vaira D. Clinical and experimental gastroenterology. 2012;5:109–112. doi: 10.2147/CEG.S25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blandizzi C, Natale G, Gherardi G, Lazzeri G, Marveggio C, Colucci R, Carignani D, Del Tacca M. Digestive diseases and sciences. 1999;44:2039–2050. doi: 10.1023/a:1026626519534. [DOI] [PubMed] [Google Scholar]
- 42.Renner M, Bergmann G, Krebs I, End C, Lyer S, Hilberg F, Helmke B, Gassler N, Autschbach F, Bikker F, Strobel-Freidekind O, Gronert-Sum S, Benner A, Blaich S, Wittig R, Hudler M, Ligtenberg AJ, Madsen J, Holmskov U, Annese V, Latiano A, Schirmacher P, Amerongen AV, D’Amato M, Kioschis P, Hafner M, Poustka A, Mollenhauer J. Gastroenterology. 2007;133:1499–1509. doi: 10.1053/j.gastro.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 43.Radl S, Klecan O, Havlicek J. J Heterocyclic Chem. 2006;43:1447–1453. [Google Scholar]
- 44.Yang J, Karver MR, Li W, Sahu S, Devaraj NK. Angewandte Chemie. 2012;51:5222–5225. doi: 10.1002/anie.201201117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Corre L, Girard AL, Aubertin J, Radvanyi F, Benoist-Lasselin C, Jonquoy A, Mugniery E, Legeai-Mallet L, Busca P, Le Merrer Y. Organic & biomolecular chemistry. 2010;8:2164–2173. doi: 10.1039/b923882d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.