

Abstract
People with Down syndrome (DS) are at an increased risk for Alzheimer's disease (AD). After 60 years of age, >50% of DS subjects acquire dementia. Nevertheless, the age of onset is highly variable possibly because of both genetic and environmental factors. Genetics cannot be modified, but environmental risk factors present a potentially relevant intervention for DS persons at risk for AD. Among them, inflammation, important in AD of DS type, is potential target. Consistent with this hypothesis, chronic peripheral inflammation and infections may contribute to AD pathogenesis in DS. People with DS have an aggressive form of periodontitis characterized by rapid progression, significant bacterial and inflammatory burden, and an onset as early as 6 years of age. This review offers a hypothetical mechanistic link between periodontitis and AD in the DS population. Because periodontitis is a treatable condition, it may be a readily modifiable risk factor for AD.
Keywords: Down syndrome, Alzheimer's disease, Infection, Inflammation, Dysbiosis, Periodontal disease, Aggressive periodontal disease, Neuroinflammation, Trisomy 21
1. Introduction
Down syndrome (DS) is the most common genetic cause of intellectual disability, and it occurs in approximately 1 to 800 births. The prevalence of DS has increased lately because of increased incidence and longer life expectancy giving rise to an elderly population with DS that is at risk for age-related comorbidities such as Alzheimer's disease (AD). More than 50% of DS subjects acquire dementia after the age of 60. However, the age of dementia onset is highly variable with both genetic and environmental factors contributing to this variability. Thus, modifiable environmental risk factors present a potentially relevant intervention for DS persons at risk for AD. Among them, inflammation is a potential target.
The purpose of this review is to explore the relationship between inflammation, DS, and dementia using knowledge gained from the study of sporadic and familial AD. It has been increasingly recognized that peripheral chronic inflammation and infections through their inflammatory and bacterial burden are involved in the pathogenesis of AD [1]. Evidence also exists linking brain inflammation to the AD pathology of DS subjects. However, it remains unknown whether inflammation is downstream of core AD pathology or is even an upstream phenomenon. DS patients are known to have multiple comorbidities throughout their life. They also have an aggressive form of periodontitis characterized by rapid progression, significant bacterial/inflammatory burden, and an onset as early as 6 years of age. This review offers a rationale for examining periodontitis in DS as a possible mechanism contributing to the high risk for AD pathology.
2. Alzheimer's disease
AD is one of the leading causes of dementia afflicting the elderly. The prevalence and incidence of AD increase with age. Eleven percent of people older than 65 and 33% of those >85 have AD.
2.1. Pathological features of AD
AD is a continuous process whose pathology starts years before the onset of dementia. The pathological hallmarks of AD are the presence of senile plaques, neurofibrillary tangles, neuronal and synaptic dysfunction, and neuronal loss. The senile plaques contain extracellular aggregates of amyloid-β (Aβ) peptide and activated glial cells, and reactive astrocytes and inflammatory molecules associate intimately with these plaques. Neurofibrillary tangles comprised phosphorylated tau proteins, and the neuronal loss leads to brain atrophy.
3. Alzheimer's disease in DS
People with DS are at significant risk of developing AD. In fact, according to the new International Working Group Criteria, DS is conceptualized as a form of preclinical AD [2]. Up to 28% of 30-year-old DS subjects develop cognitive impairments but no dementia [3]. After 30, 40, and 60 years of age, increasingly more DS people are diagnosed with dementia reaching prevalence rates of 30%, 55%, and 77%, respectively [3], [4].
3.1. Pathological features in DS
The pathological hallmarks of AD, amyloid plaques, and tangles accumulate in the brain of subjects with DS several decades earlier than in the general population with sporadic AD. The earliest amyloid depositions are thought to be diffuse, nonfibrillar, and amorphous plaques, followed by the development of the fibrillar plaques. A review of AD histopathological studies [5] in 398 DS subjects revealed that subjects younger than 10 years old lacked AD plaques and tangles. Then, in teens and in the 20–30 age range, AD pathology affected approximately 7.5% and 16% of brains, respectively. By age 40s, virtually all subjects had AD pathology. The regional distribution of the plaques and tangles in DS subjects resembled that of late-onset sporadic AD [5] although patterns resembling distribution of early-onset AD have also been described.
4. Pathogenesis of Alzheimer's disease
4.1. Role of inflammation
Inflammation is thought to play a significant role [6]. Its role can be primary [7], secondary, or a combination of both. For example, Kristic and Knuesel [7] showed that acute and chronic inflammation were able to induce AD-related pathology and cognitive decline in animal models. In this respect, in a thiamine-deficient model in which chronic inflammation and oxidative stress were early events, there was increased synthesis of Aβ and amyloid plaques [8], and antioxidants reversed the increased production of Aβ. Multiple reviews and animal studies support the concept that pro-inflammatory cytokines and lipopolysaccharide (LPS) are stimulators of Aβ production and tau phosphorylation, and Aβ and tau protein can induce increases in cytokine. However, other studies showed that inflammation could be induced secondarily by the core AD pathological processes related to Aβ cascade or tau-related neurodegeneration.
4.2. Clinical studies
Three lines of clinical evidence support the role of inflammation in AD: increased systemic inflammation, genetic data, and the presence of infectious/inflammatory peripheral conditions. Pro-inflammatory molecules including C-reactive protein (CRP), interleukin-6 (IL-6), and tumor-necrosis factor-β (TNF-β) were associated with and predicted poor cognition, cognitive decline, and dementia 2–25 years later. However, other studies failed to show these cytokines as predictors of cognitive decline. This discrepancy may be explained by the heterogeneous inflammatory responses dependent on timing and individual differences in inflammatory genotypes. For example, we have shown lower cognition in subjects with periodontal inflammation than without [9]. However, among those with periodontal inflammation, cognitive losses were greater in those having IL-1082 AA/AG genotype [10]. Because subjects with IL-10-1082 AA/AG genotype produce less IL-10, an anti-inflammatory cytokine, IL-10-1082 AA/AG genotype qualifies for a pro-inflammatory phenotype. We also found that combination of the plasma TNF-α with antibodies to specific periodontal bacteria (index of bacteria exposure and host response) increased the discriminatory accuracy between normal (NL) and AD subjects [11]. These findings consistent with Holmes [12] show that a) peripheral infectious/inflammations are important in the pathogenesis of AD, and b) perhaps, it is the combination between both peripheral infections/inflammations and the magnitude of the host response, that is critical in understanding the pathophysiology of AD.
Second, genome-wide association studies show that several genes encoding proteins of the inflammatory-immune system (PICALM, CLU, CR1, CR2, TREM2, and CD33) are associated with AD [13]. Third, peripheral infections and inflammation are associated with and predict cognitive decline and AD. Infectious agents such as cytomegalovirus [14] and herpes virus are associated with AD pathology and cognitive dysfunction/AD and increase the risk for AD. Peripheral inflammations with significant inflammatory burden such as diabetes, obesity, metabolic syndrome, and atherosclerosis are also associated with cognitive dysfunction and are now accepted risks for AD [15].
4.3. Inflammatory mechanisms
The mechanisms by which the infections and inflammations affect the progression of the AD pathology are not completely understood. Peripherally derived pro-inflammatory molecules and bacteria/bacterial products could reach the brain via systemic circulation and neural pathways [16], [17], [18] or use macrophages/monocytes system [19]. There, they increase the brain cytokine pool, promote amyloid brain deposition, induce phosphorylation of tau protein, decrease synaptic strength and neuronal degeneration, and thus contribute to cognitive decline [18].
5. Pathogenesis of AD in DS
5.1. Amyloid hypothesis
The pathogenesis of AD in DS has been recently reviewed [20], [21]. Among the hypotheses put forward to explain the AD in DS populations, amyloid-cascade hypothesis is central [20]. DS is a trisomy characterized by the triplication of chromosome 21 and amyloid precursor protein (APP) gene, the precursor of amyloid β is located on this chromosome. Thus, it is assumed that the overexpression of APP accounts for the increased AD in DS subjects. The amyloid-cascade hypothesis has its strongest evidence derived from genetic studies in presenilin 1, presenilin 2, or APP mutation carriers in which Aβ overproduction is sufficient to cause early-onset AD [4], [22].
The rare families with an extra copy of the APP gene, in which early-onset dementia develops, also support the central role of the APP increased copy number. In these families, the duplication of an area of five to eight genes including APP is sufficient to cause the disease [23]. As predicted by the APP increased copy number, these subjects have abundant brain amyloid and tangle depositions and wide spread cerebral amyloid angiopathy and intracerebral hemorrhages [23]. However, compared with dementia in people with DS, they are cognitively unimpaired before the dementia and present much narrower age range for dementia onset [24]. In contrast, although almost all DS subjects have brain amyloid accumulation by age 40, the dementia onset in DS is highly variable. The large interval between amyloid accumulation and dementia onset suggests that many genetic and environmental factors may enhance or mitigate the DS phenotype as pointed out in the literature [22], [25]. For example, mothers who gave birth to DS children before age 35 were at increased risk of dementia compared with those that gave birth after age 35, thus implicating other genetic factors in AD [4].
5.2. Inflammatory hypothesis
Another mechanism implicated in the increased risk of AD in subjects with DS is the “oxidative inflammatory” hypothesis. Wilcock and Griffin [26] proposed that several genes of the chromosome 21 favor a M1 activation of the microglia similar to M1 activation of the macrophages. Macrophages are a population of cells with diverse functions in the innate immune system. In humans, two main classes of activated macrophages have been described namely M1 and M2. Whereas LPS, granulocyte macrophage colony stimulating factor and the TH1 cytokine interferon-gamma (IFN-γ) can trigger M1 macrophage activation, M2 activation can be triggered by the TH2 cytokine IL-4, IL-13, and macrophage colony-stimulating factor. Furthermore, M1 activation leads to an inflammatory phenotype, whereas M2 activation favors an anti-inflammatory one. As described previously, the IFN-γ is one of the main M1 macrophage activators in association with LPS and TNF-α. On binding to its receptor that comprised the interferon-gamma receptor 1 (IFNGR-1) and IFNGR-2 chains, IFN activates a signal transduction pathway involving activation of transcription 1 (STAT1) and interferon regulatory factors and induces increases in cytokines such as IL-12, IL-23, IL-6, IL-1β, and TNF-α and inhibition of IL-10. The genes encoding IFNGR-2 and IFNAR-1 and IFNAR-2 genes are located on chromosome 21. Therefore, their overexpression may lead to a M1 microglial activation with subsequent increased production of pro-inflammatory cytokines [26]. Consistent with this hypothesis, the pioneering work of Griffin found that astrocytes and glial cells from DS subjects had increased expression of IL-1β, and S100 results in concurrence with the recent findings of an inflammatory phenotype in the brains of young DS subjects [27].
The genes of chromosome 21 encoding TIAM1, SOD1, and PRMT can also contribute to AD in DS subjects by increasing oxidative stress. Moreover, inflammation could further stimulate SOD1 and provoke a self-perpetuating vicious cycle with consequences on the brain.
5.3. Priming of the glia
Peripheral infections/inflammations may interact with the host immune response to induce a magnifying effect on brain biology. The priming of the glial cells may explain at least part of these effects [28]. Under homeostatic conditions, microglia have a supportive role and sample the neuro environment. When the neuro environment is challenged, the glial cells become activated. This activation is balanced and tightly regulated unless glial cells are primed. Then, the magnitude and length of response are increased and damage can incur. Glial priming can occur naturally by aging or by inflammatory/infectious conditions throughout life [7], [28], and still another priming can occur by a genetic predisposition. DS is characterized by upregulation of several genes predisposing to an inflammatory phenotype [26]; therefore, glia in DS subjects may respond with amplified effect when challenged by infections/inflammations conditions.
6. Periodontal disease
Periodontal disease (PerioD) is a polymicrobial, peripheral, chronic inflammatory disease that causes destruction of the tissues surrounding the teeth. Clinically, chronic periodontitis is characterized by the presence of gingival erythema and edema and periodontal pockets and destruction of tissue supporting the teeth. Tissue destruction is consistent with the existing local and systemic factors present. Approximately, 45% of the dentate US adults between 30 and 90 years of age have some forms of PerioD although only 8%–10% present with severe forms [29]. Two to three percent of children have chronic periodontitis and another 0.2%–2% have a severe form called aggressive periodontitis [30].
6.1. Pathogenesis of PerioD
There is an agreement that periodontal bacteria are required for the initiation, maintenance, and progression of PerioDs. Among them, Aggregatibacter actinomycetemcomitans, members of the red and orange clusters, such as Tannerella forsythia, Porphyromonas gingivalis, Treponema denticola, and Fusobacterium nucleatum, Prevotella intermedia, Prevotella nigrescens, Parvimonas micra, Streptococcus constellatus, Eubacterium nodatum, Campylobacter showae, Campylobacter gracilis, and Campylobacter rectus are considered important periodontal pathogens.
In addition to the above mentioned bacteria [31], other components of microbiota have been found to contribute to PerioD pathogenesis such as Porphyromonas endodontalis, Treponema lecithinolyticum, Treponema medium, Filifactor alocis, and Selenomonas sputigina. In addition, some bacteria such as Veillonella parvula, Actinomyces sp., or the combination of Streptococcus oralis, Streptococcus mitis, and Streptococcus intermedius are considered beneficial and may protect one from the PerioD (review [31]).
The oral microbiota constitute one of the most diverse and abundant ecosystems in our body. Approximately, 1000 bacterial species colonize the oral cavity with any particular individual holding >200 species. Up to 700 species colonize the subgingival biofilm (under the gingival line), and most are anaerobic. The diversity and abundance of the specific bacteria in the biofilm are a function of a dynamic, multidirectional communication between bacteria, environment, host genetics, and its immune system. In periodontal health, these factors lead to equilibrium consistent with a symbiotic microbial ecosystem. In PerioD, these interrelationships are compromised leading to structural and functional disturbances within the bacterial community called dysbiosis.
The mechanism by which periodontal dysbiosis occurs is far from understood. The elegant studies of Hajishengallis [32] showed that under predisposing conditions, low abundance bacteria with immune subversive capabilities, called keystone pathogens, are able to induce environmental changes that are protective and growth inducing to other bacterial species. In this altered environment, the commensal bacteria flourish, become pathobionts, and synergistically cause exaggerated local inflammation and periodontal tissue breakdown. There is considerable evidence that P. gingivalis is a keystone pathogen. Aggregatibacter actinomycetemcomitans and T. denticola have structural and physiological features that could place them in the same category. This pathogenic concept explains the presence of “disease-inducing keystone” bacteria at periodontal healthy sites, shows that the composition of the whole microbial population is health or disease relevant, and the immune system plays [32] a major role. Treatment of PerioD not only reduces bacterial counts [33] but also causes shifts in bacterial composition toward healthy microbiota.
6.2. Mechanistic pathways for AD effects
Periodontal bacteria can get into the blood stream frequently during daily procedures such as flossing, brushing, and mastication particularly when periodontitis is present. In the blood and then tissues, keystone pathogens and other bacteria can further evade and subvert the immune system and metastasize at distant sites inducing local inflammation [19]. Aggregatibacter actinomycetemcomitans, P. gingivalis, and T. denticola were recovered in atherosclerotic plaque. Brain abscesses in which oral bacteria were implicated were reported.
It has been proposed that PerioD can initiate or contribute to the AD pathogenesis through multiple pathways [18]. Infection-induced effects on AD have been reviewed critically in the literature [34], [35], [36]. A classic bacteria is Treponema pallidum whose infection causes the atrophic form of general paresis presenting with progressive dementia and brain amyloid deposits. Miklossy [34] proposed that oral bacteria including spirochetes could be possible candidates to invade the brain and contribute to AD pathology. Indeed, Riviere et al. [37] detected six different periodontal pathogen treponemes in the brains in >90% of the 16 AD cases analyzed. Moreover, P. gingivalis–derived LPS was also detected in the brains of AD patients [38]. Subjects with PerioD have also a systemic inflammation characterized by elevated levels of IL-1β, IL-6, and TNF-α and CRP (meta-analysis) inflammation that is thought to contribute to other inflammatory diseases such as cardiovascular diseases, and cardiovascular diseases are risk for AD.
Other mechanisms that have been proposed for PerioD-induced systemic pathology include reduced masticatory abilities, consequent dietary deficiencies, and increased stress response. As shown for intestinal bacteria, other mechanisms include modulation of nutrition and immune system, enhancing production of neurotransmitters and bacterial metabolic products and neuronal communications.
6.3. Clinical studies
Clinical data from our studies [9], [39] and others using various exposure indexes, study designs, and outcomes have provided evidence of a link between PerioD and AD related cognitive impairment [9], [39], [40]. For example, cross-sectional studies have reported that measures of periodontal dysbiosis were associated with cognitive impairment, cognitive decline, dementia, and AD with odd ratios of mild-to-moderate strength [41], [42], and these studies have been reviewed elsewhere [40]. Other studies did not find such an association. Similarly, two longitudinal studies showed that pocket depth and periodontal inflammation (current PerioD) predicted cognitive decline with mild-to-moderate strength (hazard ratio [HR] = 1.05, 95% confidence interval [CI] = 1.01–1.10 and odds ratio [OR] = 1.57, 95% CI = 1.01–2.45) [43], [44] although other studies did not support this link. However, when PerioD was defined by immunological parameters [45], the prediction was much stronger (OR = HR = 3.1, 95% CI = 1.5–6.4) [46]. Noticeable, Sparks et al. [45] showed that subjects with AD had high immunoglobulin G (IgG) antibodies to periodontal bacteria 10 years before conversion when the subjects were NL indicating the importance of periodontal bacteria in AD pathogenesis. Most longitudinal studies (prospective, nested case–controls) used tooth loss as a proxy of PerioD, and the results showed significant associations in at least some populations with OR between 1.05 and 2.38. Tooth loss is the ultimate outcome of PerioD, but its association with the outcome measures can be confounded by other factors. Tooth loss is used in most cohort studies because of its convenient assessment, and it can be obtained by subject report and can easily added to an existing study. However, direct measures of periodontal exposures should be used. Stewart et al. [47] recognized that there is little overlap between medical and dental research; therefore, oral examinations are present in very few medical-driven cohorts. Considering the high prevalence of PerioD in the general population (46% in adults 30 and older), even if PerioD has only a low-to-moderate effect, preventing or treating it could prevent a significant number of AD cases and, therefore, deserves unequivocal consideration. To date, there are no longitudinal studies designed specifically to assess the role of PerioD/oral health in AD incidence or cognitive decline. Likewise, there are no studies assessing the links between PerioD and AD in DS population.
7. PerioD in DS
It is well established that people with DS have increased severity and prevalence of PerioD relative to NL population or other intellectually disabled populations. Prevalence rates vary from study to study, but agreement exists that they increase with age. A thorough review of the literature [48], [49] showed that 36% of children as young as 6 years of age had pocket formation, a sign of periodontal inflammation. Up to 40% of children as young as 12 years of age had significant gingival inflammation, pocket formation, and bone loss compared with only 16% of non-DS subjects. After the age of 16, 58%–92% of DS and only approximately 20%–40% of controls with other disabilities had PerioD [50], and these results were consistent across different populations (USA, New Zealand, and Finland). Oral hygiene (OH) is a significant confounder in any investigation, and DS population has poorer OH compared with NL population. OH may even differ within DS people because of intellectual and physical disabilities. However, studies showed that OH by itself could not account for differences in the PerioD among DS, NL controls, or intellectually disabled controls [51], [52].
PerioD in DS subjects has the characteristics of an aggressive periodontitis (Fig. 1). Compared with chronic periodontitis, aggressive periodontitis affects younger population, is severe, often has rapid progression, and the amount of destruction is unexpected for a person of that age and disproportional to the presence of local factors [53]. In DS, PerioD has a significant inflammatory component [53] as shown by the elevated inflammatory markers PGE2, TNF-α, IFN-γ, and SOD1 in their gingival crevicular fluid (local inflammation) [54]. In addition, impaired immune responses are characteristic of DS phenotype such as low IgA, impaired neutrophil function, and increased inflammatory responses to bacterial accumulation [55]. Although no study investigated the DS-related periodontitis in relation to the systemic CRP, subjects with aggressive periodontitis have significantly higher levels of serum CRP and PerioD treatment decreases it.
Fig. 1.
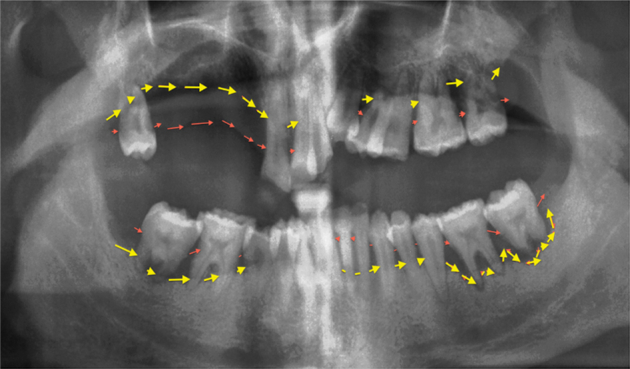
Aggressive periodontitis in subjects with Down syndrome. The panoramic radiography is from a 37-year-old man. Note: a) the bone loss marked between the two lines is severe and extensive; and 2) multiple teeth are lost due to periodontal disease.
The factors involved in the pathogenesis of PerioD in DS subjects are not completely elucidated, but as in other forms of periodontitis, bacteria play a significant role. Early colonization by periodontal pathogens such as F. nucleatum, Prevotella species, A. Actinomycetemcomitans, P. gingivalis, and spirochetes has been observed [50]. Moreover, DS children aged ≤13 harbored more frequently T. forsythia, T. denticola, P. nigrescens, and C. rectus compared with non-DS children with similar periodontal condition [49]. In adults, the differences in microbiota between DS and non-DS were not consistently found [56] although the red cluster bacteria and A. actinomycetemcomitans were present in high proportions in DS subjects with PerioD. In a larger study, initial colonizers such as S. noxia, P. acnes, S. gordonii, S. mitis, and S. oralis were found in higher proportion in DS subjects compared with the non-DS ones, whereas T. socranskii was higher compared with intellectually challenged subjects [57]. When interpreting these results, it is important to bear in mind that most of these studies investigated only a limited number of bacteria. Although Khocht study used more advanced molecular techniques, they only assessed 40 bacterial species. Therefore, the analysis of the subgingival microbiota with advanced molecular techniques is needed to understand the intricacies of the relationships between subgingival microbiota in DS and the host and the potential for systemic effects.
8. Model of periodontal effect on AD of DS
Periodontitis in DS subjects is severe and aggressive. Therefore, it can contribute to brain inflammation, neurodegeneration, and cognitive decline. Fig. 2 offers a model by which PerioD can contribute to AD pathogenesis in DS. In general, PerioD in DS occurs before the onset of AD-specific pathology and, therefore, can even be casual.
Fig. 2.

Model for periodontal disease (PerioD) contribution to Alzheimer's disease (AD) progression in people with Down syndrome (DS). The central theme of AD pathogenesis of DS is the presence of brain inflammation as illustrated by increases in pro-inflammatory cytokines, C-reactive protein, and oxidative stress (brain inflammatory pool). The PerioD-derived pro-inflammatory molecules and bacterial products can reach the brain via 1. systemic circulation and/or neural pathways, contributing to 2. brain inflammation and inflammatory vicious cycle. This would increase 3. AD-specific pathology, 4. neurodegeneration, and 5. subsequent cognitive decline. A pro-inflammatory genotype characteristic to DS would further amplify these effects through glial priming.
Periodontal bacteria colonize the oral and then periodontal environment early in life. In subjects with DS, this colonization by bacteria [49] such as P. gingivalis, T. forsythia, and T. denticola is found as early as 2 years of age compared with 4 years of age in non-DS subjects [49]. Several bacteria associated with DS-aggressive periodontitis are capable of invading tissues including A. actinomycetemcomitans, P. gingivalis, and T. denticola. P. gingivalis and possible T. denticola are keystone pathogens with an increased ability to evade the host response.
DS immune system is impaired, and defects in T-cell maturation, B-cell function, and pro-oxidative state with high levels of radical oxygen species have been described. This weakened immune system could enhance colonization by periodontal pathogens, weaken responses to these pathogens [50], and create conditions leading to periodontal bacterial dysbiosis [32] and severe local inflammatory responses. It is now accepted that PerioD is more severe (destructive) in DS than in non-DS subjects (review [30]). This localized inflammation combined with heavy bacterial burden could contribute to an elevated systemic inflammation.
Based on the previous information, hypothetically, periodontal bacteria, bacterial products, and periodontal-derived cytokines produced locally and systemically could reach the brain and amplify brain cytokine pools [58]. The keystone pathogens P. gingivalis, T. forsythia, and T. denticola have survival advantage and ability to reach distant sites such as the brain. Then, bacteria, their products, and cytokines will act on the already primed glial cells because of the DS genetic factors, resulting in an amplified neuroinflammation and progression of AD. Another potential mechanism may involve modulation of the clearance pathways affecting the amyloid breakdown such as secretory and endosomal system [20], [22].
DS subjects have also a pro-inflammatory phenotype that has been implicated in AD of DS [26]. This mechanism certainly can account independently for the pathogenesis of AD of DS. However, it can also enhance the effect of PerioD on the brain. The pro-inflammatory phenotype has been found to prime the glia, and as described earlier, primed glia responds to stimulation more dramatically than the non-primed ones [28]. Furthermore, some studies including ours showed that when peripheral infectious/inflammatory conditions are associated with a pro-inflammatory phenotype, the effect of these conditions on the brain is significantly more than predicted by either of them, thus suggesting a modifying effect [10], [11], [12], [59]. Therefore, the periodontal infectious/inflammatory burden in DS subjects could cause more and earlier brain damage than in non-DS subjects.
A sustained brain inflammation would upregulate the expression of already triplicated APP gene and contribute sooner to brain amyloid accumulation. Supportive of this model, APP expression in the brain of DS is significantly higher than predicted by the gene triplication suggesting that other factors could turn on this gene expression. Cytokines and LPS are candidates as they were found to consistently stimulate amyloid synthesis and induce cognitive impairment [7]. Our own studies showed that in NL subjects, measures of history of PerioD associated with amyloid accumulation in the brain [58] and affect tau protein hyperphosphorylation [36].
In summary, we propose that PerioD could affect the AD progression in DS subjects. PerioD occurs early in life and, therefore, preventative strategies could be instituted before the induction of AD-specific pathology. Assessing these preventive effects in AD pathogenesis requires longitudinal cohort studies. In non-DS AD, these cohort studies are difficult and expensive to implement. By comparison, examining the role of PerioD in DS may provide us with relative fast answers [60]. Moreover, because PerioD may appear before the AD-specific pathology, a causal effect may be possible to be determined.
Research in context.
-
1.
Systematic review: We searched PubMed up to October 2015, for English articles with search terms “periodontal disease,” “inflammation,” “infections,” “brain,” “amyloid,” “pathogenesis,” “cognition,” “Alzheimer's disease,” “Down syndrome,” and “trisomy 21” and reviewed the articles referenced by the articles identified in search.
-
2.
Interpretation: Although most people with Down syndrome (DS) acquire Alzheimer's disease (AD) as they age, AD onset is highly variable because of genetic and possible environmental factors. Data exist linking inflammation to AD of DS type. Therefore, peripheral inflammation/infections such as periodontal disease could contribute to AD progression in DS subjects. In contrast with the general population, DS has aggressive periodontitis with early onset and significant bacterial/inflammatory burden. Therefore, it could contribute to AD pathogenesis and even its onset. This review offers a hypothetical mechanistic link between periodontitis and AD in DS population.
-
3.
Future directions: This mechanistic hypothesis must be investigated. If true, it would have tremendous implications.
Acknowledgments
We acknowledge that many relevant publications could not be cited and apologize to all the authors we did not cite because of the Journal's constrain. This study was supported by National institutes of Health (NIH)/National Institute on Aging grants AG035137, AG032554, AG12101, AG022374, and AG13616, NIH DE023139-02, Alzheimer's Association NIRG-12-173937, and NIH/NCATS UL1 TR000038. Conflict of interest: No conflict of interest is reported for A.R.K., M.J., P.C., R.G.C., D.S., K.R.C.A., M.R., A.M., J.O.F., S.V., M.C.-I., and B.B. M.J. de Leon has a patent on an image analysis technology that was licensed to Abiant Imaging, Inc., by NYU, and has a financial interest in this license agreement, and NYU holds stock options on the company. M. de Leon has received compensation for consulting services from Abiant Imaging. Dr L. Glodzic was a principal investigator on an Investigator-Initiated project funded by Forest Laboratories and received an honorarium for serving as a consultant to Roche Pharma. Contributors: A.R.K., M.J.de.L., and J.F. wrote the manuscript. All the other authors reviewed the manuscript and contributed with the scientific literature, concepts, and modeling. All authors reviewed the manuscript for intellectual content and approved the final draft.
References
- 1.Hill J.M., Clement C., Pogue A.I., Bhattacharjee S., Zhao Y., Lukiw W.J. Pathogenic microbes, the microbiome, and Alzheimer's disease (AD) Front Aging Neurosci. 2014;6:127. doi: 10.3389/fnagi.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubois B., Feldman H.H., Jacova C., Hampel H., Molinuevo J.L., Blennow K. Advancing research diagnostic criteria for Alzheimer's disease: The IWG-2 criteria. Lancet Neurol. 2014;13:614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 3.Head E., Silverman W., Patterson D., Lott I.T. Aging and Down syndrome. Curr Gerontol Geriatr Res. 2012;2012:412536. doi: 10.1155/2012/412536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zigman W.B., Devenny D.A., Krinsky-McHale S.J., Jenkins E.C., Urv T.K., Wegiel J. Alzheimer's disease in adults with Down syndrome. Int Rev Res Ment Retard. 2008;36:103–145. doi: 10.1016/S0074-7750(08)00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mann D.M. Alzheimer's disease and Down's syndrome. Histopathology. 1988;13:125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x. [DOI] [PubMed] [Google Scholar]
- 6.Heneka M.T., Carson M.J., El Khoury J., Landreth G.E., Brosseron F., Feinstein D.L. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krstic D., Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9:25–34. doi: 10.1038/nrneurol.2012.236. [DOI] [PubMed] [Google Scholar]
- 8.Karuppagounder S.S., Xu H., Shi Q., Chen L.H., Pedrini S., Pechman D. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer's mouse model. Neurobiol Aging. 2009;30:1587–1600. doi: 10.1016/j.neurobiolaging.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamer A.R., Morse D.E., Holm-Pedersen P., Mortensen E.L., Avlund K. Periodontal inflammation in relation to cognitive function in an older adult Danish population. J Alzheimers Dis. 2012;28:613–624. doi: 10.3233/JAD-2011-102004. [DOI] [PubMed] [Google Scholar]
- 10.Kamer A., Krabbe K.S., Bruunsgaard H., Holm-Pedersen P., Mortensen E.L., Morse D.E. Periodontal inflammation effect on cognition depends on the IL-10-1082 gene polymorphism. Alzheimers Dement. 2011;7:S320–S321. [Google Scholar]
- 11.Kamer A.R., Craig R.G., Pirraglia E., Dasanayake A.P., Norman R.G., Boylan R.J. TNF-alpha and antibodies to periodontal bacteria discriminate between Alzheimer's disease patients and normal subjects. J Neuroimmunol. 2009;216:92–97. doi: 10.1016/j.jneuroim.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holmes C., Cunningham C., Zotova E., Woolford J., Dean C., Kerr S. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambert J.C., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med. 2011;13:e30–e64. doi: 10.1017/S1462399411002006. [DOI] [PubMed] [Google Scholar]
- 15.Baumgart M., Snyder H.M., Carrillo M.C., Fazio S., Kim H., Johns H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement. 2015;11:718–726. doi: 10.1016/j.jalz.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 16.Holmes C., Cotterell D. Role of infection in the pathogenesis of Alzheimer's disease: Implications for treatment. CNS Drugs. 2009;23:993–1002. doi: 10.2165/11310910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 17.Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- 18.Kamer A.R., Craig R.G., Dasanayake A.P., Brys M., Glodzik-Sobanska L., de Leon M.J. Inflammation and Alzheimer's disease: Possible role of periodontal diseases. Alzheimers Dement. 2008;4:242–250. doi: 10.1016/j.jalz.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Hajishengallis G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015;15:30–44. doi: 10.1038/nri3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartley D., Blumenthal T., Carrillo M., DiPaolo G., Esralew L., Gardiner K. Down syndrome and Alzheimer's disease: Common pathways, common goals. Alzheimers Dement. 2014;11:700–709. doi: 10.1016/j.jalz.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zigman W.B., Lott I.T. Alzheimer's disease in Down syndrome: Neurobiology and risk. Mental Retard Dev Disabil Res Rev. 2007;13:237–246. doi: 10.1002/mrdd.20163. [DOI] [PubMed] [Google Scholar]
- 22.Wiseman F.K., Al-Janabi T., Hardy J., Karmiloff-Smith A., Nizetic D., Tybulewicz V.L. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat Rev Neurosci. 2015;16:564–574. doi: 10.1038/nrn3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerriere A., Vital A. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 24.McNaughton D., Knight W., Guerreiro R., Ryan N., Lowe J., Poulter M. Duplication of amyloid precursor protein (APP), but not prion protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiol Aging. 2012;33:426.e13–426.e21. doi: 10.1016/j.neurobiolaging.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris G.P., Clark I.A., Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol Commun. 2014;2:135. doi: 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilcock D.M., Griffin W.S. Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation. 2013;10:84. doi: 10.1186/1742-2094-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilcock D.M., Hurban J., Helman A.M., Sudduth T.L., McCarty K.L., Beckett T.L. Down syndrome individuals with Alzheimer's disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer's disease. Neurobiol Aging. 2015;36:2468–2474. doi: 10.1016/j.neurobiolaging.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perry V.H., Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–224. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- 29.Eke P.I., Dye B.A., Wei L., Slade G.D., Thornton-Evans G.O., Borgnakke W.S. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol. 2015;86:611–622. doi: 10.1902/jop.2015.140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albandar J.M. Aggressive and acute periodontal diseases. Periodontol 2000. 2014;65:7–12. doi: 10.1111/prd.12013. [DOI] [PubMed] [Google Scholar]
- 31.Yost S., Duran-Pinedo A.E., Teles R., Krishnan K., Frias-Lopez J. Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis. Genome Med. 2015;7:27. doi: 10.1186/s13073-015-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014;35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teles R.P., Haffajee A.D., Socransky S.S. Microbiological goals of periodontal therapy. Periodontol 2000. 2006;42:180–218. doi: 10.1111/j.1600-0757.2006.00192.x. [DOI] [PubMed] [Google Scholar]
- 34.Miklossy J. Chronic inflammation and amyloidogenesis in Alzheimer's disease – Role of spirochetes. J Alzheimers Dis. 2008;13:381–391. doi: 10.3233/jad-2008-13404. [DOI] [PubMed] [Google Scholar]
- 35.Miklossy J. Historic evidence to support a causal relationship between spirochetal infections and Alzheimer's disease. Front Aging Neurosci. 2015;7:46. doi: 10.3389/fnagi.2015.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamer A.E.P., Tsui W., Yi L., McHugh P., Osario R., Janal M. CSF AD-related biomarkers are higher in subjects with periodontal disease. Alzheimers Dement. 2015;11:P4–P036. [Google Scholar]
- 37.Riviere G.R., Riviere K.H., Smith K.S. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer's disease. Oral Microbiol Immunol. 2002;17:113–118. doi: 10.1046/j.0902-0055.2001.00100.x. [DOI] [PubMed] [Google Scholar]
- 38.Poole S., Singhrao S.K., Kesavalu L., Curtis M.A., Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer's disease brain tissue. J Alzheimers Dis. 2013;36:665–677. doi: 10.3233/JAD-121918. [DOI] [PubMed] [Google Scholar]
- 39.Kamer A.R. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2010;74:1157. doi: 10.1212/WNL.0b013e3181d5df7f. author reply 8. [DOI] [PubMed] [Google Scholar]
- 40.Noble J.M., Scarmeas N., Papapanou P.N. Poor oral health as a chronic, potentially modifiable dementia risk factor: Review of the literature. Curr Neurol Neurosci Rep. 2013;13:384. doi: 10.1007/s11910-013-0384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamer A., Janal M.N., de Leon M. Letter to the editor regarding: Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimer's & Dementia: Diagnosis, Assessment & Disease Monitoring. 2015;1:358–386. doi: 10.1016/j.dadm.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart R., Hirani V. Dental health and cognitive impairment in an English national survey population. J Am Geriatr Soc. 2007;55:1410–1414. doi: 10.1111/j.1532-5415.2007.01298.x. [DOI] [PubMed] [Google Scholar]
- 43.Stewart R., Weyant R.J., Garcia M.E., Harris T., Launer L.J., Satterfield S. Adverse oral health and cognitive decline: The health, aging and body composition study. J Am Geriatr Soc. 2013;61:177–184. doi: 10.1111/jgs.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaye E.K., Valencia A., Baba N., Spiro A., 3rd, Dietrich T., Garcia R.I. Tooth loss and periodontal disease predict poor cognitive function in older men. J Am Geriatr Soc. 2010;58:713–718. doi: 10.1111/j.1532-5415.2010.02788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sparks Stein P., Steffen M.J., Smith C., Jicha G., Ebersole J.L., Abner E. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer's disease. Alzheimers Dement. 2012;8:196–203. doi: 10.1016/j.jalz.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noble J.M., Scarmeas N., Celenti R.S., Elkind M.S., Wright C.B., Schupf N. Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS One. 2014;9:e114959. doi: 10.1371/journal.pone.0114959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart R., Stenman U., Hakeberg M., Hagglin C., Gustafson D., Skoog I. Associations between oral health and risk of dementia in a 37-year follow-up study: The prospective population study of women in Gothenburg. J Am Geriatr Soc. 2015;63:100–105. doi: 10.1111/jgs.13194. [DOI] [PubMed] [Google Scholar]
- 48.Reuland-Bosma W., van Dijk J. Periodontal disease in Down's syndrome: A review. J Clin Periodontol. 1986;13:64–73. doi: 10.1111/j.1600-051x.1986.tb01416.x. [DOI] [PubMed] [Google Scholar]
- 49.Amano A., Kishima T., Kimura S., Takiguchi M., Ooshima T., Hamada S. Periodontopathic bacteria in children with Down syndrome. J Periodontol. 2000;71:249–255. doi: 10.1902/jop.2000.71.2.249. [DOI] [PubMed] [Google Scholar]
- 50.Khocht A., Albandar J.M. Aggressive forms of periodontitis secondary to systemic disorders. Periodontol 2000. 2014;65:134–148. doi: 10.1111/prd.12015. [DOI] [PubMed] [Google Scholar]
- 51.Cutress T.W. Periodontal disease and oral hygiene in trisomy 21. Arch Oral Biol. 1971;16:1345–1355. doi: 10.1016/0003-9969(71)90036-7. [DOI] [PubMed] [Google Scholar]
- 52.Khocht A., Janal M., Turner B. Periodontal health in Down syndrome: Contributions of mental disability, personal, and professional dental care. Spec Care Dentist. 2010;30:118–123. doi: 10.1111/j.1754-4505.2010.00134.x. [DOI] [PubMed] [Google Scholar]
- 53.Albandar J.M. Aggressive periodontitis: Case definition and diagnostic criteria. Periodontol 2000. 2014;65:13–26. doi: 10.1111/prd.12014. [DOI] [PubMed] [Google Scholar]
- 54.Tsilingaridis G., Yucel-Lindberg T., Modeer T. T-helper-related cytokines in gingival crevicular fluid from adolescents with Down syndrome. Clin Oral Investig. 2012;16:267–273. doi: 10.1007/s00784-010-0495-6. [DOI] [PubMed] [Google Scholar]
- 55.Reuland-Bosma W., van Dijk J., van der Weele L. Experimental gingivitis around deciduous teeth in children with Down's syndrome. J Clin Periodontol. 1986;13:294–300. doi: 10.1111/j.1600-051x.1986.tb02225.x. [DOI] [PubMed] [Google Scholar]
- 56.Amano A., Kishima T., Akiyama S., Nakagawa I., Hamada S., Morisaki I. Relationship of periodontopathic bacteria with early-onset periodontitis in Down's syndrome. J Periodontol. 2001;72:368–373. doi: 10.1902/jop.2001.72.3.368. [DOI] [PubMed] [Google Scholar]
- 57.Khocht A., Yaskell T., Janal M., Turner B.F., Rams T.E., Haffajee A.D. Subgingival microbiota in adult Down syndrome periodontitis. J Periodontal Res. 2012;47:500–507. doi: 10.1111/j.1600-0765.2011.01459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamer A.R., Pirraglia E., Tsui W., Rusinek H., Vallabhajosula S., Mosconi L. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol Aging. 2015;36:627–633. doi: 10.1016/j.neurobiolaging.2014.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cunningham C., Hennessy E. Co-morbidity and systemic inflammation as drivers of cognitive decline: New experimental models adopting a broader paradigm in dementia research. Alzheimers Res Ther. 2015;7:33. doi: 10.1186/s13195-015-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benejam B.L.V., Fernandez S., Carmona-Iragui M., Videla S., Fortea J. Neuropsychological assessment of adults with Down syndrome using CAMCOG-DS: A longitudinal study. Alzheimers Dement. 2014;10:P903. [Google Scholar]