

Abstract
Purpose
To investigate the safety and efficacy of the combination of lenalidomide and prednisone in patients with myelofibrosis (MF).
Patients and Methods
Forty patients with MF were treated. Therapy consisted of lenalidomide 10 mg/d (5 mg/d if baseline platelet count < 100 × 109/L) on days 1 through 21 of a 28-day cycle for six cycles, in combination with prednisone 30 mg/d orally during cycle 1, 15 mg/d during cycle 2, and 15 mg/d every other day during cycle 3. Lenalidomide therapy was continued indefinitely in patients exhibiting clinical benefit.
Results
The median follow-up was 22 months (range, 6 to 27). Responses were recorded in 12 patients (30%) and are ongoing in 10 (25%). The median time to response was 12 weeks (range, 2 to 32). According to the International Working Group for Myelofibrosis Research and Treatment consensus criteria, three patients (7.5%) had partial response and nine patients (22.5%) had clinical improvement durable for a median of 18 months (range, 3.5 to 24+). Overall response rates were 30% for anemia and 42% for splenomegaly. Moreover, 10 of 11 assessable responders who started therapy with reticulin fibrosis grade 4 experienced reductions to at least a score of 2. All eight JAK2V617F–positive responders experienced a reduction of the baseline mutant allele burden, which was greater than 50% in four, including one of whom the mutation became undetectable. Grade 3 to 4 hematologic adverse events included neutropenia (58%), anemia (42%), and thrombocytopenia (13%).
Conclusion
The combination of lenalidomide and prednisone induces durable clinical, molecular, and pathologic responses in MF.
INTRODUCTION
The median survival of patients with primary myelofibrosis (PMF) is approximately 5 years.1 Conventional therapy involves the use of chemotherapeutic agents (eg, hydroxyurea), immunomodulatory drugs (eg, thalidomide), or biologic-response modifiers (eg, androgens, erythropoietin).2 None of these therapies has been shown to prolong survival,1 and all are considered palliative. Allogeneic stem cell transplantation remains the only curative option, but only a small subset of patients benefit from this approach due to limited donor availability, poor performance status, and high mortality and morbidity.3,4
An activating point mutation at codon 617 (JAK2V617F) of the JH2 pseudokinase domain of the JAK2 is present in approximately 50% of patients with PMF,5–9 providing a target for therapeutic intervention. Several JAK2 kinase inhibitors are undergoing early stages of clinical development.10–13 Thus far, none have proven beneficial in improving ineffective erythropoiesis or the prominent bone marrow stromal fibrotic reaction, which are clinical and pathologic hallmarks of PMF. Bone marrow fibrosis is believed to be reactive to the high levels of profibrogenic and proangiogenic cytokines, such as transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), tumor necrosis factor alpha, basic fibroblast growth factor (bFGF), and vascular endothelial growth factor secreted by the neoplastic clone in the bone marrow milieu.14,15 Supporting the importance of the bone marrow microenviroment in the pathogenesis of PMF is the efficacy of compounds which, like thalidomide, are endowed with immunomodulatory cytokine inhibitory and antiangiogenic activity (IMiDs).16 Despite the activity of thalidomide in PMF,17,18 its use has been limited by its adverse toxicity profile. When used in conjunction with prednisone, the tolerance of thalidomide improves, leading to improved response rates.19 The potent thalidomide derivative lenalidomide has proven effective in patients with PMF with a toxicity profile mostly consisting of myelosuppression and rash.20 Prompted by the activity of lenalidomide therapy, we sought to evaluate the safety and efficacy of the combination of lenalidomide and prednisone in a phase II study in patients with PMF.
PATIENTS AND METHODS
Eligibility
Patients ≥ 18 years of age with PMF (according to the WHO, revised in 2001) requiring therapy, including those previously treated, relapsed, refractory, or if newly diagnosed, with intermediate- or high-risk (ie, score ≥ 1) PMF according to the Lille scoring system (risk factors: hemoglobin (Hb) < 10g/dL, WBC < 4 or > 30 × 109/L; risk groups: no factors = low, one factor = intermediate, two factors = high) or with symptomatic splenomegaly were eligible. Other eligibility criteria included: performance status ≤ 2 by the Eastern Cooperative Oncology Group scale; serum creatinine lower than 2.0 mg/dL; serum bilirubin ≤ 2.0 times the upper limit of the normal range; off-chemotherapy for 2 weeks before study entry and recovery from the toxic effects of that therapy (patients were allowed to enter the study on a stable dose, for at least 4 weeks before study entry, of anagrelide and/or hydroxyurea to control high platelet and WBC counts); negative pregnancy test in women of childbearing age, and practice of effective methods of contraception during study participation for all patients. All patients signed an informed consent form approved by the M. D. Anderson Cancer Center institutional review board.
Treatment Schedule
The initial dose schedule of lenalidomide was 10 mg/d orally, unless the platelet count was lower than 100 × 109/L, in which case the starting dose was 5 mg/d. Lenalidomide was given in 28-day cycles on a 21-day on/7-day off schedule. Lenalidomide was continued for at least 6 months unless significant toxicity was observed, to account for the delayed time to response observed with biologic agents. Thereafter, lenalidomide was continued in patients exhibiting clinical benefit, as judged by the treating physician, unless disease progression and/or toxicity warranted treatment discontinuation. Oral prednisone was given at 30 mg/d during cycle 1, 15 mg/d during cycle 2, and 15 mg every other day during cycle 3, after which it was discontinued. The dose of lenalidomide could be reduced based on adverse events, or escalated in patients with proliferative disease not controlled after an initial cycle (Table 1). Lenalidomide was interrupted for the remainder of the cycle and resumed at the next lower dose level in the event of: grade 3 rash, hypersensitivity, or grade 2 cardiac arrhythmia; grade 4 neutropenia or grade 3 if associated with fever (≥ 38.5°C); grade 3 to 4 thrombocytopenia; grade 3 to 4 thrombosis/embolism (in addition to starting systemic anticoagulation); hyperthyroidism/hypothyroidism; or other clinically significant nonhematologic toxicities. The use of recombinant erythropoietin was not allowed but filgrastim (granulocyte colony-stimulating factor) could be given to treat neutropenia secondary to the study treatment. Prophylaxis with aspirin was recommended in all patients to decrease the risk of thrombotic events.
Table 1.
Lenalidomide Treatment Schema
| Dose Level | Schedule (28-day cycle) |
|---|---|
| +1 | 15 mg/day on days 1-21 |
| 0 | 10 mg/day on days 1-21 |
| –1, starting dose if platelet count <100 × 109/L | 5 mg/day on days 1-21 |
| –2 | 5 mg every other day for 10 consecutive days |
NOTE. Lenalidomide was given at the dose schedules shown. Therapy was given for a minimum of 6 cycles and continued indefinitely in patients showing clinical benefit.
Patient Evaluation
Baseline studies included complete physical examination (monthly for the first 3 months, then every 3 months), complete blood count (every other week for the first 8 weeks, then every 4 weeks), comprehensive biochemistry panel (including liver function tests) every 4 weeks, and bone marrow aspiration and biopsy with cytogenetics (every 3 months, including staining for fibrosis), and molecular test for JAK2V617F mutation (every 3 months if present before therapy). Bone marrow fibrosis and cellularity were graded according to the European consensus guidelines.21 Responses were assessed according to the IWG-MRT criteria.22
Enzyme-Linked Immunosorbent Assays and JAK2V617F Detection
Measurements of bFGF, PDGF, and TGF-β1 levels in peripheral blood were carried out by using a commercially available kit (R&D Systems, Minneapolis, MN) thus: 100 μL of peripheral blood plasma were added to separate microplates each coated with a monoclonal antibody specific for either bFGF, PDGF, or TGF-β1. Mixtures were incubated at room temperature for 2 hours. The microplates were then washed 3 times to remove any unbound proteins. Enzyme linked polyclonal antibodies specific for each protein were added and mixtures were incubated at room temperature for 2 hours followed by another washing to remove any unbound secondary antibody. A substrate solution was added to each well. The reaction was stopped after 20 minutes and the intensity of color of each well was measured and compared with a standard curved in a Synergy HT Multi-Detection Microplate reader (BioTeck; Winooski, VT) at 450 nm wavelength.
The pyrosequencing assay to detect the 1849G>T JAK2 mutation (JAK2V617F) has been previously reported.23
Study Design
This study was a prospective open-label, single center, phase II trial with an implemented minimum value/maximum value (MinMax) two-stage design. The primary efficacy end point of the study was objective clinical response. The initial target response rate was 35%. In the event of a response rate of ≤ 20%, this would be considered unacceptable and treatment with the therapy was to be discontinued. Given the response rates stated above, if the probability of inappropriately accepting a poor therapy is 10%, a total sample size of 40 patients will result in 80% power. The statistical analysis for response rates was determined on an intention-to-treat basis. In the first stage of the design, a total of 22 patients were enrolled. If four or fewer patients responded to the combination therapy after being treated for 6 months, the study was to be terminated and the therapy would have been declared ineffective. However, as soon as five or more patients were found to respond to lenalidomide, enrollment was to be resumed.
RESULTS
Patient Characteristics
Forty patients (23 male) were enrolled between July 2006 and March 2007 (Table 2). Thirty patients (75%) had received a median of one therapy (range, 1 to 4) for PMF and 10 (25%) had not received any prior therapy. JAK2V617F was detected in 20 (56%) of 36 assessable patients. Eighteen (50%) of 36 patients with available pretreatment cytogenetic analysis had an abnormal karyotype, including del(20) in five patients, +8 in two patients, del(13) in five patients, and +9 in three patients.
Table 2.
Baseline Clinical Characteristics of Patients With Myelofibrosis Receiving Lenalidomide and Prednisone
| Characteristic | No. | % |
|---|---|---|
| Median age, years | 62 | |
| Range | 41-86 | |
| Median time from diagnosis, months | 10 | |
| Range | 0-269 | |
| Median WBC, × 109/L | 8.7 | |
| Range | 1.1-89 | |
| Median hemoglobin, g/dL | 9.8 | |
| Range | 7.8-17.3 | |
| Median platelets, × 109/L | 137 | |
| Range | 8-1,183 | |
| Previously treated | 30 | 75 |
| No. of prior therapies | 1 | |
| Range | 1-4 | |
| Hydroxyurea | 14 | 35 |
| Azacitidine | 6 | 15 |
| Corticosteroids | 5 | 12.5 |
| Anagrelide | 4 | 10 |
| Thalidomide | 4 | 10 |
| Interferon-α | 3 | 7.5 |
| Previously untreated | 10 | 25 |
| JAK2V617F mutation* | 20/36 | 56 |
| Abnormal cytogenetics | 18/36 | 50 |
Abbreviation: WBC, white blood cell.
Assessed in bone marrow by a quantitative pyrosequencing assay.
Clinical Response to Lenalidomide and Prednisone
The current median follow-up is 22 months (range, 6 to 27). Responses have been observed in 12 (30%) of 40 patients (Table 3) and are currently ongoing in 10 (25%). The overall response rates at 4 and 12 months from study entry were 23% and 30%, respectively. Responses occurred both in previously treated (n = 7) and untreated patients (n = 5). The median time to response was 12 weeks (range, 2 to 32). None of the patients achieved a complete response by the IWG criteria, which required a strict definition of complete resolution of fibrosis in the bone marrow. However, three patients (7.5%; all of them JAK2V617F negative), met all criteria for complete response except for bone marrow histologic remission, and were categorized as having a partial response (PR), which has been maintained for a median of 18 months (range, 6 to 21). One of the patients who achieved PR did so after experiencing treatment failure with thalidomide and prednisone. In addition, nine patients (22.5%; eight JAK2V617F positive, one JAK2V617F negative) achieved clinical improvement durable for a median of 18 months (range, 3.5 to 24+). Major Hb responses were achieved by seven (30%; three PR and four clinical improvement) of 23 patients with pretreatment Hb lower than 10 g/dL or transfusion dependent. Responses were also achieved by 10 (42%; two PR and eight clinical improvement) of 24 patients with splenomegaly palpable at least 5 cm below the left costal margin at study entry. Responses were observed both in patients with (eight clinical improvement) and without (three PR, one clinical improvement) the JAK2V617F mutation. None of the patients with baseline neutropenia (n = 2) or thrombocytopenia (n = 6) attained clinical improvement. Two responders lost their response after 6 and 9 months on study, respectively. At present, 10 patients remain on therapy without progression, and all 40 patients are alive.
Table 3.
Patients With Myelofibrosis Who Responded to Lenalidomide and Prednisone Therapy
| No. | Age (years) | Prior Myelofibrosis-Directed Therapy | Spleen (cm) | WBC (× 109/L) | Hemoglobin (× 109/L) | Platelets (× 109/L) | JAK2V617F allele (%) | No. of Cycles to Response | Response | Response Duration (months) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 73 | Darbepoietin | 10 | 8.4 | 7.8 | 202 | 45.7 | 7 | CI (hemoglobin, spleen) | 14+ |
| 2 | 55 | PEG-IFNα2b | 9 | 4.4 | 9.5 | 232 | 51.8 | 3 | CI (hemoglobin, spleen) | 24+ |
| 3 | 45 | IFNα | 11 | 5 | 10.6 | 181 | 0 | 8 | PR | 18+ |
| 4 | 52 | None | 15 | 12.5 | 11.6 | 156 | 81.2 | 6 | CI (spleen) | 20+ |
| 5 | 80 | None | 10 | 3 | 9.1 | 95 | 0 | 3 | PR | 21+ |
| 6 | 76 | None | 15 | 31.5 | 17.3 | 167 | 89 | 3 | CI (spleen) | 20+ |
| 7 | 71 | Hydroxyurea, erythropoietin | 12 | 18.6 | 12.2 | 358 | 0 | 2 | CI (spleen) | 19+ |
| 8 | 59 | None | 22 | 22.3 | 10.3 | 704 | 86.8 | 2 | CI (spleen) | 18+ |
| 9 | 69 | Hydroxyurea, darbepoietin, 5-azacitidine | 10 | 35.3 | 8.2 | 93 | 89.15 | 2 | CI (spleen) | 3.5 |
| 10 | 72 | Thalidomide + PRD, darbepoietin | 0 | 7.2 | 8.1 | 309 | 0 | 3 | PR | 6 |
| 11 | 68 | None | 0 | 5.4 | 9.1 | 440 | 28.35 | 4 | CI (hemoglobin) | 12+ |
| 12 | 56 | Hydroxyurea, darbepoietin | 20 | 17.4 | 9.7 | 90 | 89.6 | 1 | CI (hemoglobin, spleen) | 18+ |
Abbreviations: WBC: white blood cell; CI, clinical improvement; PEG-IFNα2b: pegylated interferon alpha 2b; PR, partial response; NA, not available; PRD, prednisone.
Cytogenetic, Molecular, and Cytokine Response
Therapy with lenalidomide and prednisone reduced the proportion of clones harboring cytogenetic abnormalities in two of seven responders with abnormal pretreatment karyotype after 7 and 8 months of treatment, respectively. Furthermore, all eight responders carrying the JAK2V617F mutation experienced a reduction of the baseline JAK2V617F allele burden, which was higher than 50% in four. In one responder, the JAK2V617F mutation became undetectable after 18 months of therapy. A significant reduction in the median baseline JAK2V617F allele burden was observed in the cohort of JAK2V617F-positive responders after 18 months of therapy (P = .03). No reductions were observed in JAK2V617F allele burden in nonresponders (Figs 1A to 1C).
Fig 1.

Dynamics of JAK2V617F mutational burden during lenalidomide and prednisone therapy. JAK2V617F mutational burden was measured by a quantitative pyrosequencing assay in bone marrow specimens at the time points specified during lenalidomide and prednisone therapy in the (A) whole cohort of patients, in (B) responders, and in (C) nonresponders. A significant reduction in JAK2V617F allele burden was observed over the baseline median value after 18 months of therapy in responders (P = .03). In contrast, no reduction in JAK2V617F allele burden was observed among nonresponders during therapy with lenalidomide and prednisone. Minimum and maximum values at each time point are depicted; median values are depicted within each column.
Since TGF-β1, PDGF, and bFGF produced by the megakaryocytes have been implicated in the pathogenesis of PMF, we also studied the impact of lenalidomide and prednisone therapy on the production of these cytokines.24 To this end, we measured the levels of FGF, PDGF, and TGF-β1, in peripheral blood by enzyme-linked immunosorbent assays at study entry and after 3 and 12 months of therapy in nine responders and 21 nonresponders (Fig 2A). The levels of all three cytokines were significantly higher in patients compared with healthy controls (P < .0001). Therapy with lenalidomide and prednisone did not decrease the levels of these cytokines. No significant differences were observed in cytokine levels between responders and nonresponders to lenalidomide and prednisone therapy.
Fig 2.
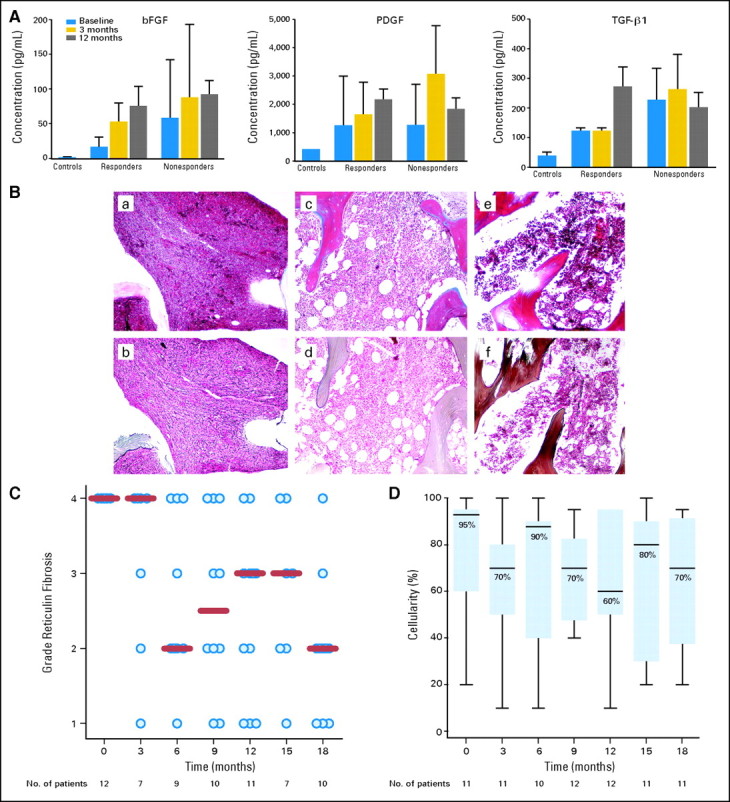
Effect of lenalidomide and prednisone therapy on cytokine levels, reticulin fibrosis and bone marrow cellularity in responders. (A) Levels of transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), and basic fibroblast growth factor (bFGF) were measured by enzyme-linked immunosorbent assays in peripheral blood before study entry and after 3 and 12 months of therapy in nine responders and 21 nonresponders to lenalidomide and prednisone. Control levels represent mean values in peripheral blood obtained from eight healthy volunteers. (B) Effects of lenalidomide and prednisone on the bone marrow of a responder carrying the JAK2V617F mutation. The upper panels (a, c, e) represent collagen trichrome stains and the lower panels (b, d, f) depict reticulin silver stains at baseline, and after 6 and 12 months of therapy. At baseline, the bone marrrow is markedly hypercellular (100%) with minimal increase in (a) collagen but with a diffuse, dense increase in reticulin fibers with (d) extensive intersections corresponding to a fibrosis score of 4. After 12 months of therapy, the bone marrow cellularity decreased to 70% with only a focal residual network of reticulin fibers in (f) perivascular areas corresponding to a fibrosis score of 1. (C) The dynamics of reticulin fibrosis are shown. All 11 assessable responders had a pretreatment reticulin fibrosis score of 4. This score was reduced to at least 2 in 10 of them during therapy (P = .007). (D) Changes in bone marrow cellularity among responders with respect to pretreatment values were not significant (P = .80).
Bone Marrow Fibrosis and Cellularity
We next evaluated the impact of the study therapy on bone marrow fibrosis. Of the 12 responders, 11 had serial bone marrow specimens available for review. All specimens showed a pretreatment reticulin fibrosis score of 4. Ten of 11 patients experienced reductions in reticulin fibrosis to at least a score of 2 (Figs 2B to 2C). This reduction was first documented after a median of 6 months of lenalidomide and prednisone therapy and was maintained or improved over the course of the study (P = .007). Furthermore, three (60%) of five responders with assessable bone marrow specimens experienced significant reductions of collagen deposition. Although two (8%) of the 12 responders experienced more than 50% reduction in bone marrow cellularity, no significant reductions were observed when the cohort of responders was considered as a whole (P = .80; Fig 2D).
Toxicity
Hematologic parameters at baseline and during therapy with lenalidomide and prednisone are presented in Table 4. Grade 3 to 4 neutropenia occurred in 23 patients (58%) and anemia in 17 patients (42%); almost all entered the study with grade 1 to 2 anemia. Grade 3 to 4 thrombocytopenia was reported in five patients (13%) and thrombocytosis in six (15%). Grade 3 to 4 nonhematologic toxicities were less frequent: fatigue in 11 (27%), diarrhea in six (15%); infection in six (15%); elevated bilirubin, edema, and rash in two each (5%); and dyspnea and nausea in one each (2%). No thrombotic complications were observed. Twenty-four patients (60%) had their lenalidomide dose reduced to dose level −1, including five (13%) who had their dose reduced to dose level −2. Only one patient (2.5%) required dose escalation while 15 (37%) remained at the initial dose level. Thirty patients (75%) discontinued the study treatment after a median of 6 months (range, 2 to 14.5) due to lack of response (n = 15), grade 3 to 4 toxicity (n = 9), patients' decision (n = 3), loss of response (n = 2), or allogeneic SCT (n = 1). No deaths or transformation to acute myeloid leukemia have been observed.
Table 4.
Baseline Hematologic Toxicities Prior to Therapy and Grade 3 to 4 Toxicities Reported in Patients With Myelofibrosis During Lenalidomide and Prednisone Therapy
| Toxicity Grade at Baseline | Neutropenia |
Anemia |
Thrombocytopenia |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient No. at Baseline | With Grade 3-4 During Therapy |
Patient No. at Baseline | With Grade 3-4 During Therapy |
Patient No. at Baseline | With Grade 3-4 During Therapy |
||||
| No. | % | No. | % | No. | % | ||||
| 0 | 38 | 23 | 60 | 7 | 1 | 14 | 26 | 3 | 11 |
| 1 | 0 | 12 | 6 | 50 | 6 | ||||
| 2 | 0 | 20 | 9 | 45 | 2 | ||||
| 3 | 0 | 1 | 2 | 2* | 100 | ||||
| 4 | 2 | 0 | 4 | ||||||
| Overall | 40 | 23 | 58 | 40 | 17 | 42 | 40 | 5 | 13 |
Grade 4 only.
DISCUSSION
Lenalidomide is a potent inhibitor of multiple proinflammatory cytokines such as tumor necrosis factor alpha and interleukin (IL) -6, secreted by activated monocytes. It also upregulates the production of IL-2, interferon-γ, IL-5, and IL-10 by T helper cells, without evidence of teratogenicity or mutagenesis.25,26 These activities, a more potent antiangiogenic activity,27 and an improved safety profile compared with the parent compound thalidomide likely accounts for the success of lenalidomide as treatment for multiple myeloma28,29 and myelodysplastic syndromes.30 Lenalidomide has also shown significant efficacy in two phase II studies involving 68 patients with symptomatic PMF, where given at 10 mg/d for 3 to 4 months resulted in an overall response rate of 29%. Anemia response was 22% (combined major and minor response as defined by Tefferi et al20 is equivalent to clinical improvement in anemia by IWG-MRT criteria used in the present study), but splenomegaly response was only 2% (major response as defined by Tefferi et al is equivalent to clinical improvement in splenomegaly as per IWG-MRT).20 Remarkably, eight patients who were either transfusion dependent or had a baseline Hb lower than 10 g/dL normalized their Hb levels.20 Although grade 3 to 4 neutropenia and thrombocytopenia occurred in 31% and 19% of patients, respectively, lenalidomide was generally well tolerated. However, a high proportion of patients lost their response after discontinuation of lenalidomide. We reasoned that the antiangiogenic, proapoptotic, and immunomodulatory effects of lenalidomide might be enhanced by the addition of prednisone to reduce marrow fibrosis and improve cytopenias. The overall response rate obtained with this combination was 30%, with major Hb responses achieved in 30% of patients with baseline Hb lower 10g/dL and reductions of splenomegaly achieved in 42% of patients with spleen size extending at least 5 cm below the left costal margin at study entry. Importantly, these responses are ongoing in 83% of responders for a median of 18 months (range, 3.5-24+). Although the design of our trial does not allow direct comparisons, the latter results are in contrast with phase II studies of single-agent lenalidomide, in which 38% of patients with major anemia response relapsed after lenalidomide discontinuation.20 Notably, while in phase II studies of single-agent lenalidomide therapy was administered only for 3 to 4 cycles in nonresponders, with intent to continue treatment until completion of either 6 or 24 cycles in responders, in our study lenalidomide was given for at least 6 months and continued in those showing clinical benefit as judged by treating physicians (not necessarily response by IWG-MRT criteria). This is of importance for two reasons. First, although the role of prednisone in this trial is difficult to establish, the response rate and the durability of the responses here reported are more likely to be a consequence of the prolonged exposure to lenalidomide, as eight of the 12 responders required three or more cycles of therapy to achieve significant clinical benefit, and only two of them have lost their response. Second, it suggests that chronic administration of lenalidomide may be required not only to maximize benefit but also to maintain the therapeutic activity of this agent in PMF.
Steady declines in the proportion of JAK2V617F-carrying clones occurred in all eight responders, including one patient for whom the mutation became undetectable, indicating marked reductions in the size of the neoplastic clone. JAK2V617F allele burden reductions were not observed in nonresponders. Our results indicate that lenalidomide is active in patients with PMF even in the absence of del(5q), as none of the responders in our study carried this karyotypic abnormality.31 Unlike patients with multiple myeloma receiving therapy with lenalidomide,32 patients with PMF treated with lenalidomide and prednisone did not exhibit an excess risk of thrombotic events. However, as with single-agent lenalidomide, grade 3 to 4 cytopenia was frequent with this combination, necessitating close monitoring of blood counts.
The bone marrow fibrosis in patients with MF is a reactive process mediated by the profibrogenic and proangiogenic cytokines (eg, TGF-β1, PDGF, bFGF) produced by clonal monocytes and megakaryocytes.33,34 As shown in mouse models,14,15 a PMF phenotype can be generated either by forced thrombopoietin expression or decreased GATA-1 expression.14,15 In contrast to the current experience with selective JAK2 inhibitors, lenalidomide and prednisone not only resulted in important anemia responses in PMF, but also markedly reduced bone marrow reticulin and collagen fibrosis. Indeed, 10 of 11 assessable patients, all with reticulin fibrosis grade 4 at study entry, exhibited reductions to at least grade 2 fibrosis. Similar reductions were also observed in collagen fibrosis in three of five responders. It could be speculated that the potent antiangiogenic activity of lenalidomide16 might have synergized with an anti-inflammatory agent, such as prednisone, to induce these remarkable responses in fibrosis. However, a correlation between marrow fibrosis reduction and reduction in cytokine levels could not be established, perhaps because the latter were measured in peripheral blood and not in megakaryocytes obtained from cultured blood CD34+ cells.24
In summary, the combination of lenalidomide and prednisone represents an active and well-tolerated regimen for patients with PMF. An important question is how biologic agents such lenalidomide and other novel IMiDs (eg, pomalidomide) compare with the emergent JAK2 kinase inhibitors. Although several have shown promising results in PMF,10,11,13 their activity appears to be limited to improvement in constitutional symptoms and reduction in spleen size. Lenalidomide, in addition to reducing spleen size, was significantly more effective at improving ineffective erythropoiesis and reducing bone marrow fibrosis and neoangiogenesis,20 the latter being an independent prognostic risk factors in PMF.35,36 The fact the IMiDs and JAK2 inhibitors tackle with different efficacies distinct aspects of the clinical and pathologic manifestations of PMF suggests the exciting prospect of combining two very active groups of agents in a disease orphan of active drug therapies for decades.
Acknowledgment
We are indebted to Neda Bell and Catriona Byrne (Celgene Corporation, Warren, NJ) who supported the undertaking of this clinical trial.
Footnotes
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical Trials repository link available on JCO.org.
Clinical trial information can be found for the following: NCT00352794.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTSOF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a [U] are those for which no compensation was received; those relationships marked with a [C] were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: None Stock Ownership: None Honoraria: None Research Funding: Srdan Verstovsek, Celgene Corporation Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Alfonso Quintás-Cardama, Srdan Verstovsek
Provision of study materials or patients: Hagop Kantarjian, Jorge Cortes, Guillermo Garcia-Manero, Alessandra Ferrajoli, Carlos Bueso-Ramos, Srdan Verstovsek
Collection and assembly of data: Alfonso Quintás-Cardama, Taghi Manshouri, Deborah Thomas, Srdan Verstovsek
Data analysis and interpretation: Alfonso Quintás-Cardama, Taghi Manshouri, Deborah Thomas, Srdan Verstovsek
Manuscript writing: Alfonso Quintás-Cardama, Srdan Verstovsek
Final approval of manuscript: Alfonso Quintás-Cardama, Hagop Kantarjian, Taghi Manshouri, Deborah Thomas, Jorge Cortes, Farhad Ravandi, Guillermo Garcia-Manero, Alessandra Ferrajoli, Carlos Bueso-Ramos, Srdan Verstovsek
REFERENCES
- 1.Hoffman R, Rondelli D. Biology and treatment of primary myelofibrosis. Hematology Am Soc Hematol Educ Program. 2007;2007:346–354. doi: 10.1182/asheducation-2007.1.346. [DOI] [PubMed] [Google Scholar]
- 2.Arana-Yi C, Quintas-Cardama A, Giles F, et al. Advances in the therapy of chronic idiopathic myelofibrosis. Oncologist. 2006;11:929–943. doi: 10.1634/theoncologist.11-8-929. [DOI] [PubMed] [Google Scholar]
- 3.Deeg HJ, Gooley TA, Flowers ME, et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood. 2003;102:3912–3918. doi: 10.1182/blood-2003-06-1856. [DOI] [PubMed] [Google Scholar]
- 4.Rondelli D, Barosi G, Bacigalupo A, et al. Allogeneic hematopoietic stem-cell transplantation with reduced-intensity conditioning in intermediate- or high-risk patients with myelofibrosis with myeloid metaplasia. Blood. 2005;105:4115–4119. doi: 10.1182/blood-2004-11-4299. [DOI] [PubMed] [Google Scholar]
- 5.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 6.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 7.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 8.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 9.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–22792. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah N, Olszynski P, Sokol L, et al. A phase I study of XL019, a selective JAK2 inhibitor, in patients with primary myelofibrosis, post-polycythemia vera, or post-essential thrombocythemia myelofibrosis. Blood. 2008:112. abstr 98. [Google Scholar]
- 11.Pardanani A, Gotlib J, Jamieson C, et al. A phase I study of TG101348, an orally bioavailable JAK2-selective inhibitor, in patients with myelofibrosis. Blood. 2008:112. abstr 97. [Google Scholar]
- 12.Moliterno A, Roboz GJ, Carroll M, et al. An open-label study of CEP-701 in patients with JAK2 V617F-positive polycythemia vera and essential thrombocytosis. Blood. 2008:112. abstr 99. [Google Scholar]
- 13.Verstovsek S, Kantarjian H, Pardanani A, et al. The JAK inhibitor, INCB018424, demonstrates durable and marked clinical responses in primary myelofibrosis (PMF) and post-polycythemia/essential thrombocythemia myelofibrosis (post PV/ETMF) Blood. 2008:112. abstr 1762. [Google Scholar]
- 14.Schmitt A, Jouault H, Guichard J, et al. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood. 2000;96:1342–1347. [PubMed] [Google Scholar]
- 15.Xu M, Bruno E, Chao J, et al. Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood. 2005;105:4508–4515. doi: 10.1182/blood-2004-08-3238. [DOI] [PubMed] [Google Scholar]
- 16.Raje N, Anderson K. Thalidomide–a revival story. N Engl J Med. 1999;341:1606–1609. doi: 10.1056/NEJM199911183412110. [DOI] [PubMed] [Google Scholar]
- 17.Abgrall JF, Guibaud I, Bastie JN, et al. Thalidomide versus placebo in myeloid metaplasia with myelofibrosis: A prospective, randomized, double-blind, multicenter study. Haematologica. 2006;91:1027–1032. [PubMed] [Google Scholar]
- 18.Marchetti M, Barosi G, Balestri F, et al. Low-dose thalidomide ameliorates cytopenias and splenomegaly in myelofibrosis with myeloid metaplasia: A phase II trial. J Clin Oncol. 2004;22:424–431. doi: 10.1200/JCO.2004.08.160. [DOI] [PubMed] [Google Scholar]
- 19.Mesa RA, Steensma DP, Pardanani A, et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood. 2003;101:2534–2541. doi: 10.1182/blood-2002-09-2928. [DOI] [PubMed] [Google Scholar]
- 20.Tefferi A, Cortes J, Verstovsek S, et al. Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006;108:1158–1164. doi: 10.1182/blood-2006-02-004572. [DOI] [PubMed] [Google Scholar]
- 21.Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128–1132. [PubMed] [Google Scholar]
- 22.Tefferi A, Barosi G, Mesa RA, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT) Blood. 2006;108:1497–1503. doi: 10.1182/blood-2006-03-009746. [DOI] [PubMed] [Google Scholar]
- 23.McClure R, Mai M, Lasho T. Validation of two clinically useful assays for evaluation of JAK2 V617F mutation in chronic myeloproliferative disorders. Leukemia. 2006;20:168–171. doi: 10.1038/sj.leu.2404007. [DOI] [PubMed] [Google Scholar]
- 24.Wang JC, Chang TH, Goldberg A, et al. Quantitative analysis of growth factor production in the mechanism of fibrosis in agnogenic myeloid metaplasia. Exp Hematol. 2006;34:1617–1623. doi: 10.1016/j.exphem.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Schafer PH, Gandhi AK, Loveland MA, et al. Enhancement of cytokine production and AP-1 transcriptional activity in T cells by thalidomide-related immunomodulatory drugs. J Pharmacol Exp Ther. 2003;305:1222–1232. doi: 10.1124/jpet.102.048496. [DOI] [PubMed] [Google Scholar]
- 26.Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer. 2004;4:314–322. doi: 10.1038/nrc1323. [DOI] [PubMed] [Google Scholar]
- 27.Tohnya TM, Hwang K, Lepper ER, et al. Determination of CC-5013, an analogue of thalidomide, in human plasma by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;811:135–141. doi: 10.1016/j.jchromb.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 28.Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357:2133–2142. doi: 10.1056/NEJMoa070596. [DOI] [PubMed] [Google Scholar]
- 29.Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123–2132. doi: 10.1056/NEJMoa070594. [DOI] [PubMed] [Google Scholar]
- 30.List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456–1465. doi: 10.1056/NEJMoa061292. [DOI] [PubMed] [Google Scholar]
- 31.List A, Kurtin S, Roe DJ, et al. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med. 2005;352:549–557. doi: 10.1056/NEJMoa041668. [DOI] [PubMed] [Google Scholar]
- 32.Hirsh J. Risk of thrombosis with lenalidomide and its prevention with aspirin. Chest. 2007;131:275–277. doi: 10.1378/chest.06-2360. [DOI] [PubMed] [Google Scholar]
- 33.Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000;342:1255–1265. doi: 10.1056/NEJM200004273421706. [DOI] [PubMed] [Google Scholar]
- 34.Dong M, Blobe GC. Role of transforming growth factor-beta in hematologic malignancies. Blood. 2006;107:4589–4596. doi: 10.1182/blood-2005-10-4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mesa RA, Hanson CA, Rajkumar SV, et al. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid metaplasia. Blood. 2000;96:3374–3380. [PubMed] [Google Scholar]
- 36.Arora B, Ho CL, Hoyer JD, et al. Bone marrow angiogenesis and its clinical correlates in myelofibrosis with myeloid metaplasia. Haematologica. 2004;89:1454–1458. [PubMed] [Google Scholar]