

Abstract
Tauopathies are a group of incurable neurodegenerative diseases, in which loss of neurons is accompanied by intracellular deposition of fibrillar material composed of hyper phosphorylated forms of the microtubule associated protein Tau. A zebrafish model of Tauopathy could complement existing murine models by providing a platform for genetic and chemical screens, in order to identify novel therapeutic targets and compounds with disease-modifying potential. In addition, Tauopathy zebrafish would be useful for hypothesis-driven experiments, especially those exploiting the potential to deploy in vivo imaging modalities. Several considerations, including conservation of specialized neuronal and other cellular populations, and biochemical pathways implicated in disease pathogenesis, suggest that the zebrafish brain is an appropriate setting in which to model these complex disorders. Novel transgenic zebrafish lines expressing wild-type and mutant forms of human Tau inCNS neurons have recently been reported. These studies show evidence that human Tau undergoes disease-relevant changes in zebrafish neurons, including somato-dendritic relocalization, hyper phosphorylation and aggregation. In addition, preliminary evidence suggests that Tau transgene expression can precipitate neuronal dysfunction and death. These initial studies are encouraging that the zebrafish holds considerable promise as a model in which to study Tauopathies. Further studies are necessary to clarify the phenotypes of transgenic lines and to develop assays and models suitable for unbiased high-throughput screening approaches. This article is part of a Special Issue entitled Zebrafish Models of Neurological Diseases.
1. Introduction
The microtubule-associated protein Tau (MAP-τ, ‘Tau’) undergoes biochemical alterations, cellular redistribution, and deposition as insoluble intraneuronal fibrils (Fig. 1), in a variety of neurodegenerative conditions that are collectively termed ‘Tauopathies’. Together, these diseases, which include Alzheimer’s disease, progressive supranuclear palsy and other conditions (Table 1), are an important cause of morbidity and mortality, with diverse clinical manifestations. No currently available treatments improve the prognosis of any of these relentlessly progressive diseases. Consequently, investigations aimed at determining the underlying pathophysiology of Tauopathies, and isolating novel therapeutic agents that prevent disease progression, are of great importance. In this review, we consider recent developments concerning the possibility that a zebrafish Tauopathy model might be useful for therapeutic target and drug discovery in vivo. After briefly reviewing current knowledge and murine models of Tauopathy, we discuss the possible advantages of a zebrafish model and whether a truly representative model encompassing key biochemical events underlying Tauopathy can be recapitulated in the zebrafish central nervous system. Finally, we review recent publications demonstrating initial proof of concept that Tauopathy zebrafish models recapitulate core features of the human disorders.
Fig. 1.
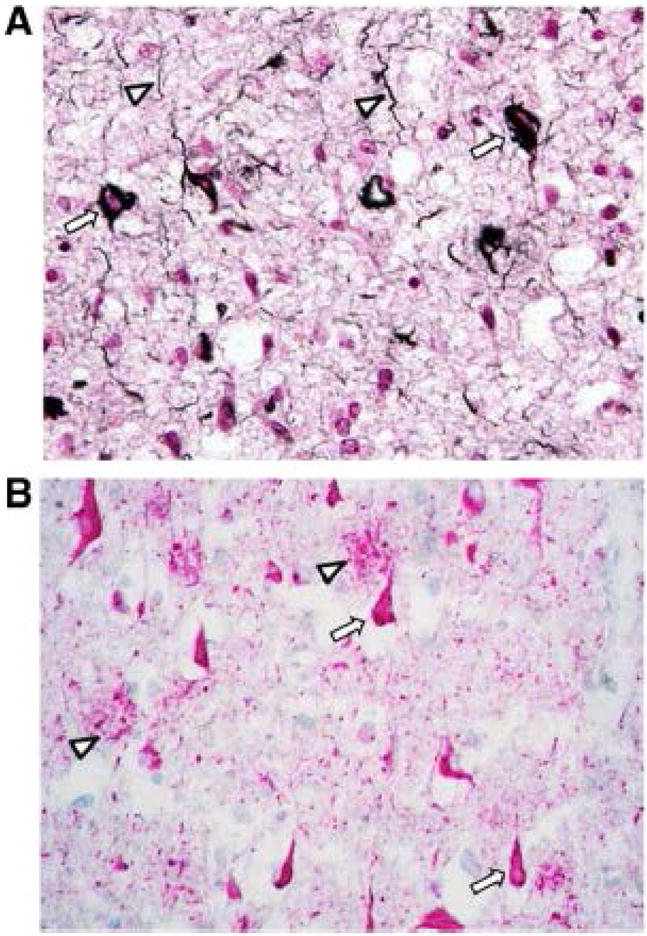
Neurofibrillary tangles in Alzheimer’s disease. A: Neurofibrillary tangles in Alzheimer’s disease prefrontal cortex are demonstrated using the Gallyas silver method [127]. NFTs are seen as numerous argyrophilic (black) fibrillar intraneuronal inclusions (arrows). Neuropil threads, axonal abnormalities also caused by accumulations of fibrillar Tau, are also seen (arrowheads). B: Neurofibrillary tangles in Alzheimer’s disease hippocampus are demonstrated by immunohistochemistry, using an antibody (AT8) that detects phospho-S202/T205-Tau. NFTs are seen as abundant immunoreactive (red) intraneuronal inclusions (arrows). Accumulations of phospho-Tau are also apparent in abnormal axonal terminals surrounding amyloid plaques (arrowheads).
Table 1.
Neurodegenerative diseases associated with prominent Tau pathology.
| Disease | Typical clinical presentations | Typical Tau pathology | Tau filaments | Tau species | Etiology |
|---|---|---|---|---|---|
| Alzheimer’s disease (AD) | Memory disorder, dysphasia frontal lobe cognitive–behavioral disorder, dementia | Flame-shaped neurofibrillary tangles | Paired helical filaments, straight filaments [123] | 4R- and 3R-Tau | Mostly unkown, few cases caused by mutations in APP, PS1 or PS2 genes |
| Progressive supranuclear palsy (PSP) | Falls, rigidity, oculomotor disorder, cognitive–executive disorder | Globose neurofibrillary tangles, tufted astrocytes | Straight filaments [124] | Predominantly 4R-Tau | Unknown, linked to H1 haplotype at MAPT locus |
| Corticobasal degeneration (CBD) | Dystonia, myoclonus, apraxia, cortical sensory loss, dementia | Ballooned neurons, pre-tangles, astrocytic plaques | Twisted ribbon filaments [125] | Predominantly 4R-Tau | Unknown, linked to H1 haplotype at MAPT locus |
| Pick’s disease (PiD) | Frontal lobe cognitive–behavioral disorder, dementia | Pick bodies | Helical filaments; straight filaments [126] | Predominantly 3R-Tau | Unknown |
| Fronto-temporal dementia and parkinsonism linked to chromosome 17 (FTDP17) | Variable Parkinsonian motor disorder and/or frontal lobe dementia | Variable | Variable | Variable (see text) | Mutations in MAPT gene encoding Tau |
2. The microtubule-associated protein Tau
Neurons rely on fast axonal transport to shuttle organelles and macromolecules over long distances, allowing their physiological distribution and turnover within axons and dendrites. Microtubules, which provide the tracks along which molecular motors rapidly transport these diverse cargos, are composed of polymerized tubulin monomers. Assembly of tubulin into microtubules is promoted by microtubule-associated proteins, the first of which to be identified was termed ‘Tau’ (τ was used to denote a factor essential for tubule formation) [1,2]. Tau is expressed widely in neurons, where it is enriched in the axonal compartment [3]. The microtubule-binding domain of Tau localizes to the C-terminal half of the protein [4,5] (Fig. 2). The N-terminal, or projection domain, contains a proline-rich region and multiple potential serine–threonine phosphorylation sites, and is thought to be involved in interactions with other cellular components.
Fig.2.

Isoforms of the microtubule-associated protein Tau. The schematic depicts the six Tau isoforms expressed in the adult human brain, labeled to the left of each protein. Positions of major protein domains are shown above the longest isoform. The N-terminal insertions encoded by exons 2 and 3, and the four tandem repeats (R1–4) in the microtubule-binding domains are indicated. Sites of known phosphorylation are shown above the longest isoform The table to the right of the figure shows the number of amino acids, predicted molecular mass and electrophoretic mobility of each protein isoform.
2.1. The MAPT gene
Tau is encoded by the MAPT gene, which is located on chromosome 17 and contains 16 exons. Alternative splicing of the primary transcript leads to a family of mRNAs, encoding different protein isoforms. In adult human brain, six isoforms are expressed, produced by alternative splicing of exons 2, 3 and 10 (exons 4A, 6 and 8 are not expressed in the CNS). Tau isoforms in the CNS contain either three or four copies of a tandem repeat containing tubulin-binding sequences, referred to as 3R- and 4R-Tau respectively (Fig. 2). Exon 10 encodes the second microtubule-binding repeat, such that inclusion of exon 10 in the mRNA results in translation of 4R-Tau, whereas exclusion of exon 10 results in a transcript encoding 3R-Tau [6,7]. Optional inclusion of exon 2, or exons 2 and 3, gives rise to N-terminal insertions of 29 (‘1N’) or 58 (‘2N’) amino acids respectively [8]. The six resulting CNS isoforms are shown in Fig. 2. Additional isoforms generated by incorporation of exon 4A (‘large Tau’) are expressed in the peripheral nervous system [9].
2.2. Functions of Tau
Association of Tau with tubulin promotes microtubule formation [1,2], and association of Tau with microtubules is thought to enhance their stability [10]. 4R-Tau binds to microtubules with greater avidity than 3R-Tau isoforms [4,11,12] (this is attributable to sequences in the first inter-repeat segment that are unique to 4R proteins, rather than to the additional copy of the microtubule-binding tandem repeat [13]), suggesting that expression of 4R-Tau may favor microtubule formation and stability. In healthy adult brain tissue, the 4R-/3R-Tau ratio is approximately 1, and disruption of this balance in the human brain may have serious consequences (see below) [14]. However, there are situations in which the 4R-/3R-Tau ratio may be altered physiologically. For example, during embryogenesis, Tau is exclusively expressed in the brain as the 3R/0N isoform [8]. It has been argued that this might promote plasticity of neuronal processes during development by reducing microtubule stability. Recent work has suggested that Tau interacts with motor proteins involved in axonal transport, provoking different functional consequences for distinct types of motor molecules [15]. Kinesin, which transports cargo towards the distal end of the axon, tended to detach from the microtubule on encountering Tau, whereas the proximally-directed motor Dynein showed more directional reversal than detachment. These effects were apparent in the presence of 3R/0N Tau, and suggest that association of Tau with microtubules may modulate axonal transport in addition to promoting microtubule stability.
2.3. Tau phosphorylation
Tau undergoes post-translational changes, including physiological serine–threonine phosphorylation [16] and O-glycosylation [17], in addition to tyrosine phosphorylation [18], SUMOylation [19] and pathological nitration [20]. Of these changes, phosphorylation has been most extensively studied, since pathological hyper phosphorylation is associated with Tauopathy [21]. Approximately 45 phosphorylation sites have been identified, which cluster in the proline rich C-terminal domains of the protein [22]. Phosphorylation of Tau, in particular at S262 and S356 within the microtubule-binding domain, promotes its detachment from microtubules, suggesting a potential mechanism in regulating microtubule stability and axonal transport [23,24]. In addition to microtubule-binding affinity, phosphorylation may have other effects, such as modulating binding to the chaperone Pin1 [25] and possibly in regulating putative signal transduction functions of Tau through phosphorylation of SH3 domains [26].
Hyper phosphorylation of Tau is a characteristic feature in Tauopathy specimens. No single phosphorylation site is specific for Tauopathy. Although some phospho-epitopes may be more readily detected in pathological specimens compared with non-Tauopathy material, most of the known phosphorylation sites have been demonstrated in normal biopsy material [16]. Hyper phosphorylation is a quantitative rather than qualitative phenomenon, defined by the degree to which Tau is phosphorylated at multiple sites. Phosphorylation is a dynamic process and Tau is rapidly de-phosphorylated post mortem, accounting for the lack of detectable phospho-epitopes in control autopsy specimens. It is unclear whether a functional abnormality of phosphatase activity is present in Tauopathy samples, or whether the failure of de-phosphorylation in Tauopathy autopsy material is attributable to changes in the properties of the hyper phosphorylated molecule as a phosphatase substrate. During normal development, Tau is present in a highly phosphorylated state [27,28], suggesting that the balance of Tau phosphorylation and dephosphorylation can change physiologically, without necessarily provoking neurodegeneration.
2.4. Tau kinases
Several cellular kinases are able to phosphorylate Tau in vitro or in cell culture (Table 2; reviewed in [22]). Many of these phosphorylate multiple residues of Tau, and individual residues can be phosphorylated by multiple different kinases, suggesting that Tau is a promiscuous kinase substrate. Consequently, it is difficult to be certain which kinase(s) are responsible for physiological or pathological phosphorylation of Tau in vivo. Kinases that have emerged as strong in vivo candidates include glycogen synthase kinase-3β (GSK3β) [29–33] and cyclin-dependent protein kinase-5 (cdk5) [34,35]; both are expressed abundantly in neurons and associate with microtubules (cdk5 is anchored to microtubules by Tau [36]). However, it is likely that more than one kinase participates in the events leading to hyper phosphorylation, since no single kinase phosphorylates all of the residues found in Tauopathy specimens. Furthermore, there are examples in vitro of cooperation between kinases; for example, cdk5-mediated phosphorylation of Tau promotes further phosphorylation by GSK3β [37,38]. Consequently, it is thought that the transition from normal to hyper phosphorylated Tau may be a hierarchical or sequential process dependent on phosphorylation by multiple different kinases. Several different protein phosphatases dephosphorylate Tau in vitro, including PP1, PP2A, PP2B and PP2C. Their specificities are overlapping and their roles in vivo are incompletely understood. PP2A has emerged as a strong candidate, since it appears to be the major brain-derived phosphatase activity directed towards Tau in the rat brain [39] and is localized to microtubules (both through an association with Tau and also directly, which may regulate its activity in vitro [40]).
Table 2.
Phylogenetic conservation of kinases implicated in tau physophorylation
| Tau Kinase | Protein accession numbers
|
% identity | % consensus | |
|---|---|---|---|---|
| Human | Zebrafish | |||
| Glycogen synthase kinase 3β (GSK3β) | NP_001139628 | NP_571456 | 94.5 | 97.4 |
| Cyclin-dependent kinase 5 (cdk5) | AAP35326 | AAH85381 | 96.1 | 98.6 |
| Mitogen activated protein kinase 1 (MAPK1, ERK1) | NP_620407 | BAB1 1813 | 90.8 | 93.5 |
| Microtubule Affinity Regulating Kinase 1 (MARK1) | CAH72462 | AA155560 | 79.3 | 85.5 |
| Casein kinase 1α (CK1α) | NP_001883 | NP_694483 | 99.3 | 99.3 |
2.5. Tau deposits in sporadic neurodegenerative diseases
Tauopathies are characterized pathologically by the presence of fibrillar deposits of hyper phosphorylated Tau. The histological appearance and distribution of Tau deposits differs between diseases, as does the ultrastructural appearance of Tau filaments that compose neurofibrillary tangles, Pick bodies and other inclusions (Table 1). Formation of neurofibrillary tangles (NFTs) likely occurs through oligomeric intermediates, and there is some evidence that these are the primary pathogenic species, rather than mature NFTs (see below). The predominant Tau isoforms deposited in neurons differ between diseases. In Alzheimer’s disease, SDS-PAGE analysis showed three major Tau immunoreactive bands of 68, 64 and 60 kDa present in detergent extracted material containing paired helical filaments [41]. After dephosphorylation of the same material, all six Tau isoforms were resolved by electrophoresis, showing that hyper phosphorylation of Tau was responsible for the alteration in its electrophoretic mobility, and that AD filaments are composed of all six Tau isoforms. Similar analyses of specimens derived from other types of Tauopathy have revealed different results. In progressive supra nuclear palsy and cortico-basal degeneration, two major Tau bands of 68- and 64 kDa are derived from hyper phosphorylation of 4R-Tau species [42,43]. Conversely, material derived from Pick’s disease brain shows two bands of 64- and 60-kDa, dephosphorylation of which reveals the presence of 3R-Tau species [43,44]. Interestingly, genetic data shows association between an extended haplotype of markers across the MAPT locus, and PSP/CBD [45]; recent data shows that this haplotype favors the splicing of exon 10+ transcripts encoding 4R-Tau [46], suggesting that genetically determined relative overproduction of 4R-Tau might be one factor in the pathogenesis of these sporadic diseases.
2.6. Hereditary fronto-temporal dementia and Parkinsonism linked to chromosome 17
Definitive evidence that the development of Tau pathology may be mechanistically important in common sporadic Tauopathies comes from the study of rare families, in which mutations within the MAPT gene are sufficient to cause a neurodegenerative disease with prominent Tauopathy [14,47]. FTDP17 is inherited as an autosomal dominant trait and can manifest as a Parkinsonian movement disorder or as a cognitive–behavioral disorder, resembling other forms of fron to-temporal dementia (reviewed in [48]). To date, 39 different pathogenic MAPT mutations have been identified in FTDP17 families. Mutations clustered around the 5′ boundary of intron 10 alter the regulation of splicing, most frequently resulting in relative over-production of exon 10+ transcripts and deposition of hyper phosphorylated 4R-Tau isoforms. Missense mutations within exon 10 also provoke a 4R-Tauopathy, by changing the biophysical properties of 4R-Tau without altering the ratio of splice isoforms; these mutations also result in 4R-Tau deposition. Finally, mutations outside exon 10 are expressed in both 3R- and 4R-Tau, but may provoke selective deposition of 3R- or 4R-, or both isoforms of Tau. A variety of mechanisms has been proposed to account for the pathogenicity of MAPT missense mutants, including promotion of Tau fibril formation, impairment of microtubule-binding and introduction or removal of key phosphorylation sites [49]. Many of the mutants show multiple functional abnormalities, such as alterations in 4R-/3R-splice ratio and reduced propensity to promote the stabilization of microtubules. Although FTDP17 is an uncommon disease, the discovery of pathogenic MAPT mutations has provided important confirmation that primary abnormalities of Tau, including alterations in the relative abundance of 3R- and 4R-isoforms, can result in neurodegeneration and Tauopathy. Given the close similarity between FTDP17 and sporadic Tauopathies in some cases, it is reasonable to conclude that abnormalities of cellular Tau metabolism may be central to neurodegeneration in common sporadic Tauopathies. Consequently, study of model systems in which Tauopathy is induced by molecular manipulations that mimic the genetic mechanisms of FTDP17 might yield important insights into pathophysiology and identify potential treatment targets for common sporadic Tauopathies.
3. Transgenic mouse models of Tauopathy
A variety of different transgenic lines, over-expressing wild-type or FTDP17 mutant human Tau, have been described (reviewed in [50,51]). Several important themes that have emerged from studies of these animals are briefly considered here.
First, dissociation between NFT formation and neuronal dysfunction or death was demonstrated in some of these models, suggesting that Tau hyper-phosphorylation, relocalization or oligomerization may be more critical for the emergence of phenotypic abnormalities in these models than formation of NFTs. Abnormalities of hippocampal synaptic protein expression and physiology preceded the formation of NFTs in mice expressing P301S mutant Tau [52]. Studies using a different model in which P301L Tau was expressed under an inducible promoter showed that suppression of transgene expression after the onset of phenotypic abnormalities prevented neuronal loss and behavioral abnormalities but did not affect neurofibrillary tangle formation [53], and there was regional dissociation between NFT formation and cell loss [54]. In addition, immunization of Tau/Aβ transgenic mice reduced soluble Tau in the brain but did not affect NFT formation — the animals showed phenotypic improvement, suggesting that neurobehavioral abnormalities were attributable to a soluble Tau species rather than NFT [55].
Second, Tau transgenic mice have provided a powerful means to test the interactions between different biochemical pathways implicated in neurodegeneration. For example, evidence of an in vivo interaction between Tau and Aβ in the pathogenesis of Alzheimer’s disease was demonstrated by intracerebral inoculation of the Alzheimer’s disease-associated amyloid peptide Aβ1–42, which enhanced the formation of NFTs in a mouse P301L Tau-expressing model [56]. Furthermore, P301L Tau/APP double transgenic animals showed exacerbation of NFT pathology in the presence of the APP transgene, whereas Aβ plaque formation was unaffected by the overexpression of mutant Tau, suggesting that β-amyloid formation or another function of APP might be upstream of Tauopathy in this model [57].
Finally, murine models have provided powerful means to test hypotheses concerning experimental treatment approaches, and biochemical interventions aimed at understanding disease pathogenesis in vivo (reviewed in [58]). For example, administration of Li+, an inhibitor of GSK3, reduced hyper phosphorylation of Tau in vivo and ameliorated some phenotypic abnormalities [31], and another kinase inhibitor targeting cdk5, GSK3 and MAPK1 delayed the onset of a motor deficit in Tau transgenic animals [59]. Other studies have deployed pharmacological interventions in order to clarify the potential therapeutic utility of interventions directed at restoring loss of Tau microtubule-stabilizing function [60], enhancing clearance of pathological Tau species from neurons by inhibiting refolding functions of the HSP90 complex to promote Tau degradation [61], and curtailing a potentially pathogenic neuroinflammatory response by targeting microglial activation using immunosuppressive drugs [52].
Together, these observations show that experimental genetic manipulations, which mimic the mechanisms underlying FTDP17 in humans, provoke phenotypic changes relevant to human Tauopathy in transgenic animals. These models have been valuable for hypothesis-driven experiments aiming to elucidate the pathogenesis of Tauopathy and for testing putative therapeutic approaches.
4. Why would a zebrafish Tauopathy model be useful?
The zebrafish is a small freshwater fish that has been used extensively in laboratory studies of vertebrate development. Fish embryos develop externally allowing direct observation of embryogenesis. Many morphological and molecular parallels are shared between fish and other vertebrates allowing insights gained in the zebrafish model to be applied to other systems. Stemming from the use of zebrafish in developmental studies, an extensive array of experimental methodologies has been developed. Straightforward techniques are available for over-expression [62,63] or targeted knockdown of genes of interest [64], allowing the consequences of altered gene function to be determined readily in vivo. The deployment of fluorescent reporters [65] allows direct observation of morphology or physiology of individual cells in vivo [66–68]. The ability of zebrafish to breed regularly and produce sizeable clutches of embryos, and the practicable housing of significant numbers of animals, has allowed development of techniques for large-scale genetic [69–72] and chemical modifier [73–75] screens. These have provided numerous novel molecular insights into development, through unbiased phenotype-driven approaches. More recently, it has been demonstrated that intact zebrafish larvae provide an in vivo neurobehavioral platform suitable for screens to find chemical modifiers of vertebrate behavior [76].
It is possible that applying these types of analysis to zebrafish models of neurodegenerative diseases would provide novel molecular insights into disease pathogenesis, and allow identification of potentially therapeutic compounds. In particular, screening studies carried out in the intact vertebrate CNS in vivo might uncover drug targets that are not present in cell culture models commonly used for discovery-driven approaches, such as pathogenic mechanisms that are not cell-autonomous, or which are only expressed in end differentiated neurons. Consequently, there has been significant recent interest in the development of zebrafish models of Tauopathy.
5. Is the zebrafish a suitable organism in which to study Tauopathy?
The proposed use of a small aquatic creature to study complex human neurobehavioral diseases naturally raises questions about relevance — will it be possible to generate a truly representative model, with potential to yield important insights into the human disorder? A variety of considerations suggest that the zebrafish may be a good model system in which to study human neurological diseases. With respect to the human CNS, the zebrafish shows conservation of basic brain organization [77] and many key neuroanatomical [78,79] and neurochemical [80,81] pathways of relevance to human disease; the zebrafish CNS contains microglia [68], cells with astrocytic properties [82,83], oligodendrocytes and myelin [84–87], and a blood–brain barrier [88], all of which have been implicated in the pathogenesis of neurological disease. These observations imply that the tissue environment in the zebrafish CNS may present appropriate conditions in which to recapitulate pathological changes representative of the human disorders. In addition, there is a striking degree of phylogenetic conservation of genes implicated in the pathogenesis of neurodegenerative diseases, and a variety of other CNS functions [89–96]. This suggests that it will be possible to provoke neurodegeneration in zebrafish through conserved biochemical pathways that are sufficiently close to those underlying human disorders that findings in the zebrafish model will yield relevant insights into diseases mechanisms. It is noteworthy that the zebrafish genome contains highly conserved orthologues of each of the kinases implicated in Tau phosphorylation (Table 2 and Fig. 3), which may be of particular relevance to Tauopathy in view of the proposed central role of hyper phosphorylation in pathogenesis. Similar considerations apply to other conserved pathways, and it seems likely that findings in zebrafish models will be representative of the mechanisms underlying the pathogenesis of human Tauopathies and will therefore be relevant to their understanding.
Fig. 3.
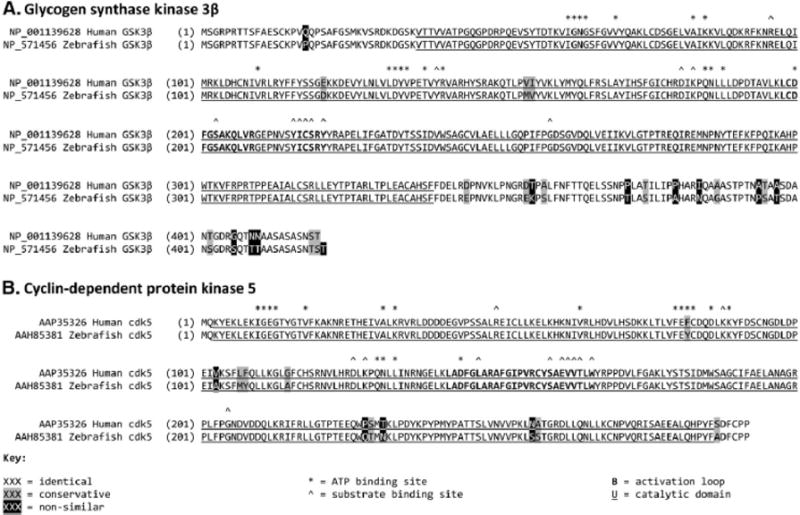
Phylogenetic conservation of kinases implicated in Tau phosphorylation. The sequence Figure shows the striking degree of amino acid conservation between human and zebrafish (A) GSK3β and (B) cdk5. Each panel shows the Human (upper) and zebrafish (lower) protein sequences, aligned using the ClustalW algorithm. Amino acids are shaded to show identical, conserved and non-similar residues. The catalytic domain of each kinase is indicated by underlining, and important Functional sites labeled as indicated.
5.1. Zebrafish paralogues of MAPT
Recent work has identified two paralogues of MAPT in zebrafish, annotated mapta and maptb [97]. Analysis of homology between zebrafish and mammalian MAP-encoding genes, and examination of synteny between zebrafish mapt and mammalian MAPT loci, strongly suggest that the two zebrafish genes arose from duplication of an ancestral mapt locus. A genome duplication event is thought to have taken place during the evolution of teleosts [98]; consequently, there are dual zebrafish paralogues of many mammalian genes [92,99]. It is possible that divergent evolution of the two mapt genes has given rise to functional sub-specialization, suggesting that the zebrafish may be a powerful tool in which to dissect multiple cellular roles of Tau. The two proteins encoded by the zebrafish genes have not yet been detected, but both mapta and maptb mRNAs were expressed in the developing CNS [97]. A complex pattern of alternative splicing of the mapta and maptb transcripts suggests that, like human Tau, zebrafish Tau isoforms with different numbers of microtubule-binding repeats are expressed in the CNS, and larger forms of Tau are expressed in the peripheral nervous system. Interestingly, mapta gives rise to transcripts encoding 4–6 microtubule-binding repeats, whereas maptb is predominantly expressed as a 3-repeat isoform, raising the fascinating possibility that conserved functions of mammalian 3R- and 4R-Tau are distributed between the two zebrafish genes [97].
6. Tools for the generation and analysis of zebrafish Tau models
Generation of transgenic Tau zebrafish requires availability of appropriate methods for introducing exogenous DNA into the germline, and resources to direct MAPT transgene expression in the desired temporal and spatial pattern.
6.1. Techniques for establishment of transgenic lines
Transgenic zebrafish can be generated by micro-injecting linearized plasmid DNA into the cytoplasm of one-cell stage embryos [100], but this gives rise to inefficient genomic integration and significant mosaicism. Two technical advances, mega nucleases and transposons, have substantially improved the efficiency by which foreign DNA can be introduced into the zebrafish genome. (i) Themeganuclease I-sce1 is an intron-encoded endonuclease from Saccharomyces cerevisiae [101], which cleaves DNA at an 18 bp sequence-specific recognition site not found within the zebrafish genome. In mega nuclease-mediated transgenesis, the transgene plasmid is designed so that the expression cassette is flanked by I-sce1 sites, and is injected into zebrafish embryos with I-sce1 enzyme. Integration of a low transgene copy number usually occurs at a single site, with substantially enhanced efficiency, and reduced mosaicism, compared with nakedness injection. This improves the rate of transmission of the transgene from the F0 to the F1 generation [102] and results in simple Mendelian transmission of transgenes in subsequent crosses [103]. (ii) Type 2 transposons are mobile DNA elements encoding a transposase, which mediates excision of the transposon from the genome and its re-insertion at another location. The Tol2 transposon, found in the genome of medaka [104], has been engineered by deletion of the transposase gene to form a no autonomous mobile element, which can insert into the genome in the presence of transposase supplied in trans [105]. In this approach to transgenesis, the transgene expression cassette is flanked by transposon elements, and injected into zebrafish embryos with mRNA encoding transposase, resulting in highly efficient, usually multiple, single-copy integration events. The technique has been used increasingly since its introduction and subsequent refinement [62], because it is necessary to inject and screen a relatively small number of embryos in order to identify stable transgenic lines.
6.2. Promoter resources for generation of neurodegeneration models
Most transgenic zebrafish lines have been constructed by expressing a cDNA under control of a characterized heterologous promoter fragment; cis-acting regulatory regions derived from zebrafish genes are usually employed for this purpose, because they may be associated with more reliable transgene expression [106,107]. For the generation of a Tauopathy model, desirable attributes for the promoter would include pan-neuronal activity and sufficient expression to provoke pathology. Three zebrafish promoter elements that drive transgene expression widely in CNS neurons have been described. (i) A neuronal enhancer derived from the gata2 promoter was expressed at high levels in the developing CNS [108]. (ii) The zebrafish huc gene encodes an RNA binding protein, HuC/D, commonly used as an early neuronal marker [109] — a 2.8 kb fragment of the proximal 5′ flanking region was sufficient to drive pan-neuronal expression in embryos [110]. (iii) The eno2 gene, encoding the neuron-specific γ-enolase isoenzyme, is expressed at low levels until 60–72 h post-fertilization, after which high level expression persists into adulthood. The eno2 regulatory region is complex, containing an untranslated first exon, and an intronic CpG island that appears important for promoter activity. A 12 kb fragment of the promoter, including the first intron, was active in driving reporter gene expression in neurons, including many populations of relevance to disease, throughout the brain and spinal cord from 36–48 h post-fertilization through adulthood [111], and in the retina and visual pathways [103] (Fig. 4). The optimal promoter for construction of Tauopathy models is unclear, and each of these promoters has been deployed in studies of transgenic Tau zebrafish (see below). It is likely that further development of promoter resources and other approaches to generation of transgenic animals will yield other useful reagents for this application.
Fig.4.
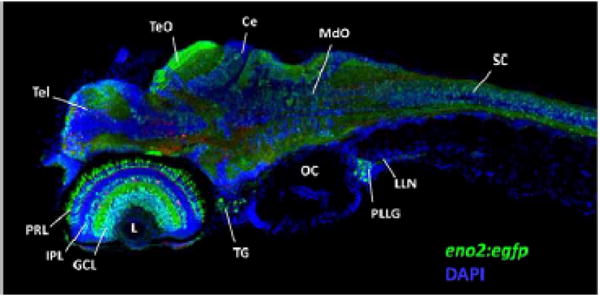
The zebrafish eno2 promoter. The micrograph shows a single confocal plane through the head region of a Tg(eno2:egfp) zebrafish larva at 5 days post-fertilization, illustrating widespread neuronal expression of the eno2 promoter. Rostral is to left, dorsal up. GFP expression appears green; the section was counterstained using a blue nuclear marker to facilitate identification of anatomical landmarks. Key: Tel, telencephalon;. TeO, optic tectum; Ce, cerebellum; MdO, medulla oblongata; SC, spinal cord; L, ocular lens; PRL, photoreceptor layer of retina; IPL inner plexiform layer of retina; GCL ganglion cell layer of retina; TG, trigeminal ganglion; OC, otic capsule; PLLG, posterior lateral line ganglion; LLN, axons of lateral line nerve.
6.3. Special considerations for expression of transgenes that provoke neurodegeneration in larvae
In order to fully exploit the potential of the zebrafish for high throughput screening approaches, it may be necessary to drive Tau expression at sufficiently high levels that disease pathogenesis occurs during larval stages of development, when zebrafish can be accommodated in 96-well plates. However, development of neurodegeneration at this early time point could provide strong selection against establishment of transgenic lines, by compromising reproductive potential or preventing survival to sexual maturity. One possible solution would be deployment of a conditionally-expressing system, for example the binary Gal4–UAS system previously employed in Drosophila genetics. This system has been successfully used in zebrafish, with recent modifications attempting to address toxicity issues caused by the viral trans-activation domain usually fused to Gal4, and inactivation of the UAS tandem repeats [112,113]. Isolation of useful driver lines for generation of neurodegenerative disease models is ongoing, and the optimal strategy for deployment of this technology is uncertain. Driver lines could be constructed by expressing the Gal4-transactivator fusion protein using small promoter fragments similar to the approach for generating cDNA transgenic animals; this has been used successfully with the huC promoter in a larval Tau model [114]. In addition, the highly efficient genomic integration of transgenes using the Tol2 transposon has allowed the deployment of approaches in which random integration of a gal4-expressing enhancer trap gives rise to robust stable expression of Gal4 under the endogenous regulatory elements of the gene of insertion [115]. Several such lines have been constructed and are currently being further evaluated.
6.4. Assays for fully exploiting transgenic models
The power of screening paradigms might be enhanced by deployment of assays that exploit unique properties of the zebrafish model. First, direct visualization of neuronal populations of interest in the zebrafish brain might allow non-invasive automated imaging to ascertain the morphology, integrity or viability of target cells as an assay end point. For example, the Tauopathies PSP and CBD are associated with severe depletion of dopaminergic neurons causing a Parkinsonian movement disorder that characterizes these conditions [116], and it would be of considerable interest to identify compounds that protect dopaminergic neurons from abnormal forms of Tau. Genetic labeling of dopamine neurons using fluorescent reporters is ongoing, and has proved to be complicated. The promoter regions of dopamine-neuron-specific genes are large and complex, such that manageable fragments of the th [117,118] or slc6a3 [119] promoters have not shown dopaminergic neuron-specific activity in vivo. Alternative approaches are being undertaken, including identification of conserved enhancers, using large genomic constructs likely to contain more cis-acting elements, or adopting an enhancer trap approach to identify genomic integrants that recapitulate endogenous gene expression patterns. The latter approach has yielded a single transgenic line in which an integration event in the vmat gene yielded zebrafish with GFP-expressing catecholaminergic neurons [67]. There is a large literature on the development of promoter resources allowing genetic labeling of other specific neuronal populations using fluorescent reporters, which is outside the scope of the present review.
Second, neuronal dysfunction presumably precedes cell death in Tauopathy, and cells in earlier stages of pathological evolution might present a more tractable therapeutic target than those close to demise. It would be of considerable value to develop assays that measure neuronal dysfunction, and which are sensitive to changes in disease progression earlier in the evolution of pathology. Since genetic and chemical screening against a zebrafish Tauopathy model would be carried out in vivo, it might be possible to use automated motor behavioral assays as a surrogate marker of neuronal function. Consequently there is much current interest in establishing reproducible and well-characterized behavioral assays for neurodegenerative phenotypes. A recent report showed that an automated motor behavioral test could be used to identify agents with neuropharmacological properties in a chemical screen [76]. Assays of motor function have been previously deployed in order to detect changes in behavior caused by alterations in neurochemistry and dopamine cell integrity secondary to Parkinson’s disease-associated toxins [81,120]. It may be possible to use a similar approach to investigate changes in behavior preceding demonstrable cell loss in a zebrafish Tauopathy model, in order to develop an assay to identify chemical modifiers of early pathological progression.
7. Current transgenic zebrafish models of Tauopathy
Three recent publications have described the first proof-of-principle experiments showing that expression of human MAPT transgenes can provoke relevant phenotypes in zebrafish. In the first publication, a 4R-Tau-GFP fusion protein was transiently over-expressed in zebrafish larvae, using a gata2 promoter element [121]. In cultured cells, the fusion protein was phosphorylated and associated with the cytoskeleton, similar to native Tau; in vitro studies showed that the fusion protein aggregated, validating its use in constructing an in vivo model of NFT formation. Zebrafish embryos microinjected with the gata2:MAPT-egfp construct, showed mosaic neuronal expression of the fusion protein. Some neurons showed accumulation of fluorescent fibrillar structures resembling neurofibrillary tangles, in the cell body and proximal axon. The human Tau-GFP fusion protein was shown to be phosphorylated in the zebrafish brain by western blot detection of a phospho-S396/S404 epitope, and expression of glycogen synthase kinase-3 in the zebrafish embryo and adult brain was demonstrated by western blot. This initial study validated the use of a GFP fusion protein to monitor evolution of tangle pathology in vivo, and suggested that human Tau is phosphorylated in the larval zebrafish, confirming the prediction that there is sufficient phylogenetic conservation of endogenous zebrafish kinases to modify the human protein. No stable lines were derived in this study.
Subsequently, stable transgenic zebrafish were constructed expressing human 4R/0N Tau [111], the most abundant Tau species deposited in progressive supranuclear palsy [43,44]. The transgene was expressed under transcriptional control of the eno2 promoter [111]. The full phenotype of these transgenic fish has not yet been reported, since the initial paper focused on characterization of the novel eno2 promoter. Abundant expression of human 4R/0N Tau was found in the CNS, persisting into adulthood (Fig. 5A). In the adult brain, refractile Tau accumulations resembling neurofibrillary tangles were found within neuronal cell bodies and proximal axons in CNS regions of pathological relevance to PSP, including the optic tectum (Fig. 5B). Stable expression of the Tau transgene into adulthood in this model will allow analyses designed to test whether age-related pathology accumulates in these transgenic lines, similar to the progressive changes seen in the human disease and recapitulated in some murine models, a potentially important step in validating the model system. Furthermore, widespread neuronal expression of the transgene may allow examination of factors involved in cellular vulnerability to Tauopathy.
Fig. 5.
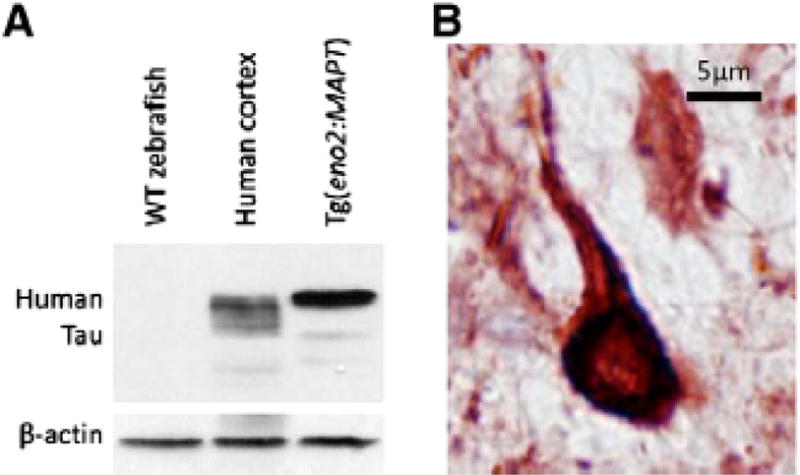
Human Tau expression in Tg(eno2:MAPT) zebrafish neurons. A: A western blot was made with protein lysate from wild-type zebrafish brain (lane 1), control post-mortem human cortex (lane 2) and Tg(eno2:MAPT) brain (lane 3). The blot was probed using an antibody to human Tau (upper panel) followed by an antibody to β-actin (lower panel). Abundant expression of human 4R-Tau is seen in transgenic zebrafish compared with the six isoforms in normal human brain [111]. B: Sections of Tg(eno2:MAPT) zebrafish brain were labeled using the antibody to human Tau used in the western blot experiment shown in panel A and a histochemical reaction yielding a red product. The micrograph shows a brainstem neuron with dense Tau immunoreactivity in the cell body and proximal axon, resembling neurofibrillary tangles shown in Fig. 1B [111].
More recently, exploitation of the Gal4–UAS system allowed generation of a Tauopathy model showing a larval phenotype [114]. This is potentially an important advance, since the development of phenotypic abnormalities at larval stages of development may allow high-throughput screening, as discussed above. Human 4R/2N-Tau, harboring a P301L mutation, was expressed from a novel bidirectional UAS promoter, allowing simultaneous expression of a red fluorescent reporter in Tau-expressing cells [114]. The high levels of mutant P301L Tau expression provoked by the huc:gal4-vp16 driver induced a transient motor phenotype during embryogenesis, likely caused by peripheral motor axonal developmental abnormalities. At later time points, after recovery of motor function, enhanced cell death was observed in the spinal cord of transgenic animals. Abnormal Tau conformers were detected using an antibody, MC1, which recognizes a discontinuous epitope present in Tauopathy tissue [122], as early as 32 h post-fertilization; by 5 weeks of age, argyophilic material was detected in the spinal cord, but the animals were otherwise phenotypically unremarkable at this time point. Subsequent loss of transgene expression prevented the examination of later pathological changes. Tau phosphorylation was detected as early as 32 h post fertilization. Phospho-Tau specific antibodies labeled cells differentially; some phospho-epitopes, such as phospho-T231/S235 and phospho-S396/S404 were detected widely in Tau-expressing neurons, whereas others such as phospho-S422 and phospho-S202/T205 were initially present in only a small subset of Tau-expressing neurons. Between 2 and 7 days post-fertilization, however, expression of all of these epitopes became widespread in Tau-expressing cells, suggesting that Tau had accumulated multiple phosphorylations in a sequential progression of biochemical changes. Tau phosphorylation was reduced by application of novel high-potency GSK3β inhibitors that were designed to target the human enzyme, confirming that extensive homology between human and zebrafish kinases allows small molecules to interact with orthologous targets from either species. This important study was the first detailed phenotypic description of a stable zebrafish Tauopathy model and showed proof of principle that biochemical changes characteristic of human Tauopathy can be recapitulated in larval zebrafish and modulated by chemical inhibitors.
8. Conclusions — what have we learned and what next?
Examination of the first publications allows preliminary assessment of the degree to which transgenic zebrafish models of Tauopathy seem representative of human disorders. Evidence so far suggests that processing of over-expressed human Tau in zebrafish CNS neurons results in similar biochemical and pathological changes to those foundin human Tauopathies. These changes include: aberrant localization of Tau to the somato-dendritic compartment; phosphorylation, with an ordered (and possibly sequential) acquisition of phospho-epitopes; adoption of abnormal conformations associated with Tauopathy; and deposition as argyrophilic material. The availability of zebrafish lines that express Tau in the brain through adulthood will allow more detailed biochemical studies of Tau in the zebrafish CNS to characterize solubility, fibril morphology and further clarify phosphorylation events. However, these data are encouraging that relevant pathways are sufficiently phylogenetically conserved between human and zebrafish to allow recapitulation of cellular processing events relating to Tau in the zebrafish model.
The pathophysiological consequences of human Tau transgene expression in zebrafish CNS neurons are less clear. This is partly because a detailed phenotype has only been reported for a single transgenic Tau zebrafish line [114]. The motor axonal developmental abnormality, motor phenotype and cell death reported in this model have an uncertain relationship to the neuronal dysfunction and neurodegeneration that characterizes human Tauopathy, on account of the onset and subsequent resolution of abnormalities during early development, and the atypical anatomical distribution in the spinal cord and motor neurons. One recurrent finding from the murine transgenic disease model literature is that phenotypes exhibited by cDNA transgenic animals are critically dependent on the promoter elements used to drive transgene expression. The temporal and spatial expression pattern of the huC promoter used in this study directed the atypical pattern of phenotypic abnormalities. However, this may be unimportant for the purposes of drug and target discovery relating to cellular mechanisms of Tau toxicity, provided that the molecular mechanisms underlying neuronal dysfunction and death are conserved between the model and the disorder. Further work, including analysis of other zebrafish Tau lines constructed using different promoter elements, will be necessary to clarify this point.
While detailed phenotypic analysis of the first cDNA transgenic Tau models is in progress, it is interesting to consider future directions in the generation of models and approaches for analysis. Use of models based on over-expression of cDNA encoding human Tau would limit discovery of novel molecular interventions to those targeting Tau modifications, Tau deposition or other abnormal effects exerted by Tau on neuronal function. However, there may be upstreamtargets related to transcription and splicing that would not be represented in cDNA models. For example, changes in the cellular 3R-/4R-Tau ratio, caused by alteration in the regulation of exon 10 splicing, underlie one form of FTDP17 and may contribute to sporadic Tauopathies, where associated haplotypes may alter3R-/4R-Tau ratios [46]. It is not clear whether the human MAPT locus would be appropriately regulated in transgenic Tau zebrafish, allowing expression of physiological levels and patterns of alternatively spliced products. Transgenic zebrafish lines harboring the entire MAPT gene would therefore be of considerable interest. Furthermore, since zebrafish evolution may have resulted in the functions of 3R- and 4R-Tau isoforms becoming distributed between two different genes, an indication of whether alterations in the ratio of these forms can be pathogenic in zebrafish might be accomplished by morpholino knockdown experiments.
Initial evidence shows that the zebrafish will most likely be a predictive platform for the discovery and assessment of Tau kinase inhibitors, although arguably the greatest potential strength of the model might be in discovering new therapeutic targets. In order to gain novel insights from screening approaches, end points that are independent of presumptions about underlying mechanisms, such as neurobehavioral analyses or cell death assays, might be deployed. Furthermore, experimental approaches that can be uniquely applied in zebrafish larvae owing to their optical transparency, for example physiological imaging modalities relating to cellular calcium levels, have the potential to yield important insights from hypothesis driven experiments that cannot be carried out using other available models.
In conclusion, the first reports show that construction of transgenic zebrafish expressing human Tau in CNS neurons is feasible and suggest that the resulting models will be relevant and useful. Consequently, there is cause for cautious optimism that novel zebrafish Tauopathy models may provide important contributions to ongoing efforts to address these common and devastating diseases.
Acknowledgments
We gratefully acknowledge research grant support from CurePSP — The Society for Progressive Supranuclear Palsy (grants #441-05 and #468-08) and NINDS (NS058369).
References
- 1.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Witman GB, Cleveland DW, Weingarten MD, Kirschner MW. Tubulin requires tau for growth onto microtubule initiating sites. Proc Natl Acad Sci U S A. 1976;73:4070–4074. doi: 10.1073/pnas.73.11.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butner KA, Kirschner MW. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–730. doi: 10.1083/jcb.115.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- 6.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 9.Goedert M, Spillantini MG, Crowther RA. Cloning of a big tau microtubule associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci U S A. 1992;89:1983–1987. doi: 10.1073/pnas.89.5.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panda D, Samuel JC, Massie M, Feinstein SC, Wilson L. Differential regulation of microtubule dynamics by three- and four-repeat tau: implications for the onset of neurodegenerative disease. Proc Natl Acad Sci U S A. 2003;100:9548–9553. doi: 10.1073/pnas.1633508100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goode BL, Feinstein SC. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol. 1994;124:769–782. doi: 10.1083/jcb.124.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Heutink P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 15.Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O’Connor M, Trojanowski JQ, Lee VM. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 17.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci. 2004;24:2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 2006;281:9919–9924. doi: 10.1074/jbc.M510127200. [DOI] [PubMed] [Google Scholar]
- 20.Horiguchi T, Uryu K, Giasson BI, Ischiropoulos H, LightFoot R, Bellmann C, Richter-Landsberg C, Lee VM, Trojanowski JQ. Nitration of tau protein is linked to neurodegeneration in tauopathies. Am J Pathol. 2003;163:1021–1031. doi: 10.1016/S0002-9440(10)63462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasegawa M, Morishima-Kawashima M, Takio K, Suzuki M, Titani K, Ihara Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J Biol Chem. 1992;267:17047–17054. [PubMed] [Google Scholar]
- 22.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 24.Gustke N, Steiner B, Mandelkow EM, Biernat J, Meyer HE, Goedert M, Mandelkow E. The Alzheimer-like phosphorylation of tau protein reduces microtubule binding and involves Ser-Pro and Thr-Pro motifs. FEBS Lett. 1992;307:199–205. doi: 10.1016/0014-5793(92)80767-b. [DOI] [PubMed] [Google Scholar]
- 25.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 26.Reynolds CH, Garwood CJ, Wray S, Price C, Kellie S, Perera T, Zvelebil M, Yang A, Sheppard PW, Varndell IM, Hanger DP, Anderton BH. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J Biol Chem. 2008;283:18177–18186. doi: 10.1074/jbc.M709715200. [DOI] [PubMed] [Google Scholar]
- 27.Kenessey A, Yen SH. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993;629:40–46. doi: 10.1016/0006-8993(93)90478-6. [DOI] [PubMed] [Google Scholar]
- 28.Brion JP, Smith C, Couck AM, Gallo JM, Anderton BH. Developmental changes in tau phosphorylation: fetal tau is transiently phosphorylated in a manner similar to paired helical filament-tau characteristic of Alzheimer’s disease. J Neurochem. 1993;61:2071–2080. doi: 10.1111/j.1471-4159.1993.tb07444.x. [DOI] [PubMed] [Google Scholar]
- 29.Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272:25326–25332. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- 30.Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272:19547–19553. doi: 10.1074/jbc.272.31.19547. [DOI] [PubMed] [Google Scholar]
- 31.Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caccamo A, Oddo S, Tran LX, LaFerla FM. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol. 2007;170:1669–1675. doi: 10.2353/ajpath.2007.061178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, Van Leuven F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. 2008;172:786–798. doi: 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- 35.Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 36.Sobue K, Agarwal-Mawal A, Li W, Sun W, Miura Y, Paudel HK. Interaction of neuronal Cdc2-like protein kinase with microtubule-associated protein tau. J Biol Chem. 2000;275:16673–16680. doi: 10.1074/jbc.M000784200. [DOI] [PubMed] [Google Scholar]
- 37.Li T, Hawkes C, Qureshi HY, Kar S, Paudel HK. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry. 2006;45:3134–3145. doi: 10.1021/bi051635j. [DOI] [PubMed] [Google Scholar]
- 38.Sengupta A, Wu Q, Grundke-Iqbal I, Iqbal K, Singh TJ. Potentiation of GSK-3-catalyzed Alzheimer-like phosphorylation of human tau by cdk5. Mol Cell Biochem. 1997;167:99–105. doi: 10.1023/a:1006883924775. [DOI] [PubMed] [Google Scholar]
- 39.Goedert M, Jakes R, Qi Z, Wang JH, Cohen P. Protein phosphatase 2A is the major enzyme in brain that dephosphorylates tau protein phosphorylated by proline-directed protein kinases or cyclic AMP-dependent protein kinase. J Neurochem. 1995;65:2804–2807. doi: 10.1046/j.1471-4159.1995.65062804.x. [DOI] [PubMed] [Google Scholar]
- 40.Sontag E, Nunbhakdi-Craig V, Bloom GS, Mumby MC. A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J Cell Biol. 1995;128:1131–1144. doi: 10.1083/jcb.128.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 42.Sergeant N, David JP, Lefranc D, Vermersch P, Wattez A, Delacourte A. Different distribution of phosphorylated tau protein isoforms in Alzheimer’s and Pick’s diseases. FEBS Lett. 1997;412:578–582. doi: 10.1016/s0014-5793(97)00859-4. [DOI] [PubMed] [Google Scholar]
- 43.Arai T, Ikeda K, Akiyama H, Shikamoto Y, Tsuchiya K, Yagishita S, Beach T, Rogers J, Schwab C, McGeer PL. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol (Berl) 2001;101:167–173. doi: 10.1007/s004010000283. [DOI] [PubMed] [Google Scholar]
- 44.Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem. 1999;72:1243–1249. doi: 10.1046/j.1471-4159.1999.0721243.x. [DOI] [PubMed] [Google Scholar]
- 45.Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- 46.Caffrey TM, Joachim C, Paracchini S, Esiri MM, Wade-Martins R. Haplotypespecific expression of exon 10 at the human MAPT locus. Hum Mol Genet. 2006;15:3529–3537. doi: 10.1093/hmg/ddl429. [DOI] [PubMed] [Google Scholar]
- 47.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Swieten J, Spillantini MG. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17:63–73. doi: 10.1111/j.1750-3639.2007.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 50.Lee VM, Kenyon TK, Trojanowski JQ. Transgenic animal models of tauopathies. Biochim Biophys Acta. 2005;1739:251–259. doi: 10.1016/j.bbadis.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 51.Denk F, Wade-Martins R. Knock-out and transgenic mouse models of tauopathies. Neurobiol Aging. 2009;30:1–13. doi: 10.1016/j.neurobiolaging.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 53.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 56.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 57.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 58.Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov. 2009;8:783–793. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci U S A. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90 – CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawakami K. Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element. Methods Cell Biol. 2004;77:201–222. doi: 10.1016/s0091-679x(04)77011-9. [DOI] [PubMed] [Google Scholar]
- 63.Soroldoni D, Hogan BM, Oates AC. Simple and efficient transgenesis with meganuclease constructs in zebrafish. Methods Mol Biol. 2009;546:117–130. doi: 10.1007/978-1-60327-977-2_8. [DOI] [PubMed] [Google Scholar]
- 64.Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 65.Peters KG, Rao PS, Bell BS, Kindman LA. Green fluorescent fusion proteins: powerful tools for monitoring protein expression in live zebrafish embryos. Dev Biol. 1995;171:252–257. doi: 10.1006/dbio.1995.1276. [DOI] [PubMed] [Google Scholar]
- 66.McLean DL, Fetcho JR. Using imaging and genetics in zebrafish to study developing spinal circuits in vivo. Dev Neurobiol. 2008;68:817–834. doi: 10.1002/dneu.20617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wen L, Wei W, Gu W, Huang P, Ren X, Zhang Z, Zhu Z, Lin S, Zhang B. Visualization of monoaminergic neurons and neurotoxicity of MPTP in live transgenic zebrafish. Dev Biol. 2008;314:84–92. doi: 10.1016/j.ydbio.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 68.Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microgliareveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133:916–927. doi: 10.1016/j.cell.2008.04.037. [DOI] [PubMed] [Google Scholar]
- 69.Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, Zwartkruis F, Abdelilah S, Rangini Z, Belak J, Boggs C. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- 70.Solnica-Krezel L, Schier AF, Driever W. Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics. 1994;136:1401–1420. doi: 10.1093/genetics/136.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Golling G, Amsterdam A, Sun Z, Antonelli M, Maldonado E, Chen W, Burgess S, Haldi M, Artzt K, Farrington S, Lin SY, Nissen RM, Hopkins N. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat Genet. 2002;31:135–140. doi: 10.1038/ng896. [DOI] [PubMed] [Google Scholar]
- 72.Amsterdam A, Burgess S, Golling G, Chen W, Sun Z, Townsend K, Farrington S, Haldi M, Hopkins N. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13:2713–2724. doi: 10.1101/gad.13.20.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov. 2005;4:35–44. doi: 10.1038/nrd1606. [DOI] [PubMed] [Google Scholar]
- 74.Stern HM, Murphey RD, Shepard JL, Amatruda JF, Straub CT, Pfaff KL, Weber G, Tallarico JA, King RW, Zon LI. Small molecules that delay S phase suppress a zebrafish bmyb mutant. Nat Chem Biol. 2005;1:366–370. doi: 10.1038/nchembio749. [DOI] [PubMed] [Google Scholar]
- 75.Molina G, Vogt A, Bakan A, Dai W, Queiroz de Oliveira P, Znosko W, Smithgall TE, Bahar I, Lazo JS, Day BW, Tsang M. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat Chem Biol. 2009;5:680–687. doi: 10.1038/nchembio.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rihel J, Prober DA, Arvanites A, Lam K, Zimmerman S, Jang S, Haggarty SJ, Kokel D, Rubin LL, Peterson RT, Schier AF. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science. 2010;327:348–351. doi: 10.1126/science.1183090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wullimann MF, Rupp B, Reichert H. Neuroanatomy of the Zebrafish Brain. Birkhauser-Verlag; Berlin: 1996. [Google Scholar]
- 78.Mueller T, Wullimann MF. An evolutionary interpretation of teleostean forebrain anatomy. Brain Behav Evol. 2009;74:30–42. doi: 10.1159/000229011. [DOI] [PubMed] [Google Scholar]
- 79.Rink E, Wullimann MF. Connections of the ventral telencephalon (subpallium) in the zebrafish (Danio rerio) Brain Res. 2004;1011:206–220. doi: 10.1016/j.brainres.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 80.Mueller T, Vernier P, Wullimann MF. The adult central nervous cholinergic system of a neurogenetic model animal, the zebrafish Danio rerio. Brain Res. 2004;1011:156–169. doi: 10.1016/j.brainres.2004.02.073. [DOI] [PubMed] [Google Scholar]
- 81.Sallinen V, Torkko V, Sundvik M, Reenilä I, Khrustalyov D, Kaslin J, Panula P. MPTP and MPP+ target specific aminergic cell populations in larval zebrafish. J Neurochem. 2008;108(3):719–731. doi: 10.1111/j.1471-4159.2008.05793.x. [DOI] [PubMed] [Google Scholar]
- 82.Bernardos RL, Raymond PA. GFAP transgenic zebrafish. Gene Expr Patterns. 2006;6:1007–1013. doi: 10.1016/j.modgep.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 83.Tomizawa K, Inoue Y, Nakayasu H. A monoclonal antibody stains radial glia in the adult zebrafish (Danio rerio) CNS. J Neurocytol. 2000;29:119–128. doi: 10.1023/a:1007156529390. [DOI] [PubMed] [Google Scholar]
- 84.Kirby BB, Takada N, Latimer AJ, Shin J, Carney TJ, Kelsh RN, Appel B. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat Neurosci. 2006;9:1506–1511. doi: 10.1038/nn1803. [DOI] [PubMed] [Google Scholar]
- 85.Yoshida M, Macklin WB. Oligodendrocyte development and myelination in GFP-transgenic zebrafish. J Neurosci Res. 2005;81:1–8. doi: 10.1002/jnr.20516. [DOI] [PubMed] [Google Scholar]
- 86.Avila RL, Tevlin BR, Lees JP, Inouye H, Kirschner DA. Myelin structure and composition in zebrafish. Neurochem Res. 2007;32:197–209. doi: 10.1007/s11064-006-9136-5. [DOI] [PubMed] [Google Scholar]
- 87.Schweitzer J, Becker T, Schachner M, Nave KA, Werner H. Evolution of myelin proteolipid proteins: gene duplication in teleosts and expression pattern divergence. Mol Cell Neurosci. 2006;31:161–177. doi: 10.1016/j.mcn.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 88.Jeong JY, Kwon HB, Ahn JC, Kang D, Kwon SH, Park JA, Kim KW. Functional and developmental analysis of the blood – brain barrier in zebrafish. Brain Res Bull. 2008;75:619–628. doi: 10.1016/j.brainresbull.2007.10.043. [DOI] [PubMed] [Google Scholar]
- 89.Bretaud S, Allen C, Ingham PW, Bandmann O. p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. J Neurochem. 2007;100:1626–1635. doi: 10.1111/j.1471-4159.2006.04291.x. [DOI] [PubMed] [Google Scholar]
- 90.Bai Q, Mullett SJ, Garver JA, Hinkle DA, Burton EA. Zebrafish DJ-1 is evolutionarily conserved and expressed in dopaminergic neurons. Brain Res. 2006;1113:33–44. doi: 10.1016/j.brainres.2006.07.057. [DOI] [PubMed] [Google Scholar]
- 91.Anichtchik O, Diekmann H, Fleming A, Roach A, Goldsmith P, Rubinsztein DC. Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J Neurosci. 2008;28:8199–8207. doi: 10.1523/JNEUROSCI.0979-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun Z, Gitler AD. Discovery and characterization of three novel synuclein genes in zebrafish. Dev Dyn. 2008;237:2490–2495. doi: 10.1002/dvdy.21569. [DOI] [PubMed] [Google Scholar]
- 93.Flinn L, Mortiboys H, Volkmann K, Koster RW, Ingham PW, Bandmann O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio) Brain. 2009;132:1613–1623. doi: 10.1093/brain/awp108. [DOI] [PubMed] [Google Scholar]
- 94.Campbell WA, Yang H, Zetterberg H, Baulac S, Sears JA, Liu T, Wong ST, Zhong TP, Xia W. Zebrafish lacking Alzheimer presenilin enhancer 2 (Pen-2) demonstrate excessive p53-dependent apoptosis and neuronal loss. J Neurochem. 2006;96:1423–1440. doi: 10.1111/j.1471-4159.2006.03648.x. [DOI] [PubMed] [Google Scholar]
- 95.Groth C, Nornes S, McCarty R, Tamme R, Lardelli M. Identification of a second presenilin gene in zebrafish with similarity to the human Alzheimer’s disease gene presenilin2. Dev Genes Evol. 2002;212:486–490. doi: 10.1007/s00427-002-0269-5. [DOI] [PubMed] [Google Scholar]
- 96.Leimer U, Lun K, Romig H, Walter J, Grunberg J, Brand M, Haass C. Zebrafish (Danio rerio) presenilin promotes aberrant amyloid beta-peptide production and requires a critical aspartate residue for its function in amyloidogenesis. Biochemistry. 1999;38:13602–13609. doi: 10.1021/bi991453n. [DOI] [PubMed] [Google Scholar]
- 97.Chen M, Martins RN, Lardelli M. Complex splicing and neural expression ofduplicated tau genes in zebrafish embryos. J Alzheimers Dis. 2009;18(2):305–317. doi: 10.3233/JAD-2009-1145. [DOI] [PubMed] [Google Scholar]
- 98.Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, Westerfield M, Ekker M, Postlethwait JH. Zebrafish hox clusters and vertebrate genome evolution. Science. 1998;282:1711–1714. doi: 10.1126/science.282.5394.1711. [DOI] [PubMed] [Google Scholar]
- 99.Chen YC, Priyadarshini M, Panula P. Complementary developmental expression of the two tyrosine hydroxylase transcripts in zebrafish. Histochem Cell Biol. 2009;132(4):375–381. doi: 10.1007/s00418-009-0619-8. [DOI] [PubMed] [Google Scholar]
- 100.Stuart GW, McMurray JV, Westerfield M. Replication, integration and stable germ-line transmission of foreign sequences injected into early zebrafish embryos. Development. 1988;103:403–412. doi: 10.1242/dev.103.2.403. [DOI] [PubMed] [Google Scholar]
- 101.Jacquier A, Dujon B. An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene. Cell. 1985;41:383–394. doi: 10.1016/s0092-8674(85)80011-8. [DOI] [PubMed] [Google Scholar]
- 102.Thermes V, Grabher C, Ristoratore F, Bourrat F, Choulika A, Wittbrodt J, Joly JS. I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech Dev. 2002;118:91–98. doi: 10.1016/s0925-4773(02)00218-6. [DOI] [PubMed] [Google Scholar]
- 103.Bai Q, Wei X, Burton EA. Expression of a 12-kb promoter element derived from the zebrafish enolase-2 gene in the zebrafish visual system. Neurosci Lett. 2009;449:252–257. doi: 10.1016/j.neulet.2008.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koga A, Suzuki M, Inagaki H, Bessho Y, Hori H. Transposable element in fish. Nature. 1996;383:30. doi: 10.1038/383030a0. [DOI] [PubMed] [Google Scholar]
- 105.Kawakami K, Shima A, Kawakami N. Identification of a functional transposase of the Tol2 element, an Ac-like element from the Japanese medaka fish, and its transposition in the zebrafish germ lineage. Proc Natl Acad Sci U S A. 2000;97:11403–11408. doi: 10.1073/pnas.97.21.11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Higashijima S, Okamoto H, Ueno N, Hotta Y, Eguchi G. High-frequency generation of transgenic zebrafish which reliably express GFP in whole muscles or the whole body by using promoters of zebrafish origin. Dev Biol. 1997;192:289–299. doi: 10.1006/dbio.1997.8779. [DOI] [PubMed] [Google Scholar]
- 107.Long Q, Meng A, Wang H, Jessen JR, Farrell MJ, Lin S. GATA-1 expression pattern can be recapitulated in living transgenic zebrafish using GFP reporter gene. Development. 1997;124:4105–4111. doi: 10.1242/dev.124.20.4105. [DOI] [PubMed] [Google Scholar]
- 108.Meng A, Tang H, Ong BA, Farrell MJ, Lin S. Promoter analysis in living zebrafish embryos identifies a cis-acting motif required for neuronal expression of GATA-2. Proc Natl Acad Sci U S A. 1997;94:6267–6272. doi: 10.1073/pnas.94.12.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim CH, Ueshima E, Muraoka O, Tanaka H, Yeo SY, Huh TL, Miki N. Zebrafish elav/HuC homologue as a very early neuronal marker. Neurosci Lett. 1996;216:109–112. doi: 10.1016/0304-3940(96)13021-4. [DOI] [PubMed] [Google Scholar]
- 110.Park HC, Kim CH, Bae YK, Yeo SY, Kim SH, Hong SK, Shin J, Yoo KW, Hibi M, Hirano T, Miki N, Chitnis AB, Huh TL. Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev Biol. 2000;227:279–293. doi: 10.1006/dbio.2000.9898. [DOI] [PubMed] [Google Scholar]
- 111.Bai Q, Garver JA, Hukriede NA, Burton EA. Generation of a transgenic zebrafish model of Tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res. 2007;35:6501–6516. doi: 10.1093/nar/gkm608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Asakawa K, Suster ML, Mizusawa K, Nagayoshi S, Kotani T, Urasaki A, Kishimoto Y, Hibi M, Kawakami K. Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc Natl Acad Sci U S A. 2008;105:1255–1260. doi: 10.1073/pnas.0704963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Distel M, Wullimann MF, Koster RW. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Proc Natl Acad Sci U S A. 2009;106:13365–13370. doi: 10.1073/pnas.0903060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S, Falting J, Distel M, Koster RW, Schmid B, Haass C. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119:1382–1395. doi: 10.1172/JCI37537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scott EK, Mason L, Arrenberg AB, Ziv L, Gosse NJ, Xiao T, Chi NC, Asakawa K, Kawakami K, Baier H. Targeting neural circuitry in zebrafish using GAL4 enhancer trapping. Nat Methods. 2007;4:323–326. doi: 10.1038/nmeth1033. [DOI] [PubMed] [Google Scholar]
- 116.Dickson D. Sporadic tauopathies: Pick’s disease, corticobasal degeneration, progressive supranuclear palsy and argyrophilic grain disease. In: Esiri M, Lee VM, Trojanowski JQ, editors. The Neuropathology of Dementia. CambridgeUniversity Press; Cambridge: 2004. pp. 227–256. [Google Scholar]
- 117.Gao Y, Li P, Li L. Transgenic zebrafish that express tyrosine hydroxylase promoter in inner retinal cells. Dev Dyn. 2005;233:921–929. doi: 10.1002/dvdy.20416. [DOI] [PubMed] [Google Scholar]
- 118.Meng S, Ryu S, Zhao B, Zhang DQ, Driever W, McMahon DG. Targeting retinal dopaminergic neurons in tyrosine hydroxylase-driven green fluorescent protein transgenic zebrafish. Mol Vis. 2008;14:2475–2483. [PMC free article] [PubMed] [Google Scholar]
- 119.Bai Q, Burton EA. Cis-acting elements responsible for dopaminergic neuronspecific expression of zebrafish slc6a3 (dopamine transporter) in vivo are located remote from the transcriptional start site. Neuroscience. 2009;164:1138–1151. doi: 10.1016/j.neuroscience.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 120.Anichtchik OV, Kaslin J, Peitsaro N, Scheinin M, Panula P. Neurochemical and behavioural changes in zebrafish Danio rerio after systemic administration of 6-hydroxydopamine and 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. J Neurochem. 2004;88:443–453. doi: 10.1111/j.1471-4159.2004.02190.x. [DOI] [PubMed] [Google Scholar]
- 121.Tomasiewicz HG, Flaherty DB, Soria JP, Wood JG. Transgenic zebrafish model of neurodegeneration. J Neurosci Res. 2002;70:734–745. doi: 10.1002/jnr.10451. [DOI] [PubMed] [Google Scholar]
- 122.Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 123.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 124.Tellez-Nagel I, Wisniewski HM. Ultrastructure of neurofibrillary tangles in Steele– Richardson–Olszewski syndrome. Arch Neurol. 1973;29:324–327. doi: 10.1001/archneur.1973.00490290064007. [DOI] [PubMed] [Google Scholar]
- 125.Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH, Weidenheim K, Dickson DW. Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol. 1994;145:1496–1508. [PMC free article] [PubMed] [Google Scholar]
- 126.Munoz-Garcia D, Ludwin SK. Classic and generalized variants of Pick’s disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol. 1984;16:467–480. doi: 10.1002/ana.410160408. [DOI] [PubMed] [Google Scholar]
- 127.Gallyas F. Silver staining of Alzheimer’s neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung. 1971;19:1–8. [PubMed] [Google Scholar]