

Abstract
Background:
Chondromyxoid fibroma (CMF) is an extremely rare, benign cartilaginous tumor that makes up <0.5% of all bone tumors, typically presenting in the second or third decade of life. CMF of the sacrum is exceedingly rare, with only seven documented cases reported in the neurosurgical literature.
Case Description:
We report a case of a 35-year-old female with a 3 month history of lower back pain after sustaining a fall on her sacrum/coccyx presenting with a progressive complaint of localized lower back pain, occasional urinary retention without incontinence, gluteal hypesthesia, and pressure below the gluteal crease. Imaging demonstrated a large, expansile enhancing soft-tissue lesion involving the sacrum, distal to the S2-3 disc space. The tumor was removed with partial sacrectomy for open en bloc resection with partial nerve sparing. The patient was found at 1.5-year follow-up with the improvement of symptoms, no recurrence, and no residual neurologic dysfunction.
Conclusion:
Sacral CMF is a rare clinical entity that may mirror more aggressive sacral pathology, including chordoma, in both clinical presentation and imaging characteristics. A review of the available literature regarding diagnosis, surgical management options, and prognosis for sacral CMF is provided.
Keywords: Chondromyxoid fibroma, chondrosarcoma, sacral chordomas, sacrectomy, sacrum
BACKGROUND
Chondromyxoid fibroma (CMF) is an extremely rare, benign cartilaginous tumor that makes up <0.5% of all bone tumors. It is most commonly found in the metaphysis of long bones and usually presents in the second or third decade of life. CMF of the sacrum is unusual and has only been reported 7 times prior with relatively benign presenting symptoms [Table 1]. The authors present a case of sacral CMF with a rather progressive clinical course in a patient presenting with neurologic symptoms occurring 3 months after a fall. Given the progressive course of presenting neurological symptoms and imaging characteristics, more aggressive pathology such as chordoma, giant cell tumor (GCT), or chondrosarcoma was initially suspected.
Table 1.
Symptoms, treatment and follow-up reported/suggested in literature for chondromyxoid fibroma of the sacrum

CASE DESCRIPTION
A 35-year-old Hispanic female presented with a 3 month history of lower back pain after sustaining a fall on her sacrum/coccyx. Her constant, localized lower back pain was accompanied by occasional urinary retention without incontinence. General physical examination demonstrated tenderness to palpation over the posterior elements of the distal lumbo-sacral spine, as well as antalgic gait. Neurologic examination demonstrated gluteal hypesthesia with diminished sensation to light touch and pressure below the gluteal crease in an S2 dermatomal pattern. Although limited by pain, the patient demonstrated, at least, 4+/5 muscle strength in bilateral hip flexion and knee extension. The rectal examination was normal.
Magnetic resonance imaging (MRI) of the sacrum with and without gadolinium demonstrated a large, expansile enhancing soft-tissue lesion involving/replacing the sacrum, distal to the S2-3 disc space, displacing the rectum ventrally. The lesion was T1 hypo and T2 hyperintense and demonstrated heterogeneous enhancement on the postcontrast sequences [Figure 1]. As the imaging was suggestive of locally aggressive pathology such as chordoma, computed tomography (CT)-guided biopsy was discussed but ultimately discouraged due to the inherent potential risk for tumor seeding along the biopsy track.
Figure 1.
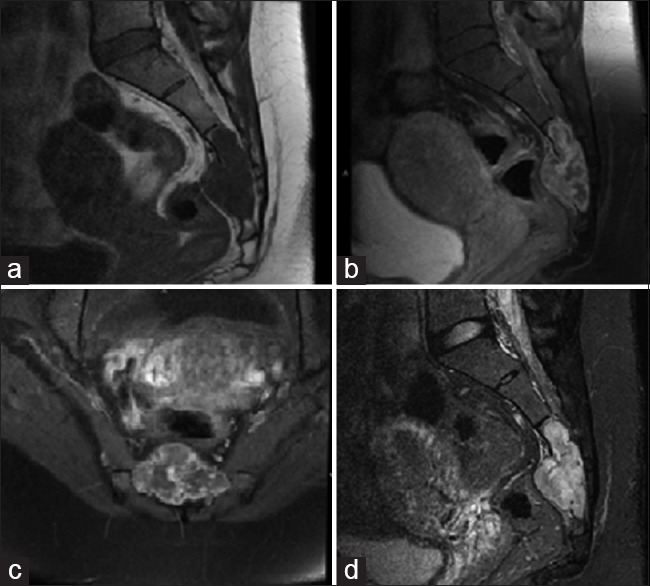
(a) Preoperative sagittal T1 magnetic resonance imaging without gadolinium. (b) Preoperative sagittal T1 magnetic resonance imaging with gadolinium. (c) Preoperative axial T1 magnetic resonance imaging with gadolinium. (d) Preoperative sagittal T2 magnetic resonance imaging
Rather, the patient elected to proceed with partial sacrectomy for en bloc resection of the mass, with possible nerve sparing. As is routine at our institution for large sacral tumors, surgery consisted of a collaborative approach with neurosurgery, colorectal surgery, and plastic surgery, through a standard posterior approach. At surgery, intra-operative findings revealed a largely white, encapsulated mass, observed to replace largely the sacrum/coccyx distal to S2-3. The mass was found to displace ventrally both the sigmoid colon and rectum. The tumor was removed en bloc, and sent to pathology as a permanent section, with great care taken not to violate the tumor capsule. The bilateral S2 nerves and left S3 nerve were spared during surgery, yet the remainder of the lower sacral nerves had to be sacrificed to achieve en bloc resection as they were found to course through the tumor itself. No bowel injury was noted. There were no intraoperative complications. Postoperative MRI/CT confirmed gross total resection [Figure 2].
Figure 2.
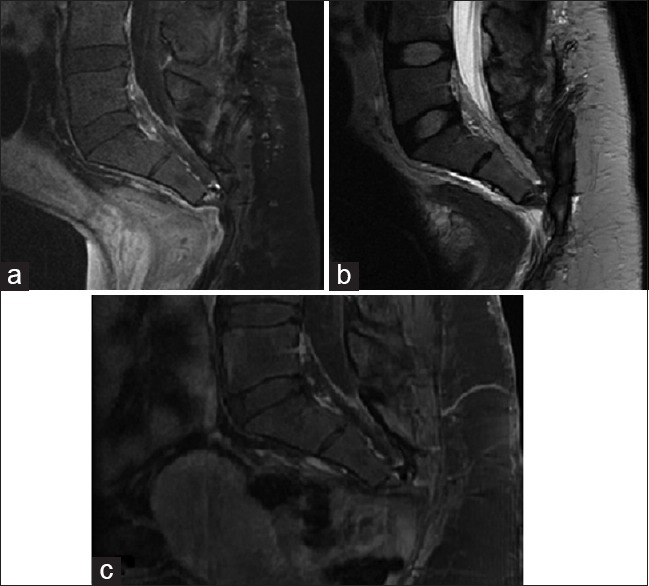
(a) Postoperative sagittal T1 magnetic resonance imaging with gadolinium. (b) Postoperative sagittal T2 magnetic resonance imaging (c) 1.5 year postoperative sagittal T1 magnetic resonance imaging with gadolinium
Pathologic specimens demonstrated a well differentiated chondroid lesion, composed of scattered, relatively bland, stromal cells/chondrocytes [Figure 3]. The dispersed chondroid matrix, was devoid of entrapped bone, as would be expected in chondrosarcoma. Higher power fields demonstrated focal areas of increased cellularity, but no other hallmarks of malignancy, such as atypia, mitoses, or pleomorphism. In addition, pathognomonic physaliferous cells were not present to suggest chordoma. Immunohistochemistry for pancytokeratin was negative, whereas smooth muscle actin was positive. Additional stains for epithelial membrane antigen (EMA), CD34 and desmin were all negative. Ki-67 demonstrated a low proliferative index. Ultimately, the final pathology returned CMF.
Figure 3.
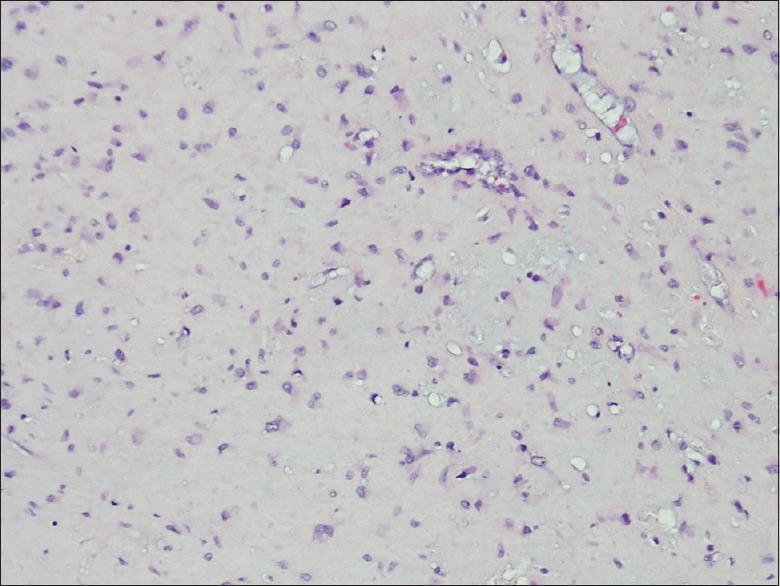
All slides stained with H and E stain. An image of slide with magnification ×400 demonstrate a well differentiated chondroid lesion composed of scattered, relatively bland, stromal cells/chondrocytes. Higher magnification view demonstrate angular and stellate cells set in bluish-pink chondromyxoid stroma. Note that the tumor lacks true hyaline cartilage matrix seen in enchondromas and chondrosarcomas
At discharge, the patient did have partial saddle anesthesia involving the lower sacral dermatomes, but was able to ambulate with assist device, and demonstrated no focal motor weakness. The patient was initially discharged with foley catheter in place and aggressive bowel regimen. She was able to ultimately wean from the foley without significant difficulty. At her recent 2 year follow-up, the patient was found to have recovered fully with regards to bowel and bladder function, ambulating independently, with minimal saddle anesthesia. Follow-up MRI showed no recurrence at 1.5 years postoperative [Figure 2].
DISCUSSION
Primary sacral bone tumors represent only a handful of cases per year in even the busiest of neurosurgical practices. The most common pathologies include chordoma, GCT, and chondrosarcoma, which are all locally aggressive tumors with high rates of local recurrence following surgical debulking/resection. Consequently, intralesional curettage for subtotal resection in an attempt to preserve normal/near-normal bowel and bladder function has largely fallen out of favor. There has been a shift in surgical philosophy/technique toward partial sacrectomy for en bloc resection of aggressive bony sacral pathology, to provide the best chance for long-term disease-free survival, and potentially a cure. Unfortunately, this aggressive surgical approach, while conferring a higher chance for tumor control/cure, also often results in higher neurologic collateral damage, in the instances where involved lower sacral nerves have to be sacrificed to achieve en bloc resection. To this end, the anatomic localization of the lesion is an important determinant of the degree of bowel/bladder dysfunction that can be expected postoperatively. It is generally accepted that lesions centered at, or distal to, the S3-4 disc space have a reasonable chance for the return of near-normal bowel/bladder function, particularly if at least one of the S3 nerve roots can be spared at the time of resection.
CMF of the sacrum, although a rare clinical entity, is an important diagnosis to consider in the differential of a sacral mass, given their similar presentation/imaging characteristics to more aggressive sacral pathology, including chordoma, GCT, and chondrosarcoma. Many of them demonstrate bone destruction, a sclerotic rim, lobulated margins, and septation.[14] Similarly, chordomas often present as well-defined, lobulated masses with internal septation and can also demonstrate lytic and sclerotic bone features on imaging.[7,10,13] Therefore, unless a lesion is seen in the proximal tibial metaphysis, CMF is rarely included in the differential diagnosis.[3]
Zillmer and Dorfman describe CMF as having atypical vertebral lesions on radiographs and show lesions in the vertebral bodies, appearing to extend beyond the confines of the periosteum. One case, in fact, demonstrated a sacral lesion associated with extensive bone destruction and infiltration into the dura. The authors suggested that the usual benign radiographic appearance of CMF may not be entirely applicable to lesions located within the vertebrae.[16] Because of these radiographic similarities, differentiating CMF from chondrosarcoma, chordoma, and GCT on the basis of imaging alone is not possible.[2] While CT-guided biopsy remains an option to establish tissue diagnosis, this has remained somewhat controversial due to the risk of tumor seeding along the biopsy tract in cases of more locally aggressive pathology, such as chordoma. When employed, the biopsy tract should be identified and resected during the more definitive procedure to lower risk of tumor contamination/recurrence.
Histologically, CMF is quite distinct from chordoma. The latter can manifest as three different histological variants: Classic, chondroid, or dedifferentiated. Classic chordomas appear soft, grey-white, lobulated tumors composed of irregular nests or groups of cells separated by a fibrous septa or pseudocapsule. They contain small, round to oval, dark staining nuclei and have numerous elongated sheets of clear cells with vacuolated cytoplasm called physaliferous or “bubble-like” cells. These cells are a pathognomonic feature of chordoma and are largely unmistakable. Mucin protein is also found, both extracellularly and intracellularly throughout the surrounding myxoid stroma. Unlike classic chordoma, chondroid and dedifferentiated subtypes demonstrate atypical features. Chondroid show both chordoma and chondrosarcoma elements while dedifferentiated contain prominent sarcomatous features. All can be pathologically identified by S-100, EMA, and cytokeratin immunoreactivity.[7,10,13] Recently, brachyury, a notochord developmental transcription factor, has also been advanced as a possible future novel biomarker.[13]
CMF is characterized by hypochromic lobules of stellate or spindle-shaped cells which typically stain positive for S-100, Sox 9, and collagen Type II.[12] These cells are surrounded by a myxoid or chondroid background often including multinuclear giant cells.[3,8,11,15,16] Other observed characteristics, although uncommon, can consist of oval nuclei, well-defined hyaline cartilage, calcification, and even bizarre-looking cells showing hyperchromatic pleomorphic nuclei.[15,16] These uncommon nuclear atypia have proven troublesome and have previously led to misdiagnoses of other, more malignant neoplasms including chondrosarcoma. Although this irregularity is rather uncommon, the most important feature distinguishing CMF from other neoplasms is the appearance of the spindle cells embedded in chondromyxoid matrix.[12] Given the predilection for the sacrum and the similarities of chordoma and other neoplasms, it is theoretically possible to obtain a histological sample of a CMF that mimics the cellularity of a sacral chordoma. Therefore, proper preparation of cell block and utilization of immunohistochemistry is paramount in securing the proper diagnosis of CMF.
Given the rather indolent growth pattern of sacral chordomas, early diagnosis is often delayed, commonly resulting in larger, more extensive tumors at the time of diagnosis. This, in turn, results in more extensive surgery, and a higher likelihood of neurologic impairment postresection. Complete en bloc resection with wide surgical margins is the optimal treatment option for sacral chordomas due to the high rate of local recurrence with subtotal resection with or without adjuvant radiation therapy. In addition, complete removal with tumor-free margins is important for prognosis as well as recurrence.[5] Chen et al. compared oncological outcomes of 168 patients from multiple studies with sacral chordoma, reporting local recurrence rates ranging from 19% to as high as 75%, with an average approximately 48%. Many of the cases showed a unique relationship between surgical margins and local recurrence. While adjuvant treatments are still controversial, molecular-targeted therapy may provide some advantages. One drug in particular, imatinib, has been shown to have antitumor activity in chordoma.[4] Postoperative adjuvant proton beam radiation therapy for sacral chordoma is routinely offered in many centers; however, it's overall role in the treatment of chordoma remains somewhat controversial.
Although follow-up and detailed documentation are rather lacking in the treatment of sacral CMF, total en bloc resection or intralesional curettage represent the current accepted surgical management options.
As sacral CMF is a rare entity, there is currently not enough data to support one approach over the other, as long-term remission/cure has been observed in both surgical groups. Radiation therapy has been utilized for tumors demonstrating progression or those deemed surgically inaccessible, without clear benefit.[1] Radiation therapy for the management of CMF is controversial due to its possible role in malignant transformation.[6] Although as rare, the sarcomatous transformation has been reported.[9] The risk of local recurrence for CMF has a broad range in the literature, ranging between 4% and 80%.[8] The longest reported follow-up for sacral CMF in the reviewed literature was a patient treated with curettage and bone grafting, who was found to be recurrence-free at 8 year follow-up.[1]
CONCLUSION
Sacral CMF is a rare clinical entity that may mirror more aggressive sacral pathology, including chordoma, in both clinical presentation and imaging characteristics. As such, CMF must be included in the differential diagnosis of all primary sacral bony lesions. Viable surgical options include en bloc resection, versus intra-lesional curettage. Although reports of long-term remission have been reported with curettage alone, given some reports of local recurrence approaching 80% in other anatomic locations, the option for more aggressive en bloc resection should be entertained. In the context of more distal sacral lesions, where near normal bowel/bladder function may be achieved with selective nerve sparing, aggressive en bloc resection may represent a more viable surgical option. There is no documented outcome benefit of further adjuvant therapy including radiation and/or chemotherapy postresection. While CT-guided biopsy to first establish tissue diagnosis is an option, this remains somewhat controversial, as it is not known if capsular disruption with biopsy may result in tumor seeding as has been described for more aggressive sacral pathology, such as chordoma.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Contributor Information
Tanya Minasian, Email: tminasian@gmail.com.
Chad Claus, Email: cclaus@westernu.edu.
Omid R. Hariri, Email: ohaririucla@gmail.com.
Zhe Piao, Email: zhe.x.piao@kp.org.
Syed A. Quadri, Email: dr.saqader@gmail.com.
Robert Yuhan, Email: robert.m.yuhan@kp.org.
Darren Leong, Email: darren.s.leong@kp.org.
Vartan Tashjian, Email: vartan.s.tashjian@nsmtp.kp.org.
REFERENCES
- 1.Ahuja SK, McCanna SP, Horn EM. Treatment strategy for chondromyxoid fibroma of the sacrum. J Clin Neurosci. 2011;18:1550–2. doi: 10.1016/j.jocn.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 2.Brat HG, Renton P, Sandison A, Cannon S. Chondromyxoid fibroma of the sacrum. Eur Radiol. 1999;9:1800–3. doi: 10.1007/s003300050925. [DOI] [PubMed] [Google Scholar]
- 3.Canale ST, Beaty J. Campbell's Operative Orthopaedics. 12th ed. Philadelphia, PA: Mosby, An Imprint of Elsevier; 2012. [Google Scholar]
- 4.Casali PG, Stacchiotti S, Sangalli C, Olmi P, Gronchi A. Chordoma. Curr Opin Oncol. 2007;19:367–70. doi: 10.1097/CCO.0b013e3281214448. [DOI] [PubMed] [Google Scholar]
- 5.Chen KW, Yang HL, Kandimalla Y, Liu JY, Wang GL. Review of current treatment of sacral chordoma. Orthop Surg. 2009;1:238–44. doi: 10.1111/j.1757-7861.2009.00027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutiérrez-González R, De Reina L, Saab A, Jiménez-Heffernan J, García-Uría J. Chondromyxoid fibroma of the lumbar spine: Case report and literature review. Eur Spine J. 2012;21(Suppl 4):S458–62. doi: 10.1007/s00586-011-2078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCormick M, Schroeder T, Benham S. Sacral chordoma: A case report with radiographic and histologic correlation and a review of the literature. WMJ. 2006;105:53–6. [PubMed] [Google Scholar]
- 8.Mehta S, Szklaruk J, Faria SC, Raymond AK, Whitman GJ. Radiologic-pathologic conferences of the University of Texas M.D. Anderson Cancer Center: Chondromyxoid fibroma of the sacrum and left iliac bone. AJR Am J Roentgenol. 2006;186:467–9. doi: 10.2214/AJR.05.1108. [DOI] [PubMed] [Google Scholar]
- 9.Rodgers WB, Kennedy JG, Zimbler S. Chondromyxoid fibroma of the ala of the sacrum presenting as a cause of lumbar pain in an adolescent. Eur Spine J. 1997;6:351–3. doi: 10.1007/BF01142685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sciubba DM, Cheng JJ, Petteys RJ, Weber KL, Frassica DA, Gokaslan ZL. Chordoma of the sacrum and vertebral bodies. J Am Acad Orthop Surg. 2009;17:708–17. doi: 10.5435/00124635-200911000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Shulman L, Bale P, de Silva M. Sacral chondromyxoid fibroma. Pediatr Radiol. 1985;15:138–40. doi: 10.1007/BF02388724. [DOI] [PubMed] [Google Scholar]
- 12.Sreedharanunni S, Gupta N, Rajwanshi A, Bansal S, Vaiphei K. Fine needle aspiration cytology in two cases of chondromyxoid fibroma of bone and review of literature. Diagn Cytopathol. 2013;41:904–8. doi: 10.1002/dc.22855. [DOI] [PubMed] [Google Scholar]
- 13.Walcott BP, Nahed BV, Mohyeldin A, Coumans JV, Kahle KT, Ferreira MJ. Chordoma: Current concepts, management, and future directions. Lancet Oncol. 2012;13:e69–76. doi: 10.1016/S1470-2045(11)70337-0. [DOI] [PubMed] [Google Scholar]
- 14.Wilson AJ, Kyriakos M, Ackerman LV. Chondromyxoid fibroma: Radiographic appearance in 38 cases and in a review of the literature. Radiology. 1991;179:513–8. doi: 10.1148/radiology.179.2.2014302. [DOI] [PubMed] [Google Scholar]
- 15.Wu CT, Inwards CY, O’Laughlin S, Rock MG, Beabout JW, Unni KK. Chondromyxoid fibroma of bone: A clinicopathologic review of 278 cases. Hum Pathol. 1998;29:438–46. doi: 10.1016/s0046-8177(98)90058-2. [DOI] [PubMed] [Google Scholar]
- 16.Zillmer DA, Dorfman HD. Chondromyxoid fibroma of bone: Thirty-six cases with clinicopathologic correlation. Hum Pathol. 1989;20:952–64. doi: 10.1016/0046-8177(89)90267-0. [DOI] [PubMed] [Google Scholar]