

Abstract
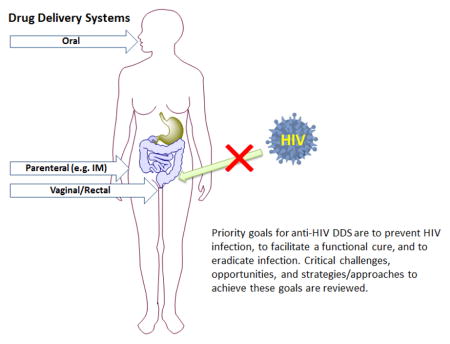
The year 2016 will mark an important milestone - the 35th anniversary of the first reported cases of HIV/AIDS. Antiretroviral Therapy (ART) including Highly Active Antiretroviral Therapy (HAART) drug regimens is widely considered to be one of the greatest achievements in therapeutic drug research having transformed HIV infection into a chronically managed disease. Unfortunately, the lack of widespread preventive measures and the inability to eradicate HIV from infected cells highlight the significant challenges remaining today. Moving forward there are at least three high priority goals for anti-HIV drug delivery (DD) research: (1) to prevent new HIV infections from occurring, (2) to facilitate a functional cure, i.e., when HIV is present but the body controls it without drugs and (3) to eradicate established infection. Pre-exposure Prophylaxis (PrEP) represents a significant step forward in preventing the establishment of chronic HIV infection. However, the ultimate success of PrEP will depend on achieving sustained antiretroviral (ARV) tissue concentrations and will require strict patient adherence to the regimen. While first generation long acting/extended release (LA/ER) DDS currently in development show considerable promise, significant DD treatment and prevention challenges persist. First, there is a critical need to improve cell specificity through targeting in order to selectively achieve efficacious drug concentrations in HIV reservoir sites to control/eradicate HIV as well as mitigate systemic side effects. In addition, approaches for reducing cellular efflux and metabolism of ARV drugs to prolong effective concentrations in target cells need to be developed. Finally, given the current understanding of HIV pathogenesis, next generation anti-HIV DDS need to address selective DD to the gut mucosa and lymph nodes. The current review focuses on the DDS technologies, critical challenges, opportunities, strategies, and approaches by which novel delivery systems will help iterate towards prevention, functional cure and eventually the eradication of HIV infection.
Keywords: HIV/AIDS, antiretroviral drugs (ARV), nanomedicine, nanotechnology, Pre-Exposure Prophylaxis (PrEP), drug delivery targeting, Long-Acting/Extended-Release (LA/ER)
Graphical Abstract
Introduction
The year 2016 will mark an important milestone - the 35th anniversary of the first reported cases of HIV/AIDS. Antiretroviral Therapy (ART) drug regimens are widely considered to be one of the greatest achievements in therapeutic drug research. In fact, ART has transformed healthcare for HIV-infected people from a terminal illness where patients quickly progress from HIV infection to AIDS and serious opportunistic infections to today, where HIV infection is widely regarded as a chronic disease. Unfortunately, the discontinuation of ART or the development of drug resistance results in rapid viral rebound. The lack of widespread successful preventive measures and the inability to eradicate HIV from infected cells highlight the significant healthcare challenges and DD opportunities that remain today.
The World Health Organization (WHO) estimates that approximately 35.0 million people were living with HIV and 2.1 million people became newly infected in 2013 [1]. Due to the global spread of HIV/AIDS it is considered a pandemic with an estimated 39 million deaths from AIDS-related causes, including 1.5 million in 2013. Further, the impact of HIV/AIDS on society and the challenges of curing it are highlighted by two facts. First, the severity of the HIV/AIDS pandemic is comparable to the plague (Yersinia pestis infection, over 75 million deaths) [1]. Second, the only infectious virus ever eradicated in humans is smallpox [2]. Fortunately, unlike past pandemics with high mortality rates, ART enables HIV-infected patients to have a near-normal lifespan and quality of life.
There is still no cure or vaccine available for HIV/AIDS. While ART regimens are considered to be highly successful, significant limitations to current approaches persist. These include the need for chronic administration, patient non-adherence to therapy, which is often exacerbated by side effects of current medications and the continued threat of drug resistance. Treating HIV during the early stages of infection is likely to be more effective than at later stages due to the vulnerability of the virus to drugs [3]. Early treatment of patients may limit the establishment of viral reservoirs and the emergence of resistant viral mutations, while preserving immune responses for controlling infection. Early treatment could move ART to the next level – a functional cure where the body is able to control the disease without drugs, despite the continued presence of the virus [4, 5]. Pre-Exposure Prophylaxis (PrEP), the treatment of non-infected individuals prior to HIV exposure, has become a high priority strategy/regimen that provides early treatment to prevent post-transmission establishment of HIV infection [6, 7]. PrEP was first used successfully in HIV-infected pregnant women undergoing ART thereby protecting the fetus from infection during pregnancy and labor/delivery [8]. Unfortunately, Truvada, the only drug product approved for chronic oral PrEP, suffers from the same limitations as chronic oral ART in terms of posology, adherence, side effects and the risk of resistance if an individual is infected in the intervening time. It is important to note that the goal of HIV eradication has thus far categorically not been met.
New DD systems and strategies are needed to facilitate a functional cure and/or enable HIV eradication. Achieving efficacious drug concentrations in HIV reservoir sites using targeting approaches and reducing cellular efflux and metabolism to prolong effective drug concentrations in target cells remain significant DD challenges. In addition, the urgent need for long acting/extended release (LA/ER) treatments to reduce patient compliance issues and complement those technologies in late stage development has become clear. LA/ER strategies for PrEP have numerous desirable attributes that include infrequent dosing and long dosing intervals making administration convenient for patients; the possibility of directly observed therapy and better long-term adherence; use in difficult to treat populations such as adolescents or those with ongoing substance abuse; use in patients reporting pill fatigue; and protection of patient privacy by eliminating the risk of disclosing pill taking to family and co-workers. The current review focuses on DD technologies, critical challenges, opportunities, strategies, and approaches that will help iterate towards infection prevention, functional cure and eventually the eradication of HIV infection.
Importance of Understanding HIV Pathogenesis to Enable Effective Anti-HIV Drug Delivery
Unlike traditional pathogens that coevolved with mankind for millions of years, HIV is an exotic pathogen that crossed species from African primates to humans in at least five independent events (hence HIV-1 groups M, N, O and P and HIV-2) starting approximately a century ago [9]. Consequently, humans have not had sufficient time to develop an adequate immune response. While broadly neutralizing antibodies eventually develop in 10 to 30% of patients, the overwhelming majority of these antibodies (~ 99%) are not potent enough to stop the progression of disease. In addition, the induction of these antibodies through vaccination has been unsuccessful [10].
HIV is a member of the lentivirus genus in the retroviridae family. The high mutation rate allows HIV to evade destruction by host immune responses. The provirus form of HIV can hide in the genome of latently-infected CD4+ memory T cells for years without revealing any sign of infection to host immune surveillance. It also does not provide an obvious target for eradicative treatment. Natural eradication of any pathogen relies on host immunity. However, HIV is unique among retroviral pathogens because its main target is CD4+ helper T cells, which serve as key coordinators within the immune system. As the CD4+ T cell pool is progressively depleted, host immune function becomes weaker, making eradication increasingly difficult. Compounding the reduction in CD4+ T-cells is the establishment of cellular HIV reservoirs, defined in the context of eradication as a cell type or anatomical site that allows persistence of replication-competent HIV-1 on a timescale of years in patients on optimal ART [11–13]. Anatomically, HIV reservoirs are mainly located in the mucosa of the alimentary, respiratory and genital tracts, the brain/central nervous system (CNS), and some lymph nodes (LNs) [12, 14]. Cellular reservoirs include latently infected CD4+ memory T cells but may also include other infected cells such as hematopoietic stem cells and macrophages [11]. Some reservoir sites are also considered viral sanctuary sites where drug concentrations are suboptimal due to biological barriers (e.g., blood-brain barrier) that protect the anatomical sites (e.g., brain/CNS). This allows HIV to replicate unhindered [14].
HIV establishes persistent infection by two mechanisms: reservoir formation and ongoing replication. The gut mucosa plays a pivotal role in HIV pathogenesis. The gut mucosa harbors approximately 60% of the body’s immune cells, including the majority of CD4+ T cells. Many of the CD4+ T cells are activated due to the bacteria present in the gut lumen, enabling viral replication. Furthermore, these CD4+ T cells express the HIV co-receptor CCR5, which is needed for HIV entry [15, 16]. Accordingly, at all stages of HIV infection, viral replication and CD4+ T cell depletion predominantly occurs in the gut mucosa, even under apparently successful ART [17, 18]. Following CD4+ T cell depletion, bacterial products breach the gut epithelial barrier. This leads to further local and systemic CD4+ T cell activation, and the acceleration of viral replication and immune cell depletion [19]. Very recently, whole-body immunoPET scans of simian immunodeficiency virus (SIV)-infected macaques confirmed that persistent viral replication occurs in strategic mucosal sites, such as the gut, irrespective of ART [20]. Thus, ART cannot suppress low level, persistent viral replication in mucosal sites, making them high priority targets for DD.
Targeting HIV mucosal transmission earlier is likely to be more effective than treating at later stages of infection [3]. CD4+ T-cells are scattered throughout the colorectal and vaginal submucosa in small numbers. Innate immunity suppresses initial infection, resulting in a small founder population of infected cells that is highly vulnerable to ART [21]. During the first few days after transmission, there is a dynamic balance between the shrinkage of the infected founder cells resulting from viral and host innate immunity killing and expansion of these cells by spreading of infection to uninfected cells. The basic reproductive rate of the founder cells is the ratio of expansion to shrinkage and when the ratio falls below 1 the founder population of the infected cells shrinks, resulting in aborted infection. Once locally expanded HIV spreads to the draining LNs, systemic infection is established. The viral vulnerability creates prevention opportunities since it would not be necessary to completely inactivate all HIV or kill all infected cells. Rather, reducing the founder population to below the critical threshold value may suffice. This window of opportunity lasts about one week from the time of vaginal exposure to the establishment of HIV infection whereas after colorectal exposure, it may be significantly shorter (Fig. 1) [3]. Although this estimate is based on data from non-human primates and vaginal SIV challenge, the mechanism provides a logical basis and rationale for the concept of a window for establishing HIV infection in humans, post exposure to HIV, and an opportunity for PrEP DDS to deliver ART and prevent the establishment of HIV infection.
Fig 1. Approximate timeline for establishing HIV infection after vaginal exposure to HIV[3].

This timeline suggests a window of opportunity for PrEP therapy. Given the difficult logistics of diagnosing and initiating treatment immediately post transmission, PrEP delivers effective drug concentrations for at risk populations prior to exposure (adapted from [3]).
In a critical proof-of-concept study, Li et al [22] demonstrated that inhibition of the recruitment of CD4+ T cells alone was sufficient to prevent vaginal transmission in a SIV–macaque model. They found that mucosal inflammatory signaling in conjunction with the innate immune responses to infection greatly fueled CD4+ T cell recruitment. The signaling involves macrophage inflammatory protein-3α cytokine, plasmacytoid dendritic cells (pDC) and pDC-produced CCR51 chemokine. The surfactant glycerol monolaurate is an antimicrobial compound that has no effect on the SIV life cycle per se but is known to inhibit immune activation and chemokine/cytokine production in vitro. Topical application of glycerol monolaurate to the vaginal mucosa protected rhesus macaques from acute infection despite repeated intra-vaginal exposure to high doses of SIV. The data thus suggest that limiting local expansion to below a critical threshold alone can be effective in preventing viral infection. The concept of early treatment for prevention has also been supported in other studies using non-human primate models. For example, agents that block viral binding, co-receptor-mediated entry and reverse transcription have been shown to protect against SHIV and SIV vaginal and rectal challenges in the rhesus macaque model [3]. The implications for DD are clear: (1) constant, therapeutically effective drug concentrations need to be maintained in the vaginal or colorectal mucosa of high risk individuals prior to viral exposure, (2) methods to inactivate HIV prior to mucosal entry may have value, and (3) approaches to control the number and rate of responding immune cells to the colorectal and vaginal mucosa after HIV exposure are warranted.
Drugs for Prevention and Treatment of HIV Infection
There are currently 37 Food and Drug Administration (FDA)-approved ARV drug products used clinically in the US for the treatment of HIV/AIDS [23]. Although a myriad of highly potent drugs have been approved and widely used, the HIV drug pipeline still shows robust growth in volume as well as in molecular target diversity [24]. Existing ARVs can be classified by the site of action (i.e., extracellularly in the vaginal/colorectal lumen or mucosa or intracellularly in HIV-susceptible immune cells). Extracellular acting ARVs, which interfere directly with the entry process, target either the HIV co-receptor (CCR5) [25, 26] on host cells or gp41, the viral transmembrane glycoprotein on HIV. The vast majority of ARVs act inside host cells by preventing viral proliferation. Four classes of intracellular acting ARVs have been introduced including Nucleoside/nucleotide Reverse Transcriptase Inhibitors (NRTI), Non-nucleoside Reverse Transcriptase Inhibitors (NNRTI), Protease Inhibitors (PI), and HIV Integrase Strand Transfer Inhibitors (InSTI). The vast majority of ARVs have solubility, stability and/or permeability limitations that ultimately lead to a high degree of pharmacokinetic (PK) variability in humans [27–31]. National Institute of Allergy and Infectious Diseases (NIAID) -supported researchers established ART regimens, a combination of at least two drugs, in order to reduce the incidence and rate of viral resistance. These treatment regimens are multi-class drug combination products. Truvada, an orally administered fixed combination drug product containing tenofovir disoproxil fumarate (TDF) and emtricitabine, represents the first potent combination drug product approved for preventing HIV infection.
Intensive ARV research has resulted in drugs with high potency and the discovery of new mechanisms of action (Table 1). Besides introducing newer generations of NRT, NNRT InSTI and protease inhibitors, novel mechanisms of action are being explored, e.g. therapeutic reclamation of apoptotic proficiency-based (TRAP) are moving to the clinical trials stage. As of today, 59 HIV PrEP clinical trials are listed in the AVAC database (avac.org). However, most trials involve Truvada. Even though new potent ARVs are needed, it appears that a paradigm shift from drug discovery to drug delivery is occurring in the field of HIV therapy.
Table 1.
Recent drugs in use or development (MOA: mechanism of action; TRAP: therapeutic reclamation of apoptotic proficiency).
| Name | MOA | Status | Source (Reference, trial number) |
|---|---|---|---|
| Tenofovir disoproxil fumarate (TDF) | RT inhibition | Approved 2012, Truvada | [32] |
| Emtricitabine, FTC | RT inhibition | ||
| Tenofovir alafenamide (TAF, GS-7340) | RT inhibition | Investigational prodrug of TFV | [33] |
| Rilpivirine (TMC-278) | NNRT inhibition | Recruiting | [34] |
| Cabotegravir (GSK 744) (S/GSK1265744) | InST inhibition | Not yet recruiting | [35, 36] |
| Maraviroc | CCR5 antagonism | Ongoing trial | NCT01505114 [37] |
| Ciclopirox | TRAP | Recruiting | [38, 39] |
| Deferiprone |
Drug Delivery Systems for HIV Prevention
Currently, there are few options for preventing HIV infection. PrEP DDS in development are administered either locally through rectal or vaginal administration, or systemically by means of oral or parenteral administration, providing therapeutically relevant drug concentrations in vaginal and colorectal tissues [7, 40–45]. By any route of administration, the key requirements are the delivery and maintenance of effective ARV concentrations at vulnerable mucosal sites during periods of HIV exposure since HIV transmission through the colorectal and vaginal mucosae is responsible for the vast majority of new HIV infections [46, 47].
In 1995, Tsai et al. [48] published a seminal paper demonstrating that subcutaneous administration of TFV could prevent SIV transmission in macaques. Following this report, the use of vaginally- and orally-administered TFV and TDF, the orally bioavailable prodrug of TFV [49], to prevent HIV infection in high-risk populations became the focus of many investigations [42]. To date, numerous clinical trials have been conducted investigating TDF and other ARVs as potential oral PrEP agents. Of these studies, one treatment regimen was successful, leading to the only FDA-approved and licensed treatment for HIV prevention. This product, Truvada, consists of two drugs, TDF and emtricitabine (FTC) in a once a-day oral dose form [42].
One early investigation involving Truvada was the Partner’s PrEP study where it was found to reduce HIV acquisition with a 75% rate of effectiveness. However, in the FEM-PrEP and VOICE (Vaginal and Oral Interventions to Control the Epidemic) clinical trials that followed, it was not found to be effective. A lack of patient compliance was determined to be one of the primary factors in the failure of these studies. Similar results were seen in other clinical trial including the iPrex study in which Truvada reduced HIV transmission by only 42% overall [42, 50]. It was determined that only 18% of patients were taking the drug at the prescribed daily regimen [42]. When the analysis was narrowed to only patients with high adherence to the regimen (i.e., where patients steady-state plasma TFV concentrations > 40 ng/mL), the therapies were found to be 88% and 91% effective for the groups that received TDF and TDF/FTC, respectively [51]. Therefore, while a number of trials have reported that systemic oral PrEP is only 39% to 75% effective, subsequent correction of the data for medication regimen adherence, as indicated by measureable drug blood levels, showed that effectiveness was greater than 90% in patients that took their medication consistently [40, 41, 43, 52, 53]. This and other pharmacokinetic (PK) studies show a clear relationship between PrEP efficacy and patient adherence as well [42, 52]. For rectal and vaginal PrEP formulations there is also a relationship between the size, texture and color of a formulation, perceptibility (user sensory perceptions including leakiness/messiness) and patient adherence [54, 55].
This body of work highlights the two major challenges influencing patient adherence to PrEP regimens: the chronic nature of administration/frequency of use (oral, rectal and vaginal) and perceptibility (rectal and vaginal). Chronic oral PrEP also raises concerns of toxicity and the potential for ARV resistance due to widespread systemic exposure. Finally, additional studies have also revealed suboptimal drug concentrations in the target mucosal tissues even though relatively high drug concentrations are found in the blood [56]. Alternatively, microbicides are applied topically to the vagina or rectum and act locally to prevent HIV transmission. Therefore, the development of DD strategies and DDS for colorectal and vaginal mucosal PrEP that maximize local tissue drug concentrations and minimize systemic exposure are a high priority.
Microbicides have been extensively investigated as potential preventative options in numerous DDS/dose forms such as gels, creams, films, foams, quick-dissolving tablets and intravaginal rings (IVR) [41, 42, 44, 57–59]. Highlights of significant microbicide/local PrEP DDS are described below.
Vaginal Gels
Microbicides were first explored as a means of offering broad protection against most or all sexually transmitted infections in the early 1990’s. The first generation of microbicides, were membrane solubilizing surfactants including the nonionic spermicide nonoxynol-9 (N-9), an OTC vaginal contraceptive known to destroy the membrane of cells [7, 40, 44]. Unfortunately, N-9 failed at the clinical trial stage where it was found to increase HIV transmission due to the development of inflammatory genital tract lesions in the epithelial layer of the vagina. Additionally, the product did not adequately cover the vaginal mucosa evenly, pointing to major formulation challenges for the future for vaginal PrEP products [41, 43, 52]. Since the publication of these reports, N-9 and similar surfactants have been excluded, with few exceptions, from the developmental pipeline [41].
Second generation microbicides incorporated polymers into the application design. Early formulations of this period were aimed at reducing the toxicity of nonoxynol-9. Polymers were used to minimize mucosal irritation and prevent environmental changes in the vagina that may increase risk of infection. However, sufficient efficacy was not achieved. Thus with the widely accepted dismissal of surfactants, efforts focused on polyanionic compounds. This group of polymers was specifically chosen because of their ability to act as entry inhibitors to prevent HIV from entering target immune cells in the vagina [7]. Several polyanionic polymer-based microbicides advanced to clinical trials including Carraguard, Cellulose Sulfate and PRO2000. Unfortunately, all were proven ineffective [60].
Third generation topical PrEP focused on microbicides using intracellularly acting ARVs to prevent HIV infection [42]. Karim et al. [61] presented the results of a study (CAPRISA 004), which was carried out to assess the safety and effectiveness of a 1% TFV gel for HIV prevention. They demonstrated that the gel, if applied both before and after sex, reduced HIV incidence by 39% overall and by greater than 54% for those who used the gel consistently [57]. TFV was the first ARV found to be effective as a microbicide. However, although it was found to be safe and effective in preventing HIV infection, patient adherence was once again an obvious concern with these treatments [61].
Haaland et al. [62] reported another clinical trial studying an ARV microbicide candidate, UC781. UC781 is a NNRTI with poor oral bioavailability. Although it was unsuccessful as a potential oral agent, it was believed to be a promising choice as a topically applied microbicide. A carbopol gel formulation of 0.1% UC781 was found to be effective in vitro and in vivo in a macaque model. When evaluated in a phase III clinical trial, the study was quickly terminated due to the insolubility and instability of UC781; similar limitations were reported after oral administration. This study showed the importance of evaluating microbicide effectiveness after exposure to the human female genital tract and to semen. It is possible that semen may inhibit the antiviral activity of UC781 in vivo albeit this interaction with semen plasma was investigated in vitro during preclinical studies and found to have no significant effect.
Intravaginal Rings (IVR)
In order to overcome patient adherence issues, IVRs have been proposed to deliver LA/ER ART [42, 59]. IVRs are typically formed from elastically deformable polymers such as thermostat silicones, poly(ethylene-co-vinyl acetate), or polyurethanes. The drug is usually mixed or dissolved in the polymer matrix during the formulation process and incorporated into the injection molding or hot-melt extrusion. Once the DDS is exposed to the vaginal lumen, a concentration gradient is initiated, allowing for the surface drug to diffuse into the contacting tissue. The rate of drug release depends on numerous factors such as drug solubility, partition coefficient, and diffusion coefficient of drug in IVR polymer and of drug in vaginal fluid.
Nel, et al. [63] reported a clinical trial investigating the safety and PK of an IVR that delivered the NNRTI dapivirine. Twenty-four women were treated with dapivirine (25 mg) silicone elastomer matrix IVR, dapivirine (25 mg) silicone elastomer reservoir IVR, or a silicone elastomer placebo IVR. IVRs were used for 28 consecutive days and plasma and vaginal fluid samples were collected on day 1 and day 28 of the trial. The matrix IVR and reservoir IVR were able to achieve significant drug levels in vaginal fluid with maximum drug levels of 6 mM and 42 μM respectively. This data is encouraging because the reported vaginal fluid and mucosal tissue drug levels surrounding the IVR location were more than 1000-times the in vitro 50% effective concentration (EC50) against the wild-type HIV-1. This suggests that the IVR investigated may be able to achieve sufficient drug concentrations within vaginal mucosal tissue to prevent HIV infection [59, 63].
Several other studies have been conducted investigating IVRs incorporating dapirivine [64–66]. Nel et al. presented results of a clinical trial for the Dapivirine Vaginal Ring-004 (25 mg dapivirine) at the 22nd Conference on Retroviruses and opportunistic infections. Patients wore the IVR for periods between 4 to 12 weeks at a time. For patients that wore the IVR for 12 weeks consistently, mean vaginal fluid concentrations were found to be more than 4000-times the in vitro IC99 in cervical tissues at the end of the study. This ring is currently being evaluated for safety and efficacy in phase III clinical trials (International Partnership for Microbicides website: last accessed May 30, 2015).
Recently, a 90-day TFV reservoir ring inspired by the success of the 1% TFV gel was investigated [67]. They hypothesized that the IVR would be able to provide a more controlled and sustained vaginal drug concentrations in the cervicovaginal area. Polyurethane tubings of various hydrophobicities were filled with high densities semisolid paste of TFV, glycerol and water. In vitro, a more rapid release of TFV was found with increasing polyurethane hydrophilicity allowing for systematic control of drug release. IVRs were evaluated in two 90-day in vivo sheep studies where pharmacokinetics and safety of TFV was evaluated. The polyurethane IVR was found to be capable of delivering 10 to 30 mg of TFV daily for up to 90 days consistently. Previous matrix IVRs typically had high fractions of undissolved drug that was never released from the ring. They also commonly showed a decrease in drug release rates with time as well as a decrease in material stiffness as a result of drug release. This study is significant because it highlights a tunable membrane that can be controlled to achieve and maintain a desired TFV loading, release rate and ring stiffness by altering material properties. The polyurethane TFV IVR also showed no significant toxicological effects. However, in comparison to the control, there was slight to moderate increase in inflammatory infiltration within vaginal epithelium. Although clinical studies are needed to confirm the safety and potential efficacy of this therapy, it shows promising results for the future of IVR for HIV PrEP [67].
Patient adherence to PrEP regimens remain a concern and developing better ways of determining adherence are needed. Boyd et al. [68] describes the development of a technology to address the pivotal issue of inaccurate self-reporting of patient adherence during clinical trials. They report a temperature-monitoring microbicide-releasing vaginal ring that uses DST nano-T temperature loggers to monitor user adherence. While still in the pre-clinical stages, the IVR was tested in macaques where it showed high sensitivity to fluctuations in vaginal temperatures and the ability to detect periods of IVR removal with accuracy. This introduces the concept of multipurpose IVR to treat patients as well as monitor their use and significant events that may affect efficacy such as sexual arousal, vaginal intercourse, and menstrual cycle). Such technologies would markedly enhance the accuracy of product evaluation during clinical testing.
Vaginal Films
Another type of mucosal PrEP DDS being explored is the use of vaginal films for local mucosal PrEP [66, 67, 69, 70]. Polymeric vaginal films are a solid dosage form that offers several additional advantages over other microbicide dose forms such as gels and creams. Some of these advantages are (1) accuracy of dose administration in the absence of an applicator; (2) rapid drug release upon exposure to vaginal fluid; (3) discreet use and minimal product volume; (4) improved drug stability; (5) convenient storage; and (6) minimal leakage. In addition, when compared to soft-gel capsules and tablets (not discussed in this review), an acceptability study reported that greater than 80% of participants surveyed, found the films to be acceptable forms of treatment. 77% to 91% of women in the study also stated that they would definitely use any of these products if they were confirmed to be effective at preventing HIV transmission [6, 69, 70].
Until recently, most published studies proved the feasibility of formulating polymeric vaginal films containing a single anti-HIV drug candidate. For example, a vaginal film reported by Akil et al. [71] comprised of only dapivirine was shown to be effective in blocking HIV-1 both in vitro using P4/R5 and MT-2 cells as models of HIV infection. The film was also shown to be effective ex vivo in polarized and cervical explant models. Recently, research has given increased focus on developing films capable of co-delivering multiple drugs [70, 72]. Another paper by Akil et al. [70] describes the utilization of vaginal films to deliver different combinations of dapivirine, maraviroc, and TFV. Films were manufactured by a solvent casting method and solid phase solubility was used to select appropriate polymers to be used in the formulation. Cellulose polymers and polyvinyl alcohol were used in the development of three combinational film products. These vaginal films were determined to be stable for up to 12 months at ambient temperatures with rapid drug release reported (>50% of each drug) within the first 30 minutes in vitro. The results from this study show the potential of vaginal films to incorporate multiple antiretroviral agents, which is of great importance considering the prevalence in use of ART for both prevention and eradication. The vaginal films were also well tolerated by both the women and their partners, which makes them promising candidates for microbicide therapies.
A comprehensive summary of clinical trials for topical vaginal microbicides can be found in a recent paper by Antimisiaris et al. [73]. To date, there have not been any products approved for use in humans. This demonstrates a great need for new technologies and innovative ideas moving forward. Recently, there has been an increased interest in LA/ER ARV drug combinations for microbicides to enhance efficacy and user adherence. Nanotechnology-based drug delivery systems are a promising option to advance the field of microbicides by their ability to (1) facilitate drug/virus interactions, (2) increase synergy for drug cocktails (3) penetrate mucosa tissue, (4) provide sustained and triggered drug release, (5) target HIV susceptible cells, (6) improve drug solubility and permeability, and (7) serve as a drug barrier along the epithelial cell lining [74–76]. Recent review articles published earlier this year discuss the benefits and potential drawbacks of exploring nanotechnology-based solutions to improve the efficacy of vaginal microbicides [73, 77].
Colorectal Microbicides
Until recently, most efforts at microbicide development and initiated clinical trials focused on vaginal microbicides despite the major role colorectal transmission plays in the establishment of new infections. Men and women who practice unprotected receptive anal intercourse (RAI) are at a much higher risk of contracting HIV from an infected partner than those who engage in unprotected vaginal sex. Unprotected RAI is estimated to result in 10–100 times more incidences per exposure than unprotected vaginal intercourse [78]. A major cause of this is the structure of the colorectal mucosa (shown in Fig 2) [79]. The colorectal epithelial barrier is a thin single-cell columnar layer above the lamina propria that is much easier to penetrate than the multi-layer barrier found within the vagina. Once HIV traverses the epithelial layer and enters the lamina propria, it has direct access to an extensive population of activated resident CD4+ T cells, macrophages, dendritic cells, and other lymphocytes that are highly susceptible to HIV. There are also individual LNs that serve as a primary route for HIV to enter the systemic circulation [56]. Due to distinct anatomical differences between the vagina and colorectum, microbicide technologies developed specifically for one route of administration cannot be incautiously translated to an application for the other.
Fig 2. Colorectal Mucosal Barriers and HIV Transmission Pathway.

A single epithelial layer provides a barrier between the intestinal lumen and the lamina propria. The cells most susceptible to/or responsible for propagating early HIV infection, CD4+ T-cells, macrophages, dendritic cells, Langerhans cells and M-cells are located in the Lamina Propria. The lymphatics play an important role in the dissemination of HIV into the systemic circulation. (Image adapted from [79] with permission)
Since colorectal microbicides were recently reviewed in this journal, a summary of the formulation factors, DDS issues and challenges will be presented and the reader is referred elsewhere for a more general discussion of broader issues [56]. Colorectal microbicide dose forms commonly include gels, enemas and suppositories. The use of these traditional dose forms present numerous delivery challenges to PrEP treatment and this is an area that would greatly benefit from the development of new DDS technologies and techniques. The challenges can be generally categorized as follows: (1) administration methods as it relates to mucosal coverage, (2) patient acceptability and adherence as it relates to volume administered and retention, (3) coital dependence and persistence of therapeutic effect, and (4) chronic safety/toxicity of the DDS/formulation.
Administration methods as it relates to mucosal coverage
Complete coverage of the colorectal musoca is important for providing protection from HIV transmission. It is suggested that a colorectal microbicide should cover the entire rectum, sigmoid colon and descending colon to up to the splenic flexure to provide sufficient protection. Careful consideration must be taken to choose the ideal delivery vehicle and applicator to ensure the spread of therapeutic agent to all areas that may become exposed to the virus [80–82].
Patient acceptability and adherence as it relates to volume administered and retention
While there is no standard volume of product that is used for colorectal treatments, the volume should be sufficient enough to cover the entire area that may become exposed to HIV during intercourse. One study [83] reported that participants tolerated up to 35 mL of a rectal microbicide gel, and once this volume was exceeded, there is increased anal leakage and discomfort. The smallest effective volume for treatment is preferred [56, 83]. As has been proven many times, patient acceptability will have a major influence on efficacy [54, 56].
Coital dependence and persistence of therapeutic effect
Coital dependence of an application (requiring dosing close to the time of sexual intercourse) may be a disadvantage when it relates to adherence. It requires that the users anticipate when they will have sex and be able to privately apply the treatment in advance. If the therapy is only active for a few hours, this may place a limitation on the timespan in which a person has to use the application. Ideally, a LA/ER PrEP approach with sustained drug activity would allow for more flexibility in application and increase adherence and efficacy [44].
Chronic safety/toxicity of the DDS/formulation
Microbicide formulations need to preserve the activity and stability of drugs while also ensuring that the excipient concentrations used pose no significant toxicity. Product acceptability is strongly dependent on the safety and comfort so it is important that products are nontoxic especially for chronic treatment.
In parallel to these four primary challenges, the physicochemical properties of the delivery vehicle play a key role in addressing these overcoming these challenges and increasing overall therapeutic efficacy. The ideal delivery vehicle will differ depending on the route of administration and vehicle type. In order to achieve sufficient levels of coverage, retention, safety, drug release and therapeutic efficacy, many properties of the formulation have to be optimized to compliment the biological environment that is being treated. Important properties to consider include pH, viscosity, osmolality, mucoadhesivity, yield stress and shear rate [56, 84, 85].
Although it has not always been a main focus of PrEP research, interest in colorectal PrEP has existed since the development of the first generation of microbicides. Nonionic surfactant N-9 was one of the first microbicides tested for colorectal application. Similarly to the results for vaginal administration, N-9 was found to cause significant epithelial sloughing and a potential increase in the risk of infection [56, 86]. Since then, five ARV phase 1 clinical trials have been completed for rectal microbicides. Most of these trials have also focused on TFV gel formulations and one study investigated the UC781 gel that was also explored as a vaginal application [42]. Anton, et al. [86] described the first phase 1 double-blind, placebo-controlled randomized clinical trial for UC781 gel as a rectal microbicide. Men and women were treated rectally with two concentrations of UC781 (0.1% and 0.25%) in gels formulated with Carbomer 974P, methylcellulose, glycerin, purified water and paraben preservatives. This class of gels is similar to thickened solutions with a limited ability to spread evenly or be retained on the mucosa. Both dosages were found to be safe with no epithelial sloughing from rectal lavage and no changes in histology.
Another gel investigated as a rectal microbicide was the vaginal formula of the 1% TFV gel used in the CAPRISA 004. This gel was evaluated in a phase 1 rectal study [RMP-02/MTN-006] where it was shown to induce mild to moderate gastrointestinal side effects such as pain, bloating and diarrhea. These symptoms were believed to be caused by the osmolality of the vaginal formulation (3111 mOsmol/kg) [87, 88]. Following this study, a second gel was developed to address the osmolality of the 1% TFV gel. The gel formulation included a reduced amount of glycerin compared to the 1% TFV vaginal formulation and was studied in the MTN-007 phase 1 rectal microbicide trial. With a lower osmolality (836 mOsmol/kg), it was determined to be better tolerated by the participants of the trial [58, 88]. Recently, McGowan et al. [88] reported a randomized phase 1 clinical trial comparing the safety, acceptability, PK, pharmacodynamics of the 1% TFV gel, the reduced glycerin gel, and a third gel never tested before in a phase 1 rectal study (Charm-01). The third gel was a rectal-specific formulation made with an even lower concentration of glycerin than the already reduced glycerin formulation. An additional carbopol was also added to create a nearly iso-osmolar product (479 mOsmol/kg). After a single dose of each treatment, all three gels were found to be safe and acceptable to participants. Ex vivo colorectal studies were also performed and all formulations were found to result in a significant suppression of HIV-1 viral replication. However, due to low participation and limited product availability, Charm-01 was not fully enrolled and significant conclusions regarding which product should advance to later stage trials as a rectal microbicide could not be made. Currently the reduced glycerin gel is being evaluated in an International Phase 2 expanded safety study expected to be completed by early 2016.
Nano-sized DDS are showing promise for colorectal microbicide development. The majority of information published involves Vivagel® (Starpharma Pty Ltd). Vivagel is a carbomer-based gel loaded with SPL7013, a dendrimer reported to have inhibitory activity against both X4 and R5 strains of HIV-1. A 5% SPL703 gel reduced HIV-1 infection by greater than 85% in colorectal explants ex vivo but it was also shown to cause epithelial shedding. The toxicity of the treatment was attributed to the vehicle and not the dendrimer [56, 89]. Das Neves et al. reports the use of poly(ethylene oxide)-modified poly(ε-caprolactone) (PEO-PCL)-based nanoparticles (NPs) as a potential nanocarrier (NC) for the NNRTI dapivirine. 200 nm PEO-PCL NPs loaded with dapivirine were shown to increase drug uptake into Caco-2 intestinal cells in vitro [56, 90, 91]. The development of colorectal microbicides is in the very early stages. An editorial published by Sarmento and das Neves called for more research in this area and describes the growing field in more detail [92].
There are numerous rectally administered products and excipients that have been collectively approved for human use for literally hundreds of years. However, given the unique application to rectal microbicides, the use of traditional rectal products is not very promising. A technology covering the concept of a colorectal dose form/DDS that could spread uniformly along the descending colon from the splenic flexure to the rectum and be retained for a reasonable period of time is virtually nonexistent. Even if such a technology existed, the coital dependence of efficacy due to physical abrasion of the colorectal tissue and the persistence of effect would be questionable. The obvious modifications to traditional rectal dose forms/administration such as large volume enemas to deliver adequate volumes to ensure initial complete colorectal mucosal coverage would not be well received by patients. This will certainly result in unacceptable leakage volume and extremely poor patient use/adherence no less a transient effect due to poor mucosal retention. Hence, the dependence on old, approved rectal formulations that were not designed for the purpose of rectal microbicides is a critical issue facing the DD field.
Drug Delivery Systems for HIV Treatment
Although a variety of DDSs have been explored for treating HIV/AIDS, they have been, for the most part extensively covered in previous reviews [76, 93–97]. In the following sections, DDSs that focus on resolving the remaining DD challenges are highlighted.
Gut Mucosa and Lymph Nodes: Targeting the critical sites of HIV infection
The current understanding of HIV pathogenesis informs future DD strategies for eradicating HIV infection from the gut and LNs. Viral replication and CD4+ T cell depletion predominantly occurs in the gut mucosa at all stages of HIV infection even under apparently successful ART making DD to the gut mucosa a high priority [17, 18]. In a 2012 study [98], 30 acute HIV infected patients (viremia+/HIV IgG−) were identified/recruited from screening 24,430 high-risk persons and treated with mega-HAART (five drugs at the entry, RT and integration steps) for 24 weeks. The mega-HAART regimen resulted in a viral reservoir reduction and partial CD4+ T cell count recovery in the gut mucosa. However, even in these patients, the viral reservoir persisted emphasizing the need for a gut-targeted DDS to achieve a functional cure for early diagnosed patients.
The mucosal mucus layer is the first barrier that a DDS will encounter. Mucus properties, function, barrier to NP delivery and mucoadhesive and mucopenetrating polymeric NPs have been extensively reviewed [99]. The mucus layer covering the alimentary and reproductive tracts consists of interwoven fibers and water-filled spaces in between. NPs must penetrate through this layer to reach the mucosa. The Saltzman group has found that certain polymers (e.g., PEG and PVP) are capable of altering the mucus structure to facilitate polymeric NP, or even monocytes, penetration of the mucus layer [100]. While the Hanes group reported that short (~ 2 kDa) PEG polymers displayed on NPs are able to penetrate mucus layer, apparently mimicking the surface property of viruses that infect human epithelia (reviewed in [99]), the Sinko group demonstrated size dependence of particle translocation through mucus [101, 102]. Mucoadhesive NPs can be used in HIV topical PrEP while mucopenetrating NPs are more suitable for oral DD. In the latter case, however, mucopenetrating alone is not sufficient; the NP must be able to penetrate the gut epithelial barrier. In this regard, an oral NP targeting the neonatal Fc receptor has achieved some degree of success [103].
Lipid-formulated DDS that exploit the gut lymphatic route for absorption are becoming increasingly popular [104]. Lipids are cost effective in comparison to synthetic polymers and the relative ease of manufacture, which avoids the use of organic solvents and collectively this makes this technology worthy of further investigation [105]. In separate rats dosed intraduodenally with a solid lipid NP formulation of lopinavir, cumulative lopinavir concentrations in lymph fluid were 9.68 μg for the nanoformulation compared to 1.97 μg for the conventional formulation 6 hours after administration [106]. Negi et al reported similar observations using another solid lipid NP of lopinavir that was fabricated using a hot self nano-emulsification technique [107, 108]. Although the gut lymphatic-targeting mechanism of lipid-formulated drugs is not clear, it is believed to be similar to dietary fat absorption. Dietary fats translocate across the gut in the form of nano-sized chylomicrons and since the chylomicrons are too big to enter the blood microvasculature they by default enter the close-ended/one way terminal capillary lymphatics (i.e., the lacteals) through endothelial intercellular gaps. This also suggests that lymphatic-targeted NPs have the potential to be retained in gut lamina propria if their size exceeds the size of the chylomicrons. Solid lipid NPs have been reported to exhibit low drug loading capacity and low lipid chemical stability, which may limit their use for some drug delivery applications [109]. Other technologies that result in the formation of micellar suspensions have also been shown to improve the plasma concentrations of various ARVs after oral administration but lymph penetration was not assessed as the potential mechanism [110, 111].
Since LNs are an important immune induction site, a HIV replication site and a reservoir, DD to LNs is also a high priority. Unfortunately, with the exception of a very early attempt in 2001 [112], literature reports suggesting or proving the feasibility of specifically targeting the gut mucosa using DDS are lacking. This 2001 study achieved a four-fold higher AZT gut mucosa concentrations 90 minutes after oral dosing in rat using a poly(isohexylcyanoacrylate) NP formulation compared to free AZT. The enhancement mechanism was proposed to be mucoadhesion. DD to the LNs has proven to have a disproportionally larger effect on controlling HIV as compared to the blood compartment (i.e., systemic therapy). Critical progress made in targeting LNs is described in the following sections.
The Ho lab pioneered LN targeted HIV DD. They found that in HIV-1 positive patients, indinavir (IDV) concentrations in LN mononuclear cells were about 25–35% that of mononuclear cells in blood. LN targeting enhanced IDV delivery in a HIV-2-infected macaque model [113]. The DDS consisted of a lipid-associated IDV complex NP (50–80 nm in diameter). The subcutaneously (SC) injected lipid/drug NPs became trapped in the draining and systemic LNs where they were taken up by resident macrophages. The LN macrophages served as a drug depot slowly releasing IDV. IDV concentrations in both peripheral and mesenteric LNs were 250–2270% higher than plasma while in humans soluble lipid-free drug administration reached a concentration <35% compared to the plasma. SC injection at IDV-equivalent 20 mg/kg daily for 30–33 weeks resulted in significantly reduced viral RNA load and increased CD4+ T-cell counts. Although the exact fate of SC-injected lipid-associated IDV NPs is not well characterized, it is likely that lipid–IDV NPs were trapped in LNs throughout the lymphatic system. The Ho group further optimized a lipid NP formulation [114]. Compared to previous formulations, new lipid-IDV NP provided 6-fold higher IDV concentrations in LNs of the macaque/SIV model and enhanced drug exposure in blood. Recently, they reported that they had further developed a lipid NP formulation incorporating newer HIV drugs [115]. With the new generation of lipid-drug formulation, they reported a lipid-drug NP containing a combination of lopinavir, ritonavir, and TFV [116]. These NPs produced over 50-fold higher intracellular lopinavir, ritonavir and TFV concentrations in LNs compared to free drug. Enhanced plasma and intracellular drug levels persisted for 7 days after a single SC dose, exceeding that achievable with current oral therapies. These results underscore the benefit of targeting an anatomic site critical in HIV infection as compared to systemic administration.
Targeting cell receptors on HIV infected T-cells and macrophages
As evidenced by the lack of progress in HIV vaccine development, targeting HIV proteins has been difficult. Host cell surface receptors are more readily targeted regardless of HIV infection status. The major roadblock for targeting CD4+ T cells and macrophages is the limited availability of targeting ligands. Most potential targeting ligands are small molecule chemicals whereby conjugation to a DD carrier tends to reduce the ability of the ligand to bind to a target cell receptor. In addition, there is a tendency to “decorate” rather than engineer NC surfaces. Ligand display configuration (spacing, linker dimensions, density, etc.) will impact NC-receptor binding and cellular uptake. Little has been published in this area in general and specifically as it relates to HIV/AIDS. A priority must be put on the discovery of new cell targeting ligands for NCs, their optimal display on drug-loaded NCs. In addition, techniques to avoid endosomal entrapment and the avoidance of off-target uptake and effects will have to be carefully considered. While there have been some reviews regarding active DDS targeting in HIV/AIDS, this area remains largely undeveloped and, from a critical in vivo perspective, feasibility is largely unproven [93, 96, 117].
There are numerous potential cell targets on CD4+ T-cells and macrophages. However, only a few have been exploited due to concerns about the potential inference with host immune function or the lack of suitable ligands that can be conjugated to NCs. Most targeting ligands have been peptides as the peptidyl nature makes it amenable to conjugation to a NC. DV3 is a 10-mer, D peptide derived from a natural CXCR7 ligand vMIP-II. It has been shown to be able to bind to CXCR4 and to exhibit an anti-HIV activity of EC50 0.439 μM [118]. Recently, a derivative of DV3 named DV1-K-(DV3) was reported [119]. It was created by chemically linking DV1 (a 21-mer peptide also derived from vMIP-II) and DV3. This peptidyl ligand of CXCR4 shows an anti-EC50 4 nM, which is more potent than the natural CXCR4 ligand SDF-1a. T140 is a 14-mer peptide derived from a horseshoe crab protein named polyphemusin that is a CXCR4 antagonist with anti-HIV activity of EC50 3.3 nM [120]. Remarkably, a cyclic pentapeptide derivative of T140 named FC131 retains most T140’s potency and one of FC131 analogs is even more potent than T140 with an EC50 1.4 nM [121]. Floudas’ group has identified a few short (10 to 15 residue) peptides as HIV CXCR4 entry and HIV fusion inhibitors, respectively [122, 123]. New and better performing derivatives of these peptides are being tested in our lab (to be published). Tuftsin is a tetrapeptide with the sequence of TKPR that utilizes an unidentified receptor on macrophages. It was displayed on efaviranz-packed dendrimers and the resultant dendrimer demonstrated macrophage targeting specificity and moderate anti-HIV activity [124]. Other potential ligands for NCs have also been explored, including a three-residue short peptide fMLF [125] and the monosaccharide mannose [126, 127].
Cell Depot-based DDS
Significant progress has been made in using human cells as DD depot sites. These approaches have utilized macrophages as drug carriers. HIV drugs were first loaded into NCs [128] or red blood ghost cells [129] that become engulfed by macrophages ex vivo or in vivo. The macrophages then migrated to LNs or additionally to other reticuloendothelial system (RES) tissues, acting as a cellular depot in these critical sites of HIV infection where the drugs slowly diffuse out over a period of days to weeks resulting in sustained high local drug concentrations in the LNs or other RES tissue.
In a feline immunodeficiency virus (FIV) model [129], the membrane-impermeable HIV drug zalcitabine-TP was first loaded ex vivo into autologous red blood cells and the plasma membrane of these red blood cells was then chemically modified so macrophages would recognize and engulf them. In a 7-month experimental FIV infection, zalcitabine-loaded erythrocytes protected the majority of peritoneal macrophages and reduced the infection of circulating lymphocytes. The Gendelman group has developed LA/ER nanoformulations of ritonavir (RTV), indinavir (IDV) and efavirenz (EFV) (nanoART) [128]. They reasoned that circulating macrophages traveling across the blood-brain barrier could enhance nanoART brain delivery [130]. An HIV-1 encephalitis (HIVE) rodent model was used. IDV NPs (a nanoART) were loaded into murine bone marrow macrophages (BMM, IDV-NP-BMM) ex vivo and the IDV-NP-BMM was administered intravenously. Rhodamine-labeled IDV-NP showed up in areas of HIVE. Continuous IDV release was observed for 14 days. IDV-NP-BMM treatment led to reduced HIV-1 replication in HIVE brain regions. Mechanistic studies [131] showed that nanoART-macrophage interactions enhanced phagocytosis, secretory functions, and cell migration, which could be exploited to increase macrophage nanoART loading capacity. Aouadi et al. [132] studied anti-inflammatory siRNA delivery utilizing yeast ghost cells. Yeast ghost cells were made by chemically treatment of yeast cell wall so that the cell surface was left with only beta1 3-D-glucan, for which macrophages have a special receptor. siRNA was then loaded into the ghost cells. The ghost cells can be efficiently absorbed orally through M-cells and, once crossed M-cells, avidly phagocytosed by macrophages in the Peyer’s Patches. Interestingly, macrophages in the Peyer’s Patches were able to migrate into blood circulation and settle at various LNs. Oral gavage of mice with the siRNA-loaded ghost cells containing as little as 20 μg/kg of siRNA directed against tumor necrosis factor alpha (TNF-α) depleted its messenger RNA in macrophages recovered from the peritoneum, spleen, liver and lung, and lowered serum TNF-α levels. Although not developed an HIV application, this approach is readily translatable to the delivery siRNA against HIV infection.
In addition to treating HIV/AIDS using pharmacotherapy, vaccination and even gene delivery have been attempted. Steinbach has extensively reviewed protein and oligonucleotide DDSs for vaginal microbicides against viral sexually transmitted diseases [133]. These biological agents include antibodies for passive immunization in the vagina, various proteins or plasmid DNA encoding viral proteins as antigens for induction of vaginal immunity, siRNA to downregulate the expression of viral and host proteins involved in infection, and other proteins and peptides as antagonists in virus-host interactions. Often, these agents are non-covalently or covalently complexed/linked to polymers or other molecules in vaginal formulations and the formulations are incorporated into various vaginal devices. These studies are generally at the early stages but they have established proof-of-concept. Typically, natural infection is the most effective vaccination. The fact that not a single patient among millions infected with HIV has spontaneously recovered, underlines the inefficiency of a conventional vaccination strategy for HIV. Current vaccine development focuses on new approaches, such as broadly neutralizing antibodies and unconventional vaccines with recombinant DNA technology [134, 135]. In 2009, a report of a cure in the so-called “Berlin Patient” by using bone marrow transplantation of stems cells homozygous for a deletion in the CCR5 gene proved for the first time that eradication of HIV is possible [136]. Unfortunately, this strategy has proven to be unrealistic for most patients leading to efforts to develop gene therapy based strategies to disrupt the gene encoding CCR5 or other host proteins, as well as to disrupt the provirus [137]. Another strategy being explored involves purging the latent reservoir by activating latent provirus while eliminating new infections with ART [138, 139]. So far none of these strategies has achieved eradication in patients.
Long-Acting/Extended-Release (LA/ER) Parenteral (Injectable) DDS for PrEP and Treatment
The development of LA/ER injectable ARV DDS that could be administered infrequently (i.e., monthly or less), either in the clinic or at home represents an important potential solution to many of the problems associated with chronic ART and PrEP. There are numerous challenges facing LA/ER injectable DDS including identifying the optimal dosing interval, number and volume of injections per visit, the classes and properties of drugs that can be combined, and what to do if a patient becomes pregnant after the drug is administered. However, benefits to patients include infrequent dosing and long dosing intervals making administration convenient; the possibility of directly observed therapy and better long-term adherence; use in difficult to treat populations such as adolescents or those with ongoing substance abuse; use in patients reporting pill fatigue; and protection of patient privacy by eliminating the risk of disclosing pill taking to family and co-workers. There is also a clear rationale for the benefit of using LA/ER ARV formulations in pregnant women, postpartum women (including those who are breast feeding), and infants/children who cannot take typical adult oral formulations. In fact, patient surveys disclose great enthusiasm for trying LA/ER injectable nanoformulations for treatment. One recently published survey found that 84% of patients currently taking oral therapy definitely or probably would try such an intervention if the dosing frequency was once-monthly or less frequent [140]. Additional applications that would benefit from LA/ER ARV formulations include their possible temporary use as parenteral ART for those unable to take oral medications. This could include patients requiring gastrointestinal surgery, those who are comatose and/or intubated, those with significant nausea and vomiting, patients developing severe mucositis from cancer chemotherapy, and very young pediatric patients unable to reliably swallow pills. Treatment options for such patients are extremely limited at present, resulting in significant risk of treatment failure as a consequence of intercurrent illness [141].
Injectable nanoformulation technologies previously applied to approved LA/ER treatments for chronic schizophrenia, such as paliperidone [142] or LA/ER injectable contraception, are now being evaluated for treating HIV [143]. The LA/ER formulation consists of a solid drug particle nanosuspension produced by wet bead milling of large fragments of drug in aqueous solution in the presence of a surfactant until the particles are within the nanometer size range. Since these NPs are composed of pure drug and not a mixed matrix including polymers and other excipients, which typically comprise 50% or more of the NP formulation, higher loading into the formulation of up to 200 mg/ml is possible along with a reduced injection volume. A formulation of the nanosuspension is administered to form an IM depot. Given typical IM injection volumes once monthly dosing is feasible for potent drugs. Due to the physicochemical and dosing limitations as well as insufficient antiviral potency of currently used ARVs, it was not feasible to apply this approach until the development of two potent ARVs, rilpivirine and cabotegravir (S/GSK1265744). Both compounds are poorly water-soluble but possess sufficient potency and favorable PK properties to support once monthly dosing. The combination subnanomolar potency and a plasma elimination half-life of approximately 40 hours makes cabotegravir an ideal candidate for this approach. One concern with IM depots of poorly soluble drugs is the relatively high variability in bioavailability that has been observed in other applications. This would be a concern if the lower end of the deviation in blood/tissue concentrations falls below the IC90 of HIV-1.
Two formulations, a LA/ER injectable NNRTI rilpivirine (LA-RPV), and an InSTI, LA/ER injectable cabotegravir (LA-744), have been developed and are currently undergoing clinical testing [45, 144, 145]. The target-dosing interval for LA-RPV, based on its PK properties, is once monthly, and for LA-744 is once every three months for prevention applications and once monthly if combined with LA-RPV. Although LA-RPV and LA-744 represent important first steps in developing LA/ER ARV’s, they are not well matched in terms of PK properties. Differences in half-life mean that missed dosing may negatively impact resistance since LA-RPV has a shorter half-life. In the first study [146] investigating LA-RPV, the reported plasma and genital tract concentrations in 60 women and six men given intramuscular (IM) depot injections of three different doses of LA-RPV (300, 600, and 1200 mg) showed that RPV concentrations in females were between 26 and 77 % of plasma concentrations in cervico-vaginal fluid and between 45 and 379 % of plasma concentrations in cervicovaginal tissue, while in males, they were between 33 and >77 % of plasma concentration in rectal fluid and tissue. In a second study [45], LA-744 (200 mg/ml) was evaluated after abdominal SC (100 mg, 200 mg, and 400 mg) or gluteal IM (100 mg, 200 mg, 400 mg and 800 mg) administration. The highest doses for each route were split due to injection volume considerations. Due to the low solubility of the NPs and inherent low tissue perfusion of muscle, the mean absorption half-times ranged from 21 to 50 days as compared to 40 h following single dose oral administration. With the exception of the 100 mg doses, all treatment groups remained above the IC90 of wild-type HIV-1 for at least 6 months. In two additional cohorts, (400 mg and 2 × 200 mg IM) were studied in 4 males and 4 females. Median individual tissue:plasma drug concentration ratios ranged from 16 to 25% in the female genital tract and were 8% in rectal tissue in men suggesting that at the dose tested the formulation would fall short as a PrEP agent. Furthermore, a recent study conducted in rhesus macaques showed that two IM doses 1 month apart protected eight out of eight animals from multiple rectal exposures to simian/human immunodeficiency virus (SHIV), while all eight animals receiving placebo injections acquired infection [147–149].
Since all currently used ART regimens involve the administration of at least three drugs in combination, there is an urgent need for other LA/ER technologies to complement those in late stage development. Additionally, LA/ER technologies that do not require injection need to be developed. It is clear that high drug potency is a prerequisite for compatibility with the LA/ER approach since it determines the plasma concentrations that need to be achieved (and sustained). Ultimately, dose is the limiting factor for LA/ER delivery because it directly influences the volume of the depot required to provide sufficient drug concentrations, which is limited by anatomical considerations as well as patient acceptability. However, release rate is also a key determinant of success since too fast a release rate will result in “dose dumping” where high initial concentrations that are rapidly cleared, whereas too slow a release rate will not provide sufficiently high plasma concentrations for therapeutic effect. A recent study employed physiologically-based pharmacokinetic modeling to determine which release rate/dose combinations would be required to provide either weekly or monthly therapeutic concentrations for a range of commonly used existing ART drugs [150]. Importantly, these data do not determine that the predicted dose/release characteristics are achievable, but they do set a benchmark to inform the target product profile for developing LA/ER medicines with other ART drugs. The National Institutes for Health have recently funded a Long-Acting/Extended Release Antiretroviral Resource Program (LEAP), which once launched will aim to foster further development of such medicines. LEAP will also provide services, which include a PBPK modelling core to help guide development of LA/ER drugs and/or regimens.
Future Perspectives, Gaps and Opportunities
The tripartite goals of preventing new HIV infections, achieving a functional cure and complete eradication have become even more important since ART transformed HIV infection into a chronically managed disease. In recent years, great strides have been made in understanding how novel DDS may benefit HIV therapy in a disease-specific manner. Central to this has been the explicit demonstration in pre-clinical species of pharmacologically beneficial behaviors. Indeed, some of these benefits have also been translated to studies in humans and this is true for PrEP and LA/ER development discussed in this review. However, there remain a significant number of unmet needs and this is particularly true for those technologies that are at early stages of translation. In broad terms, considerations for successful future development can be divided into those that relate to the drug delivery system itself, as well as those that relate to the drug, pharmacological rationale and the ultimate clinical application.
While numerous benefits have been achieved and demonstrated pre-clinically, there remain significant challenges in terms of the intricacies of manufacture. The development of complex DDS such as NCs is challenging at the bench no less in scale for manufacture. Translation at all stages represents the biggest hurdle for large-scale fabrication, testing and manufacture of DDS. For many technologies, there is a current paucity of knowledge regarding how the physical properties and constituents of the DDS specifically relate to the pharmacological behavior. Drug loading potential will be a key determinant of success for many technologies because of its impact upon format size or volume required for injectable depot medicines. Moreover, many technologies are only compatible with drugs that exhibit specific physiochemical properties and this may pose challenges to holistic development of enabled-regimens consisting of drugs from different classes. For conventional oral medicines, the framework within which drugs are developed has been extensively researched over the past decades. This framework has resulted in the Biopharmaceutics Classification System (BCS) for oral drugs and a solid understanding of the chemical space that is optimal for oral drug delivery that is provided by Lipinski’s rule of 5 (and variations thereof). However, there are gaps in knowledge that specifically relate to many newer DDS as well as the chemical compatibility of drugs with the DDS and with specific applications (e.g. LA/ER depot delivery).
One of the significant gaps in HIV/AIDS DDS research is targeting to sites of infection in order to achieve effective ART concentrations. Another significant gap is the ability to prolong ART effectiveness in order to reduce the frequency of administration and burden on patients. Classic sustained release technologies currently being utilized in LA/ER PrEP have proven the feasibility of the approach. However, significant improvements are needed. This suggests a major opportunity for DD research – the marriage of targeting technologies and techniques to sustain not just the release of ART but, more importantly, the effect.
Since the overwhelming burden of HIV disease continues to fall upon individuals in resource-limited settings, there are societal implications that need to be considered if novel technologies are to impact significantly on the HIV pandemic. Some novel DDS challenge convention in terms of route of delivery, and consideration needs to be given to compatibility with existing global healthcare systems and acceptability of the approach to patients with different cultural backgrounds. Clearly, it is imperative that early technological development and selection of technologies for further development consider cost, and compatibility of the platform with sterile production of the medicine at scale.
Acknowledgments
Supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases Awards R37 AI051214, R01 AI117776 and R24 AI118397.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Antoinette G. Nelson, Email: antgnelson@gmail.com.
Xiaoping Zhang, Email: xzhang@rci.rutgers.edu.
Usha Ganapathi, Email: ganapaus@rci.rutgers.edu.
Zoltan Szekely, Email: zoltan@rci.rutgers.edu.
Charles W. Flexner, Email: flex@jhmi.edu.
Andrew Owen, Email: aowen@liverpool.ac.uk.
Patrick J. Sinko, Email: sinko@rutgers.edu.
References
- 1.WHO [Internet] 2015 [updated July 2015; ]. Available from: http://www.who.int/mediacentre/factsheets/fs360/en/
- 2.Tognotti E. The eradication of smallpox, a successful story for modern medicine and public health: What lessons for the future? J Infect Dev Ctries. 2010;4(5):264–266. doi: 10.3855/jidc.1204. [DOI] [PubMed] [Google Scholar]
- 3.Haase AT. Targeting early infection to prevent HIV-1 mucosal transmission. Nature. 2010;464(7286):217–223. doi: 10.1038/nature08757. [DOI] [PubMed] [Google Scholar]
- 4.Sáez-Cirión A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, GI, Lecuroux C, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLOS Pathog. 2013;9(3):e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salgado M, Rabi SA, O’Connell KA, Buckheit RW, III, Bailey JR, Chaudhry AA, et al. Prolonged control of replication-competent dual-tropic human immunodeficiency virus-1 following cessation of highly active antiretroviral therapy. Retrovirology. 2011;8(97) doi: 10.1186/1742-4690-8-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckheit RW, Jr, Watson KM, Morrow KM, Ham AS. Development of Topical Microbicides to Prevent the Sexual Transmission of HIV. Antiviral Res. 2010;85(1):142. doi: 10.1016/j.antiviral.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garg A, Nuttall J, Romano J. The future of HIV microbicides: challenges and opportunities. Antivir Chem & Chemother. 2009;19(4):143–150. doi: 10.1177/095632020901900401. [DOI] [PubMed] [Google Scholar]
- 8.Senise JF, Castelo A, Martinez M. Current treatment strategies, complications and considerations for the use of HIV antiretroviral therapy during pregnancy. AIDS Rev. 2011;13(4):198–213. [PubMed] [Google Scholar]
- 9.Tebit DM, Arts EJ. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect Dis. 2011;11(1):45–56. doi: 10.1016/S1473-3099(10)70186-9. [DOI] [PubMed] [Google Scholar]
- 10.Gruell H, Klein F. Opening Fronts in HIV Vaccine Development: Tracking the development of broadly neutralizing antibodies. Nat Med. 2014;20(5):478–479. doi: 10.1038/nm.3567. [DOI] [PubMed] [Google Scholar]
- 11.Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37(3):377–388. doi: 10.1016/j.immuni.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarmati L, D’Ettorre G, Parisi SG, Andreoni M. HIV Replication at Low Copy Number and its Correlation with HIV Reservoir: A Clinical Perspective. Curr HIV Res. 2015;13(3):250–257. doi: 10.2174/1570162X13666150407142539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Svicher V, Ceccherini-Silberstein F, Antinori A, Aquaro S, Perno CF. Understanding HIV compartments and reservoirs. Curr HIV/AIDS Rep. 2014;11(2):186–194. doi: 10.1007/s11904-014-0207-y. [DOI] [PubMed] [Google Scholar]
- 14.Cory TJ, Schacker TW, Stevenson M, Fletcher CV. Overcoming pharmacologic sanctuaries. Curr Opin HIV AIDS. 2013;8(3):190–195. doi: 10.1097/COH.0b013e32835fc68a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siewe B, Landay A. Key Concepts in the Early Immunology of HIV-1 Infection. Curr Infect Dis Rep. 2012;14(1):102–109. doi: 10.1007/s11908-011-0235-3. [DOI] [PubMed] [Google Scholar]
- 16.Walker B, McMichael A. The T-cell response to HIV. Cold Spring Harb Perspect Med. 2012;2(11) doi: 10.1101/cshperspect.a007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veazey RS, Lackner AA. Getting to the guts of HIV pathogenesis. J Exp Med. 2004;200(6):697–700. doi: 10.1084/jem.20041464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavarelli M, Scarlatti G. HIV-1 infection: the role of the gastrointestinal tract. Am J Reprod Immunol. 2014;71(6):537–542. doi: 10.1111/aji.12245. [DOI] [PubMed] [Google Scholar]
- 19.Assimakopoulos SF, Dimitropoulou D, Marangos M, Gogos CA. Intestinal barrier dysfunction in HIV infection: pathophysiology, clinical implications and potential therapies. Infection. 2014;42(6):951–959. doi: 10.1007/s15010-014-0666-5. [DOI] [PubMed] [Google Scholar]
- 20.Santangelo PJ, Rogers KA, Zurla C, Blanchard EL, Gumber S, Strait K, et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiretroviral therapy-treated macaques. Nat MEthods. 2015;12(5):427–432. doi: 10.1038/nmeth.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tebit DM, Ndembi N, Weinberg A, Quinones-Mateu ME. Mucosal transmission of human immunodeciciency virus. Curr HIV Res. 2012;10(1):3–8. doi: 10.2174/157016212799304689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458(7241):1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.FDA-Approved HIV Medicines [Internet] 2015 [updated May 2015; ]. Available from: https://aidsinfo.nih.gov/education-materials/fact-sheets/21/58/fda-approved-hiv-medicines.
- 24.Wong A. The HIV pipeline. Nature Reviews Drug Discovery. 2014;13:649–650. doi: 10.1038/nrd4364. [DOI] [PubMed] [Google Scholar]
- 25.Cox BD, Prosser AR, Sun Y, Li Z, Lee S, Huang MB, et al. Pyrazolo-Piperidines Exhibit Dual Inhibition of CCR5/CXCR$ HIV Entry and Reverse Transcriptase. ACS Med Chem Lett. 2015;6:753–757. doi: 10.1021/acsmedchemlett.5b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chong H, Qui Z, Su Y, He Y. The N-terminal T-T Motif of a Third-Generation HIV-1 Fusion Inhibitor Is Not Required for Binding Affinity and Antivitral Activity. J Med Chem. 2015:1–43. doi: 10.1021/acs.jmedchem.5b00109. Just Accepted Manuscript. [DOI] [PubMed] [Google Scholar]
- 27.Gogtay JA, Malhotra G. Reformulation of existing antiretroviral drugs. Curr Opin HIV AIDS. 2013;8(6):550–555. doi: 10.1097/COH.0000000000000006. [DOI] [PubMed] [Google Scholar]
- 28.Falcon RW, Kakuda TN. Drug interactions between HIV protease inhibitors and acid-reducing agents. Clin Pharmacokinet. 2008;47(2):75–89. doi: 10.2165/00003088-200847020-00001. [DOI] [PubMed] [Google Scholar]
- 29.Adams JL, Greener BN, Kashuba AD. Pharmacology of HIV integrase inhibitors. Curr Opin HIV AIDS. 2012;7(5):390–400. doi: 10.1097/COH.0b013e328356e91c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kakuda TN, Falcon RW. Effect of food and ranitidine on saquinavir pharmacokinetics and gastric pH in healthy volunteers. Pharmacotherapy. 2006;26(8):1060–1068. doi: 10.1592/phco.26.8.1060. [DOI] [PubMed] [Google Scholar]
- 31.Owen A. The impact of host pharmacogenetics on antiretroviral drug disposition. Curr Infect Dis Rep. 2006;8(5):401–408. doi: 10.1007/s11908-006-0052-2. [DOI] [PubMed] [Google Scholar]
- 32.Press Announcements: FDA approves first drug for reducing the risk of sexually acquired HIV infection [Internet] 2012 [updated July 2012; ]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312210.htm.
- 33.Callebaut C, Stepan G, Tian Y, Miller MD. In Vitro Virology Profile of Tenofovir Alafenamide, a Novel Oral Prodrug of Tenofovir with Improved Antiviral Activity Compared to Tenofovir Disoproxil. Antimicrob Agents Chemother. 2015 doi: 10.1128/AAC.01152-15. Accepted manuscript posted online 6 July 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson AG, Else LJ, Mesquita PM, Egan D, Back DJ, Karolia Z, et al. A compartmental pharmacokinetic evaluation of long-acting rilpivirine in HIV-negative volunteers for pre-exposure prophylaxis. Clin Pharmacol Ther. 2014;96(3):314–323. doi: 10.1038/clpt.2014.118. [DOI] [PubMed] [Google Scholar]
- 35.Andrews CD, Heneine W. Cabotegravir long-acting for HIV prevention. Curr Opin HIV AIDS. 2015;10(4):258–263. doi: 10.1097/COH.0000000000000161. [DOI] [PubMed] [Google Scholar]
- 36.Pharmacokinetic Study of Cabotegravir Long-acting in Healthy Adult Volunteers [Internet] 2015 [updated June 2015; ]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02478463.
- 37.Evaluating the Safety and Tolerability of Antiretroviral Drug Regimens Used as Pre-Exposure Prophylaxis to Prevent HIV Infection in At-Risk Men Who Have Sex With Men and in At-Risk Women [Internet] 2015 [updated August 2015; ]. Available from: https://clinicaltrials.gov/ct2/show/NCT01505114.
- 38.Evaluation of the Safety, Efficacy, and Pharmacokinetics of Intraveneous Deferiprone in HIV-Positive Subjects [Internet] 2015 [updated July 2015; ]. Available from: https://clinicaltrials.gov/ct2/show/NCT02456558?term=deferiprone+hiv&rank=2.
- 39.Hanauske-Abel H, Saxena D, Palumbo PE, Hanauske A, Luchessi AD, Cambiaghi TD, et al. Drug-Induced Reactivation of Apoptosis Abrogates HIV-1 Infection. PLoS ONE. 2013;8(9):e74414. doi: 10.1371/journal.pone.0074414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friend D, Kiser PF. Assessment of topical microbicides to prevent HIV-1 transmission: Concepts, testing, lessons learned. Antiviral Res. 2013;99(3):391–400. doi: 10.1016/j.antiviral.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 41.Karim SSA. Microbicides for the Prevention of HIV Infection. 2nd South African AIDS Conference; Durban, South Africa. 2005. pp. 30–40. [Google Scholar]
- 42.Mcgowan I. An Overview of Antiretroviral Pre-Exposure Prophylaxis of HIV Infection. Am J Reprod Immunol. 2014;71(6):624–630. doi: 10.1111/aji.12225. [DOI] [PubMed] [Google Scholar]
- 43.Omar RF, Bergeron MG. The future of microbicides. International Journal of Infectious Diseases. 2011;15:e656–e660. doi: 10.1016/j.ijid.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Shattock RJ, Rosenberg Z. Microbicides: Topical Prevention against HIV. Cold Spring Harb Perspect Med. 2012;2(2):1–17. doi: 10.1101/cshperspect.a007385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spreen WR, Margolis DA, Pottage JC. Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr Opin HIV AIDS. 2013;8:565–571. doi: 10.1097/COH.0000000000000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson CG, Cohen MS, Kashuba ADM. Antiretroviral Pharmacology in Mucosal Tissues. J Acquir Immune Defic Syndr. 2013;63(0–2):S240–S247. doi: 10.1097/QAI.0b013e3182986ff8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hendrix CW. The clinical pharmacology of antiretrovirals for HIV prevention. Curr Opin HIV AIDS. 2012;7(6):498–504. doi: 10.1097/COH.0b013e32835847ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsai C, Follis KE, Sabo A, Beck TW, Grant RF, Bischofberger N, et al. Prevention of SIV Infection in Macaques by (R)-9-(2-Phosphonylmethoxypropyl)adenine. Science. 1995;270:1197–1199. doi: 10.1126/science.270.5239.1197. [DOI] [PubMed] [Google Scholar]
- 49.Gallant JE, Deresinski S. Tenofovir Disoproxil Fumarate. Clin Infect Dis. 2003;37(7):944–950. doi: 10.1086/378068. [DOI] [PubMed] [Google Scholar]
- 50.Grant R, Lama JR, Anderson PL, McMahan V, Lui AY, Vargas L, et al. Preexposure Chemoprophylaxis for HIV Prevention in Men Who Have Sex with Men. N Engl J Med. 2010;363(27):2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donnell D, Baeten J, Bumpus N, Brantley J, Bangsberg D, Haberer J, et al. HIV Protective Efficacy and Correlates of Tenofovir Blood Concentrates in a Clinical Trial of PrEP for HIV Prevention. J Acquir Immune Defic Syndr. 2014;66(3):340–348. doi: 10.1097/QAI.0000000000000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herold BC, Scordi-Bello I, Cheshenko N, Marcellino D, Dzuzelewski M, Francois F, et al. Mandelic Acid Cendensation Polymer: Novel Candidate Microbicide for Prevention of Human Immunodeficiency Virus and Herpes Simplex Virus Entry. J Virol. 2002;76(22):11236–11244. doi: 10.1128/JVI.76.22.11236-11244.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gagné N, Cormier H, Omar R, Désormeaux A, Gourde P, Tremblay M, et al. Protective Effect of a Thermoreversible Gel Against the Toxicity of Nonoxynol-9. Sexually Transmitted Diseases. 1999;26(3):177–183. doi: 10.1097/00007435-199903000-00009. [DOI] [PubMed] [Google Scholar]
- 54.Morrow KM, Underhill K, van den Berg, Jacob J, Vargas S, Rosen RK, Katz DF. User-Identified Gel Characteristics: A Qualitative Exploration of Perceived Product Efficacy of Topical Vaginal Microbicides. Arch Sex Behav. 2014;43:1459–1467. doi: 10.1007/s10508-013-0235-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morrow KM, Fava JL, Rosen RK, Vargas S, Shaw JG, Kojic EM, et al. Designing Preclinical Perceptibility Measures to Evaluate Topical Vaginal Ge Formulations: Relating User Sensory Perceptions and Experiences to Formulaton Properties. AIDS Res Hum Retroviruses. 2014;30(1):78–91. doi: 10.1089/aid.2013.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nunes R, Sarmento B, das Neves J. Formulation and delivery of anti-HIV rectal microbicides: Advances and challenges. Journal of Controlled Release. 2014;(194):278–294. doi: 10.1016/j.jconrel.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 57.Abdool Karim S, Baxter C. Current Topics in Microbiology and Immunology. Springer-Verlag; 2014. Microbicides for Prevention of HIV Infection: Clinical Efficacy Trials; pp. 97–115. [DOI] [PubMed] [Google Scholar]
- 58.Dezzutti CS, Rohan LC, Wang L, Uranker K, Shetler C, Cost M, et al. Reformulated tenofovir gel for use as a dual compartment microbicide. J Antimicrob Chemother. 2012;67:2139–2142. doi: 10.1093/jac/dks173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kiser PF, Johnson TJ, Clark JT. State of the Art in Intravaginal Ring Technology for Topical Prophylaxis of HIV Infection. AIDS Rev. 2012;(14):62. [PubMed] [Google Scholar]
- 60.Singh O, Garg T, Rath G, Goyal AK. Microbicides for the Treatment of Sexually Transmitted HIV infections. Journal of Pharmaceutics. 2014;2014:1–18. doi: 10.1155/2014/352425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abdool Karim Q, Abdool Karim SS, Frohlich J, Grobler A, Baxter C, Mansoor L, et al. Effectiveness and Safety of Tenofovir Gel, an Antiretroviral Microbicide, for the Prevention of HIV Infection in Women. Science. 2010;329(5996):1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haaland R, Evans-Strickfaden T, Holder A, Pau C, McNicholl J, Chaikummao S. UC781 microbicide gel retains anti-HIV activity in cervicovaginal lavage fluids collected following twice-daily vaginal application. Antimicrob Agents Chemother. 2012;56(7):3592–3596. doi: 10.1128/AAC.00452-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nel A, Smythe S, Young K, Malcolm K, McCoy C, Rosenberg Z, et al. Safety and Pharmacokinetics of Dapivirine Delivery From Matrix and Reservoir Intravaginal Rings to HIV-Negative Women. J Acquir Immune Defic Syndr. 2009;51(4):416–423. doi: 10.1097/qai.0b013e3181acb536. [DOI] [PubMed] [Google Scholar]
- 64.Nel A, Haazen W, Nuttall J, Romano J, Rosenberg Z, van Niekerk N. A safety and pharmacokinetic trial assessing delivery of dapivirine from a vaginal ring in healthy women. AIDS. 2014;28:1479–1487. doi: 10.1097/QAD.0000000000000280. [DOI] [PubMed] [Google Scholar]
- 65.Nuttall JP, Thake DC, Lewis MG, Ferkany JW, Romano JW, Mitchnick MA. Concentrations of dapivirine in the rhesus macaque and rabbit following once daily intravaginal administration of a gel Formulation of [14C]dapivirine for 7 days. Antimicrob Agents Chemother. 2008;52(3):909–914. doi: 10.1128/AAC.00330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akil A, Devlin B, Cost M, Rohan LC. Increased Dapivirine Tissue Accumulation through Vaginal Film Codelivery of Dapivirine and Tenofovir. Mol Pharmaceutics. 2014;12:1533–1541. doi: 10.1021/mp4007024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson TJ, Clark MR, Albright TH, Nebeker JS, Tuitupou AL, Clark JT, et al. A 90-Day Tenofovir Reservoir Intravaginal Ring for Mucosal HIV. Antimicrobial Agents and Chemotherapy. 2012;56(12):6272. doi: 10.1128/AAC.01431-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boyd P, Desjardins D, Kumar S, Fetherston SM, Le-Grand R, Dereuddre-Bosquet N, et al. A Temperature-Monitoring Vaginal Ring for Measuring Adherence. PLoS ONE. 2015;10(5):1–19. doi: 10.1371/journal.pone.0125682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nel AM, Mitchnick LB, Risha P, Muungo LTM, Norick PM. Acceptability of Vaginal Film, Soft Gel Capsule, and Tablet as Potential Microbicide Delivery Methods Among African Women. Journal of Women’s Health. 2011;20(8):1207–1214. doi: 10.1089/jwh.2010.2476. [DOI] [PubMed] [Google Scholar]
- 70.Akil A, Agashe H, Dezzutti CS, Moncla BJ, Hillier SL, Devlin B, et al. Formulation and Characterization of Polymeric Films Containing Combinations of Antiretrovirals (ARVs) for HIV Prevention. Pharm Res. 2015;32:458–468. doi: 10.1007/s11095-014-1474-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akil A, Parniak MA, Dezzutti CS, Moncla BJ, Cost M, Li M, et al. Development and Characterization of a Vaginal Film Containing Dapivirine, a Non-nucleoside Reverse Transcriptase Inhibitor (NNRTI), for Prevention of HIV-1 Sexual Transmission. Drug Deliv Transl Res. 2011;1(3):209–222. doi: 10.1007/s13346-011-0022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang W, Hu M, Shi Y, Gong T, Dezzutti CS, Moncla B, et al. Vaginal Microbicide Film Combinations of Two Reverse Transcriptase Inhibitors, EFdA and CSIC, for Prevention of HIV-1 Sexual Transmission. Pharm Res. 2015 doi: 10.1007/s11095-015-1678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Antimisiaris SG, Mourtas S. Recent advances on anti-HIV vaginal delivergy systems development. Adv Drug Deliv Rev. 2015 doi: 10.1016/j.addr.2015.03.015. ( http://dx.doi.org/10.1016/j.addr.2015.03.015) [DOI] [PubMed]
- 74.Chaowanachan T, Krogstad E, Ball C, Woodrow KA. Drug Synergy of Tenofovir and Nanoparticle-Based Antiretrovirals for HIV Prophylaxis. PLoS ONE. 2013;8(4):e61416. doi: 10.1371/journal.pone.0061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.das Neves J, Araújo F, Andrade F, Michiels J, Ariën KK, Vanham G, et al. In vitro and ex vivo evaluation of polymeric nanoparticles for vaginal and rectal delivery of the anti-HIV drug dapivirine. Mol Pharm. 2013;10(7):2793–2807. doi: 10.1021/mp4002365. [DOI] [PubMed] [Google Scholar]
- 76.Sharma P, Garg S. Pure drug and polymer based nanotechnologies for the improved solubility, stability, bioavailability and targeting of anti-HIV drugs. Adv Drug Deliv Rev. 2010;62(4–5):491–502. doi: 10.1016/j.addr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 77.das Neves J, Nunes R, Machado A, Sarmento B. Polymer-based nanocarriers for vaginal drug delivery. Adv Drug Deliv Rev. 2015 doi: 10.1016/j.addr.2014.12.004. ( http://dx.doi.org/10.1016/j.addr.2014.12.004) [DOI] [PubMed]
- 78.Hladik F, Hope TJ. HIV infection of the genital mucosa in women. Curr HIV/AIDS Rep. 2009;6(1):20–28. doi: 10.1007/s11904-009-0004-1. [DOI] [PubMed] [Google Scholar]
- 79.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nature Review Immunology. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 80.Hendrix CW, Fuchs EJ, Macura KJ, Lee LA, Parsons TL, Bakshi RP, et al. Quantitative Imaging and Sigmoidoscopy to Assess Distribution of Rectal Microbicide Surrogates. Clinical Pharmacology & Therapeutics. 2008;83(1):97–105. doi: 10.1038/sj.clpt.6100236. [DOI] [PubMed] [Google Scholar]
- 81.Cao YJ, Caffo BS, Fuchs EJ, Lee LA, Du Y, Li L, et al. Quantification of the spatial distribution of rectally applied surrogates for microbicide and semen in colon with SPECT and magnetic resonance imaging. Br J Clin Pharmacol. 2012;74(6):1013–1022. doi: 10.1111/j.1365-2125.2012.04267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Louissant NA, Nimmagadda S, Fuchs EJ, Bakshi RP, Cao Y, Lee LA, et al. Distribution of Cell-free and Cell-associated HIV surrogates in the Colon Following Simulated Receptive Anal Intercourse in Men who Have Sex with Men. J Acquir Immune Defic Syndr. 2012;59(1):10–17. doi: 10.1097/QAI.0b013e3182373b5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carballo-Diéguez A, Exner T, Dolezal C, Pickard R, Lin P, Mayer KH. Rectal Microbicide Acceptability: Results of a Volume Escalation Trial. Sexually Transmitted Diseases. 2007;34(4):224–229. doi: 10.1097/01.olq.0000233715.59239.83. [DOI] [PubMed] [Google Scholar]
- 84.Morrow KM, Hendrick C. Clinical Evaluation of Microbicide Formulations. Antiviral Res. 2010;88(Suppl 1):S40–S46. doi: 10.1016/j.antiviral.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mcgowan I, Dezzutti C. Rectal Microbicide Development. Curr Top in Microbiol Immunol. 2014;383:117–136. doi: 10.1007/82_2013_325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anton PA, Saunders T, Elliott J, Khanukhova E, Dennis R, Adler A, et al. First Phase 1 Double-Blind, Placebo-Controlled, Randomized Rectal Microbicide Trial Using UC781 Gel with a Novel Index of Ex Vivo Efficacy. PLoS ONE. 2011;6(9) doi: 10.1371/journal.pone.0023243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anton PA, Cranston RD, Kashuba ADM, Hendrix CW, Bumpus N, Richardson-Harman N, et al. RMP-02/MTN-006: A Phase 1 Rectal Safety, Acceptability, Pharmacokinetic, and Pharmacodynamic Study of Tenofovir 1% Gel Compared with Oral Tenofovir Disoproxil Fumarate. AIDS Res Hum Retroviruses. 2012;28(11):1412–1421. doi: 10.1089/aid.2012.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mcgowan I, Cranston RD, Duffill K, Siegel A, Engstrom JC, Nikiforov A, et al. A Phase 1 Randomized, Open Label, Rectal Safety, Acceptability, Pharmacokinetic, and Pharmacodynamic Study of Three Formulations of Tenofovir 1% Gel (the CHARM-01 Study) PLoS ONE. 2015;10(5):e0125363. doi: 10.1371/journal.pone.0125363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Telwatte S, Moore K, Johnson A, Tyssen D, Sterjovski J, Aldunate M, et al. Virucidal Activity of the Dendrimer Microbicite SPL7013 Against HIV-1. Antiviral Res. 2011;90(3):195. doi: 10.1016/j.antiviral.2011.03.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.das Neves J, Michiels J, Arien KK, Vanham G, Amiji M, Bahia MF, et al. Polymeric Nanoparticles Affect the Intracellular Delivery, Antiretroviral Activity an Cytotoxicity of the Microbicide Drug Candidate Dapivirine. Pharm Res. 2012;(29):1468. doi: 10.1007/s11095-011-0622-3. [DOI] [PubMed] [Google Scholar]
- 91.das Neves J, Araujo F, Andrade F, Michiels J, Arien KK, Vanham G, et al. In Vitro and Ex Vivo Evaluation of polymeric Nanoparticles for Vaginal and Rectal Delivery of Anti-HIV Drug Dapivirine. Mol Pharmaceutics. 2013;10:2793. doi: 10.1021/mp4002365. [DOI] [PubMed] [Google Scholar]
- 92.Sarmento B, das Neves J. Nanosystem formulations for rectal microbicides: a call for more research. Therapeutic Delivery. 2012;3(1):1–4. doi: 10.4155/tde.11.139. [DOI] [PubMed] [Google Scholar]
- 93.Ramana LN, Anand AR, Sethuranam S, Krishnan UM. Targeting strategies for delivery of anti-HIV drugs. J Control Release. 2014;192:271–283. doi: 10.1016/j.jconrel.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wong HL, Chattopadhyay N, Wu XY, Bendayan R. Nanotechnology applications for improved delivery of antiretroviral drugs to the brain. Adv Drug Deliv Rev. 2010;62(4–5):503–517. doi: 10.1016/j.addr.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 95.Gupta U, Jain NK. Non-polymeric nano-carriers in HIV/AIDS drug delivery and targeting. Adv Drug Deliv Rev. 2010;62(4–5):478–490. doi: 10.1016/j.addr.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 96.Date AA, Destache CJ. A review of nanotechnological approaches for the prophylaxis of HIV/AIDS. Biomaterials. 2013;34(26):6202–6228. doi: 10.1016/j.biomaterials.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.das Neves J, Amiji MM, Bahia MF, Sarmento B. Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv Drug Deliv Rev. 2010;62(4–5):458–477. doi: 10.1016/j.addr.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 98.Ananworanich J, Schuetz A, Vandergeeten C, Sereti I, de Souza M, Rerknimitr R, et al. Impact of Multi-Targeted Antiretroviral Treatment on Gut T Cell Depletion and HIV Reservoir Seeding during Acute HIV Infection. PLoS ONE. 2012;7(3):e33948. doi: 10.1371/journal.pone.0033948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2012;64:557–570. doi: 10.1016/j.addr.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Willits RK, Saltzman WM. Synthetic polymers alter the structure of cervical mucus. Biomaterials. 2001;22:445–452. doi: 10.1016/s0142-9612(00)00197-6. [DOI] [PubMed] [Google Scholar]
- 101.Norris DA, Sinko PJ. Effect of size, surface charge, and hydrophobicity on the translocation of polystyrene microspheres through gastrointestinal mucin. J Appl Polym Sci. 1997;63(11):1481–1492. [Google Scholar]
- 102.Norris DA, Puri N, Sinko PJ. The effect of physical barriers and properties on the oral absorption of particulates. Adv Drug Deliv Rev. 1998;34(2–3):135–154. doi: 10.1016/s0169-409x(98)00037-4. [DOI] [PubMed] [Google Scholar]
- 103.Pridgen EM, Alexis F, Kuo TT, Levy-Nissenbaum E, Karnik R, Blumberg RS, et al. Transepithelial Transport of Fc-Targeted Nanoparticles by the Neonatal Fc Receptor for Oral Delivery. Nanomedicine. 2013;5(213):213ra167. doi: 10.1126/scitranslmed.3007049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nature Reviews: Drug Discovery. 2007;6:231–248. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 105.Harde H, Das M, Jain S. Solid lipid nanoparticles: an oral bioavailability enhancer vehicle. Expert Opinion on Drug Delivery. 2011;8(11):1407–1424. doi: 10.1517/17425247.2011.604311. [DOI] [PubMed] [Google Scholar]
- 106.Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. European Journal of Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical Sciences. 2011;42(1–2):11–18. doi: 10.1016/j.ejps.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 107.Ravi PR, Vats R, Dalal V, Murthy AN. A hybrid design to optimize preparation of lopinavir loaded solid lipid nanoparticles and comparative pharmacokinetic evaluation with marketed lopinavir/ritonavir coformulation. The Journal of Pharmacy and Pharmacology. 2014;66(7):912–926. doi: 10.1111/jphp.12217. [DOI] [PubMed] [Google Scholar]
- 108.Negi JS, Chattopadhyay P, Sharma AK, Ram V. Development of solid lipid nanoparticles (SLNs) of lopinavir using hot self nano-emulsification (SNE) technique. European Journal of Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical Sciences. 2013;48(1–2):231–239. doi: 10.1016/j.ejps.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 109.Bunjes H. Lipid nanoparticles for the delivery of poorly water-soluble drugs. The Journal of pharmacy and pharmacology. 2010;62(11):1637–1645. doi: 10.1111/j.2042-7158.2010.01024.x. [DOI] [PubMed] [Google Scholar]
- 110.Chiappetta DA, Hocht C, Taira C, Sosnik A. Oral pharmacokinetics of the anti-HIV efavirenz encapsulated within polymeric micelles. Biomaterials. 2011;32(9):2379–2387. doi: 10.1016/j.biomaterials.2010.11.082. [DOI] [PubMed] [Google Scholar]
- 111.McDonald TO, Giardiello M, Martin P, Siccardi M, Liptrott NJ, Smith D, et al. Antiretroviral solid drug nanoparticles with enhanced oral bioavailability: production, characterization, and in vitro-in vivo correlation. Advanced Healthcare Materials. 2014;3(3):400–411. doi: 10.1002/adhm.201300280. [DOI] [PubMed] [Google Scholar]
- 112.Dembri A, Montisci MJ, Gantier JC, Chacun H, Ponchel G. Targeting of 3′-azido 3′-deoxythymidine (AZT)-loaded poly(isohexylcyanoacrylate) nanospheres to the gastrointestinal mucosa and associated lymphoid tissues. Pharm Res. 2001;18(4):467–473. doi: 10.1023/a:1011050209986. [DOI] [PubMed] [Google Scholar]
- 113.Kinman L, Brodie SJ, Tsai CC, Bui T, LArsen K, Schmidt A, et al. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: a proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr. 2003;34(4):387–397. doi: 10.1097/00126334-200312010-00005. [DOI] [PubMed] [Google Scholar]
- 114.Kinman L, Bui T, Larsen K, Tsai CC, Anderson D, Morton WR, et al. Optimization of Lipid-Indinavir Complexes for Localization in Lymphoid Tissues of HIV-Infected Macaques. J Acquir Immune Defic Syndr. 2006;42(2):155–161. doi: 10.1097/01.qai.0000214822.33905.87. [DOI] [PubMed] [Google Scholar]
- 115.Duan J, Freeling JP, Koehn J, Shu C, Ho RJ. Evaluation of atazanavir and darunavir interactions with lipids for developing pH-responsive anti-HIV drug combination nanoparticles. J Pharm Sci. 2014;103(8):2520–2529. doi: 10.1002/jps.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Freeling JP, Koehn J, Shu C, Sun J, Ho RJ. Long-acting three-drug combination anti-HIV nanoparticles enhance drug exposure in primate plasma and cells within lymph nodes and blood. AIDS. 2014;28(17):2625–2627. doi: 10.1097/QAD.0000000000000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gunaseelan S, Gunaseelan K, Deshmukh M, Zhang X, Sinko PJ. Surface modifications of nanocarriers for effective intracellular delivery of anti-HIV drugs. Adv Drug Deliv Rev. 2010;62(4–5):518–531. doi: 10.1016/j.addr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou N, Luo Z, Luo J, Fan X, Cayabyab M, Hiraoka M, et al. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J Biol Chem. 2002;277(20):17476–17485. doi: 10.1074/jbc.M202063200. [DOI] [PubMed] [Google Scholar]
- 119.Xu Y, Duggineni S, Espitia S, Richman DD, An J, Huang Z. A synthetic bivalent ligand of CXCR4 inhibits HIV infection. Biochem Biophys Res Commun. 2013;435(4):646–650. doi: 10.1016/j.bbrc.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tamamura H, Omagari A, Oishi S, Kanamoto T, Yamamoto N, Peiper SC, et al. Pharmacophore Identification of a Specific CXCR4 Inhibitor, T140, Leads to Development of Effective Anti-HIV Agents with Very High Selectivity Indexes. Bioorg Med Chem Lett. 2000;10:2633–2637. doi: 10.1016/s0960-894x(00)00535-7. [DOI] [PubMed] [Google Scholar]
- 121.Inokuchi E, Oishi S, Kubo T, Ohno H, Shimura K, Matsuoka M, et al. Potent CXCR4 Antagonists Containing Amidine Type Peptide Bond Isosteres. ACS Med Chem Lett. 2011;2:477–480. doi: 10.1021/ml200047e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tamamis P, Floudas CA. Molecular recognition of CCR5 by an HIV-1 gp120 V3 loop. PLoS ONE. 2014;9(4):e95767. doi: 10.1371/journal.pone.0095767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bellows ML, Taylor MS, Cole PA, Shen L, Siliciano RF, Fung HK, et al. Discovery of entry inhibitors for HIV-1 via a new de novo protein design framework. Biophys J. 2010;99(10):3445–3453. doi: 10.1016/j.bpj.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dutta T, Agashe HB, Garg M, Balakrishnan P, Kabra M. Poly (propyleneimine) dendrimer based nanocontainers for targeting of efavirenz to human monocytes/macrophages in vitro. J Drug Target. 2007;15(1):89–98. doi: 10.1080/10611860600965914. [DOI] [PubMed] [Google Scholar]
- 125.Pooyan S, Qui B, Chan MM, Fong D, Sinko PJ, Leibowitz MJ, et al. Conjugates bearing multiple formyl-methionyl peptides display enhanced binding to but not activation of phagocytic cells. Bioconj Chem. 2002;13(2):216–223. doi: 10.1021/bc0100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen P, Zhang X, Jia L, Prud’homme RK, Szekely Z, Sinko PJ. Optimal structural design of mannosylated nanocarriers for macrophage targeting. J Control Release. 2014;194:341–349. doi: 10.1016/j.jconrel.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.D’Addio SM, Baldassano S, Shi L, Cheung L, Adamson DH, Bruzek M, et al. Optimization of cell receptor-specific targeting through multivalent surface decoration of polymeric nanocarriers. J Control Release. 2013;168(1):41–49. doi: 10.1016/j.jconrel.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nowacek AS, Miller RL, McMillan J, Kanmogne G, Kanmogne M, Mosley RL, et al. NanoART synthesis, characterization, uptake, release and toxicology for human monocyte-macrophage drug delivery. Nanomedicine (Lond) 2009;4(8):903–917. doi: 10.2217/nnm.09.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Magnani M, Rossi L, Fraternale A, Silvotti L, Quintavalla F, Piedimonte G, et al. Feline immunodeficiency virus infection of macrophages: in vitro and in vivo inhibition by dideoxycytidine-5′-triphosphate-loaded erythrocytes. AIDS Res Hum Retroviruses. 1994;10(9):1179–1186. doi: 10.1089/aid.1994.10.1179. [DOI] [PubMed] [Google Scholar]
- 130.Dou H, Grotepas CB, McMillan JM, Destache CJ, Chaubal M, Werling J, et al. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J Immunol. 2009;183(1):661–669. doi: 10.4049/jimmunol.0900274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martinez-Skinner AL, Veerubhotla RS, Liu H, Xiong H, Yu F, McMillan JM, et al. Functional proteome of macrophage carried nanoformulated antiretroviral therapy demonstrates enhanced particle carrying capacity. J Proteome Res. 2013;12(5):2282–2294. doi: 10.1021/pr400185w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, et al. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458(7242):1180–1184. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Steinbach JM. Protein and oligonucleotide delivery systems for vaginal microbicides against viral STIs. Cell Mol Life Sci. 2015;72:469–503. doi: 10.1007/s00018-014-1756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang HB, Mo QH, Yang Z. HIV vaccine research: the challenge and the way forward. J Immunol Res. 2015;2015:503978. doi: 10.1155/2015/503978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jardine J, Julien JP, Menis S, Ota T, Kalyuzhniy O, McGuire A, et al. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340(6133):711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hütter G, Nowak D, Mossner M, Ganepola S, Müßig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360(7):692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 137.Peterson CW, Younan P, Jerome KR, Kiem HP. Combinational anti-HIV gene therapy: using a multipronged approach to reach beyond HAART. Gene Ther. 2013;20(7):695–702. doi: 10.1038/gt.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. 2014;111(31):11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLOS Pathog. 2014;10(10):e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Williams J, Sayles HR, Meza JL, Sayre P, Sandkovsky U, Gendelman HE, et al. Long-acting parenteral nanoformulated antiretroviral therapy: interest and attitudes of HIV-infected patients. Nanomedicine (Lond) 2013;8:1807–1813. doi: 10.2217/nnm.12.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Swindells S, Flexner C, Fletcher CV, Jacobson JM. The critical need for alternative antiretroviral formulations and obstacles to their development. J Infect Dis. 2011;204:669–674. doi: 10.1093/infdis/jir370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Adams CE, Fenton MKP, Quraishi S, David AS. Systematic meta-review of depot antipsychotic drugs for people with schizophrenia. Brit J Psychiatry. 2001;179:290–299. doi: 10.1192/bjp.179.4.290. [DOI] [PubMed] [Google Scholar]
- 143.Westhoff C. Depot-medroxyprogesterone acetate injection (Depo-Provera): a highly effective contraceptive option with proven long-term safety. Contraception. 2003;68:75–87. doi: 10.1016/s0010-7824(03)00136-7. [DOI] [PubMed] [Google Scholar]
- 144.Dolgin E. Long-acting HIV drugs advanced to overcome adherence challenge. Nature Med. 2014;20:323–324. doi: 10.1038/nm0414-323. [DOI] [PubMed] [Google Scholar]
- 145.Flexner C, Saag M. The antiretroviral drug pipeline: prospects and implications for future treatment research. Curr Opin HIV/AIDS. 2013;8:572–578. doi: 10.1097/COH.0000000000000011. [DOI] [PubMed] [Google Scholar]
- 146.Jackson A, Else L, Tjia J, et al. Rilpivirine-LA formulation: pharmacokinetics in plasma, genital tract in HIV-females and rectum in males [Abstract no. 35]. 19th Conference on Retroviruses and Opportunistic Infections; Seattle WA. 2012. [Google Scholar]
- 147.Andrews CD, Spreen WR, Mohri H, Moss L, Ford S, Gettie A, et al. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science. 2014;343(6175):1151–1154. doi: 10.1126/science.1248707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Andrews CD, Yueh YL, Spreen WR, St Bernard L, Boente-Carrera M, Rodriguez K, et al. A long-acting intergrase inhibitor protects female macaques from repeated high-dose intravaginal SHIV challenge. Sci Transl Med. 2015;7(270):270ra4. doi: 10.1126/scitranslmed.3010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Radzio J, Spreen W, Yueh YL, Mitchell J, Jenkins L, García-Lerma JG, et al. The long-acting integrase inhibitor GSK744 protects macagues from repeated intravaginal SHIC challenge. Sci Transl Med. 2015;7(270):270ra5. doi: 10.1126/scitranslmed.3010297. [DOI] [PubMed] [Google Scholar]
- 150.Rajoli RK, Back DJ, Rannard S, Freel Meyers CL, Flexner C, Owen A, et al. Physiologically Based Pharmacokinetic Modeling to Inform Development of Intramuscular Long-Acting Nanoformulations for HIV. Clin Pharmacokinet. 2015;54(6):639–650. doi: 10.1007/s40262-014-0227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]