

Abstract
Purpose of Review:
This article presents a practical and informative approach to the evaluation of a patient with a rapidly progressive dementia (RPD).
Recent Findings:
Prion diseases are the prototypical causes of RPD, but reversible causes of RPD might mimic prion disease and should always be considered in a differential diagnosis. Aside from prion diseases, the most common causes of RPD are atypical presentations of other neurodegenerative disorders, curable disorders including autoimmune encephalopathies, as well as some infections, and neoplasms. Numerous recent case reports suggest dural arterial venous fistulas sometimes cause RPDs.
Summary:
RPDs, in which patients typically develop dementia over weeks to months, require an alternative differential than the slowly progressive dementias that occur over a few years. Because of their rapid decline, patients with RPDs necessitate urgent evaluation and often require an extensive workup, typically with multiple tests being sent or performed concurrently. Jakob-Creutzfeldt disease, perhaps the prototypical RPD, is often the first diagnosis many neurologists consider when treating a patient with rapid cognitive decline. Many conditions other than prion disease, however, including numerous reversible or curable conditions, can present as an RPD. This chapter discusses some of the major etiologies for RPDs and offers an algorithm for diagnosis.
INTRODUCTION
Neurologists generally are familiar with the differential diagnoses of slowly progressive neurodegenerative dementias, many of which are discussed individually in this issue of Continuum. The general approach to a patient with dementia is discussed in the article “The Mental Status Examination in Patients With Suspected Dementia” by Murray Grossman, MD, FAAN, and David J. Irwin, MD,1 in this issue of Continuum, but the diagnosis of rapidly progressive dementias (RPDs) entails a different diagnostic approach. Although there is no clear definition for the time frame of an RPD, the author typically uses the term to refer to conditions that progress from onset of first symptom to dementia (decline in more than one cognitive domain with functional impairment) in less than 1 to 2 years, although most occur over weeks to months. This article presents some of the major adult-onset causes of RPDs and proposes some diagnostic algorithms.
FINDINGS FROM JAKOB-CREUTZFELDT DISEASE AND RAPIDLY PROGRESSIVE DEMENTIA REFERRAL CENTERS
Perhaps the prototypical RPDs are prion diseases, such as Jakob-Creutzfeldt disease, which was covered in detail in the article “Prion Diseases” by Michael D. Geschwind, MD, PhD,2 in the December 2015 Continuum issue and thus is not discussed in detail in this article. (As discussed in the December 2015 article, Jakob-Creutzfeldt disease was commonly referred to for many decades as Jakob or Jakob-Creutzfeldt disease until Clarence J. Gibbs, a prominent researcher in the field, started using the term Creutzfeldt-Jakob.3 However, only two of five of Jakob’s cases and not even Creutzfeldt’s case had what we would now consider prion disease. Thus, in the author’s opinion, the disease should be called Jakob or Jakob-Creutzfeldt disease,4 which is what this article uses to discuss the disease.) Many patients referred to various national prion referral centers with suspected Jakob-Creutzfeldt disease turn out not to have prion disease. Over the past 14 years, more than 2500 suspected prion disease cases have been referred to the RPD program and dementia clinic at the University of California, San Francisco (UCSF). The distribution of diagnoses for these referrals through June 1, 2015, is shown in Figure 7-1A.5,6 Of all RPD cases referred to the UCSF center (Figure 7-1A), about one-fourth came in person for an inpatient or outpatient evaluation (Figure 7-1B); for those patients not evaluated in person, a detailed records review was conducted and communicated with the families, patients, or their physicians. As a center with expertise in prion diseases, there is a referral bias toward these diagnoses. At the UCSF center, the non–Jakob-Creutzfeldt disease/non–prion disease group comprises 25% of all referrals (both records review or in-person evaluation) and 44% of those evaluated in person. Figure 7-1B shows the diagnostic distribution for the subset of referred patients who were evaluated in person at our center over about the past 13 years. Many cases referred for potential sporadic Jakob-Creutzfeldt disease were found to have nonprion diagnoses when evaluated in person at our center.5
Figure 7-1.
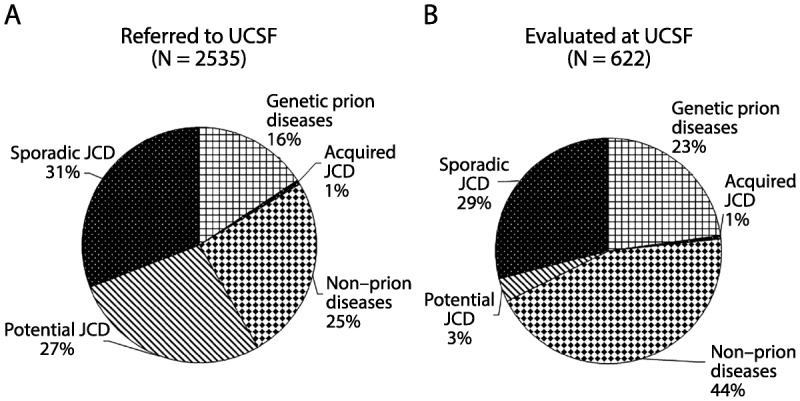
Major diagnostic categories of patients with rapidly progressive dementia (RPD) referred to, versus evaluated at, the University of California, San Francisco (UCSF) rapidly progressive dementia program over 13 years. A, Diagnostic distribution of patients with RPD referred to UCSF over about a 13-year period, most of whom had extensive medical record review, but only about one-fourth of whom were evaluated in person at UCSF. Almost one-third of cases referred to (as well as evaluated at) UCSF were diagnosed with sporadic Jakob-Creutzfeldt disease. In more than one-fourth of referred cases, although a sporadic Jakob-Creutzfeldt disease diagnosis (potential sporadic Jakob-Creutzfeldt disease) was suspected, not enough information existed to make a probable Jakob-Creutzfeldt disease diagnosis.5,6 Acquired Jakob-Creutzfeldt disease includes iatrogenic and infectious forms of prion disease. The genetic prion diseases category included patients who had confirmed mutations (autosomal dominant) in the prion protein gene, PRNP, or were from families with genetic prion disease. Whereas many of the genetic prion diseases presented similarly to sporadic Jakob-Creutzfeldt disease, as an RPD, a significant minority had clinical presentations more similar to other more slowly progressive diseases, such as Alzheimer disease or atypical parkinsonian or ataxic syndromes.2 One-fourth of cases were diagnosed with a nonprion etiology for their RPD. B, Diagnostic distribution of patients with RPD evaluated in person at UCSF. A larger percentage of nonprion RPDs and genetic prion diseases is evident; the latter is a bias partly because of the UCSF research program in genetic prion diseases and antibody-mediated encephalopathies.
JCD = Jakob-Creutzfeldt disease.
Table 7-1 shows the types of diagnoses of nonprion RPDs referred to UCSF, the German Jakob-Creutzfeldt disease surveillance unit, and the US National Prion Disease Pathology Surveillance Center (NPDPSC), based on publications from these three major prion referral centers.7,8 The UCSF cohort was during an approximately 8-year period and includes some previously published cases.5 In many cases, Jakob-Creutzfeldt disease was suspected by the referring physicians. Overall, the most common nonprion RPD diagnostic categories were nonprion neurodegenerative diseases. Neurologic autoimmune conditions were very common even in some of these cohorts, which are a few years old, predating the discovery of some of the more recently discovered antibodies associated with autoimmune encephalopathies. It is likely that antibody-mediated encephalopathies today are an even larger percentage of nonprion RPDs than shown in Table 7-1. Several patients from the UCSF center had leukoencephalopathies of unclear etiology. For reviews of these disorders, refer to the article “Autoimmune Encephalopathies and Dementias” by Andrew McKeon, MD,9 and to the article “Adult-Onset Leukoencephalopathies” by Deborah L. Renaud, MD,10 in this issue of Continuum. Despite CSF pleocytosis being extremely rare in prion disease, several patients with meningoencephalitis were referred as suspected Jakob-Creutzfeldt disease; in only a few patients with encephalitis was the agent identified (human immunodeficiency virus [HIV], Lyme disease, and enterovirus).5 In the German center publication, 34% of patients initially suspected of having Jakob-Creutzfeldt disease by referring physicians were ultimately found by pathology or clinical follow-up to have other diagnoses. Importantly, many patients had not only treatable, but potentially reversible conditions.
Table 7-1.
Diagnostic Breakdown of NonJakob-Creutzfeldt Disease Rapidly Progressive Dementia Referrals to Three Jakob-Creutzfeldt Disease Referral Centersa
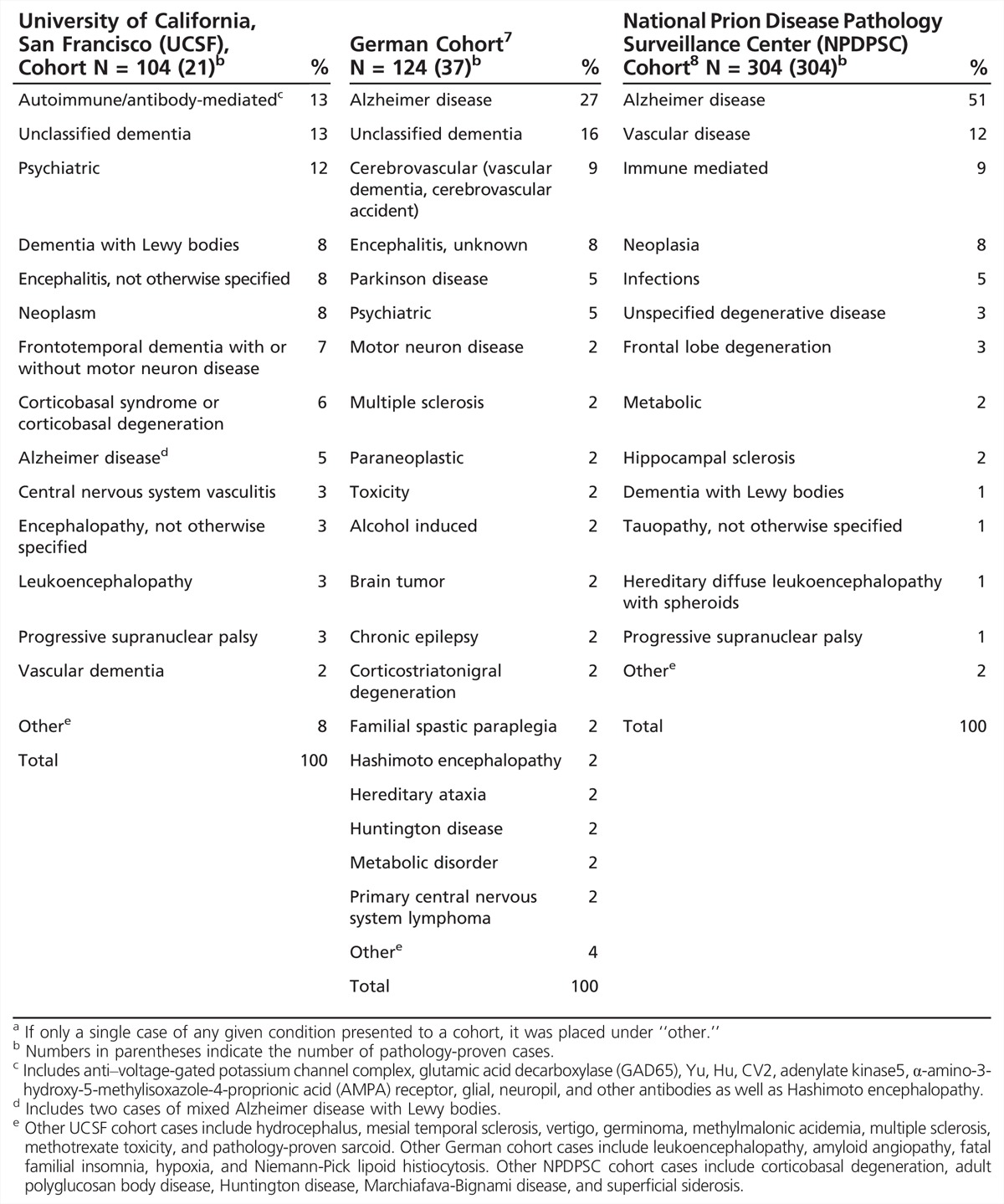
In the United States, to help monitor for potential new forms of human prion disease, the Centers for Disease Control and Prevention funds the NPDPSC to perform brain autopsies on all referred suspected prion cases. In an important study, they reviewed 1106 autopsies performed over about a 10-year period for those in whom prion disease was not identified (N = 352). Among the 304 in whom they confirmed another, nonprion pathologic diagnosis, 73% had what they deemed as “untreatable” conditions, but 23% had “treatable” (potentially curable) dementias.8 Of the 71 “treatable” cases they identified, 37% were immune mediated, 35% were neoplasms, 20% were infections, and 8% were metabolic disorders.8 Although one advantage of this study from the NPDPSC is that all cases were autopsy proven, this is also a shortcoming as they do not include patients with RPD who did not die or did not come to autopsy. This study also would not include RPD cases diagnosed premortem or those who might have recovered (from treatment or other reasons).
Two retrospective studies examined the causes of RPD in patients admitted to two European tertiary medical centers (Table 7-2).11,12 In the first, at a tertiary referral center in Greece, RPD was defined as progression to dementia from first symptom onset in less than 1 year. The authors of this study also excluded cases with acute cognitive impairment in the context of a confusional state due to acute infectious, metabolic, or toxic causes (eg, acute viral encephalitis, acute hyponatremia, hypoglycemia, and recent use of anticholinergic drugs). Over a 3-year period, they identified 68 subjects with RPD meeting their criteria. The largest single category were nonprion neurodegenerative diseases (47.1%), followed by secondary dementias (26%). Some of the larger single diagnoses were Alzheimer disease (AD) (17.6%), followed by frontotemporal dementia (FTD) (16.2%), and equally represented (among 13.2% of the cohort) vascular dementia, Jakob-Creutzfeldt disease, and various other neurodegenerative diseases (multiple system atrophy, dementia with Lewy bodies [DLB], Parkinson disease dementia, progressive supranuclear palsy, and corticobasal syndrome). Secondary dementias included four patients with normal pressure hydrocephalus, two with neurosyphilis, and one each with scleroderma, sarcoidosis, systematic lupus erythematosus, central nervous system (CNS) primary vasculitis, limbic encephalitis, HIV encephalitis, CNS tumor, Q fever, vitamin B12 deficiency, multiple sclerosis, drug-induced dementia, and dementia in the context of chronic psychosis. The mean age at onset ± standard deviation (SD) was 65.5 ± 10.0 (median 66.7; range 35.3 to 82.8) years of age. For non–Jakob-Creutzfeldt disease causes, the mean time to dementia was about 8 (range 1 to 11) months, but for Jakob-Creutzfeldt disease it was 3 (range 1 to 5) months.11
Table 7-2.
Causes of Rapidly Progressive Dementia in Two European Tertiary Care Hospitals
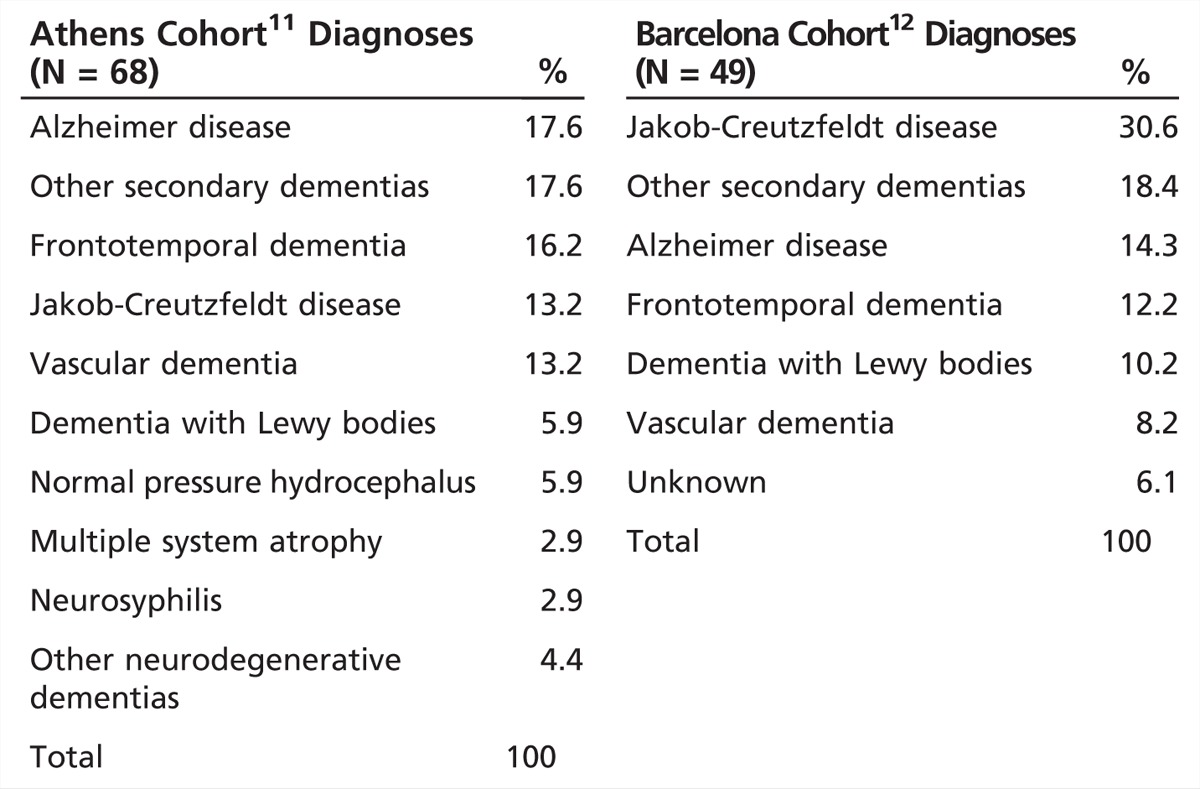
The second retrospective study reviewed all admissions for RPD from 1994 to 2009 at a major hospital in Barcelona, Spain (Table 7-2).12 The study authors applied similar diagnostic criteria to the study from Athens, Greece.11 Among 49 subjects identified, similar to the Athens study, the largest group was nonprion neurodegenerative diseases (36.8%), but Jakob-Creutzfeldt disease was the second most common (30.6%), followed by secondary dementias (18.4%). Regarding the distribution of each of the nonprion neurodegenerative diseases in the cohort, 14% were AD, 12% were FTD, and 10% were DLB. The percent due to vascular disease was 8.25%, not too dissimilar from the Athens cohort.11 The mean age of onset was 72.4 ± 11.6 (range 41 to 86) years of age, and the time from onset to evaluation was 4.6 ± 3.8 (range 1 to 12) months. Among the 19 cases followed to death, mean survival time was 8.6 ± 9.5 months overall, but the prion cases had significantly shorter survival than the nonprion cases, of 3.3 ± 2.4 (range 1 to 8) versus 14 ± 11.1 (range 5 to 34) months (P=0.04).12
DIAGNOSTIC APPROACH TO RAPIDLY PROGRESSIVE DEMENTIAS
The diagnostic approach for evaluating standard dementia has been presented in the article “The Mental Status Examination in Patients With Suspected Dementia” by Murray Grossman, MD, FAAN, and David J. Irwin, MD,1 in this issue of Continuum. For evaluating patients with RPDs, many of the same principles apply, but because of the rapidity of decline, and as many causes of RPD are treatable, it is essential to have a systematic, comprehensive approach to the diagnostic plan.
Initial Evaluation of a Rapidly Progressive Dementia
RPDs are rather rare and can be difficult to diagnose. Establishing the time course through a thorough history is a critical first step to the evaluation of an RPD. It is important to rule out delirium in any initial evaluation. The first symptoms of a dementia can help with diagnosis. This is particularly true of the neurodegenerative dementias, which often start in specific neuroanatomic regions.13,14 Autoimmune encephalopathies early on often affect the limbic system and, thus, typically present with memory loss or behavioral changes. Viral encephalopathies or acute demyelinating encephalomyelitis may be preceded by a flulike illness. The evaluation of an RPD typically occurs in stages in which several tests are performed in parallel. For an initial RPD evaluation, recommended blood, urine, CSF, imaging, and other tests are shown in Table 7-3.15 Not all of these tests will need to be done, depending on the clinical scenario and the results of tests that have already returned.
Table 7-3.
Recommended Initial Screening Tests for Evaluation of a Rapidly Progressive Dementia
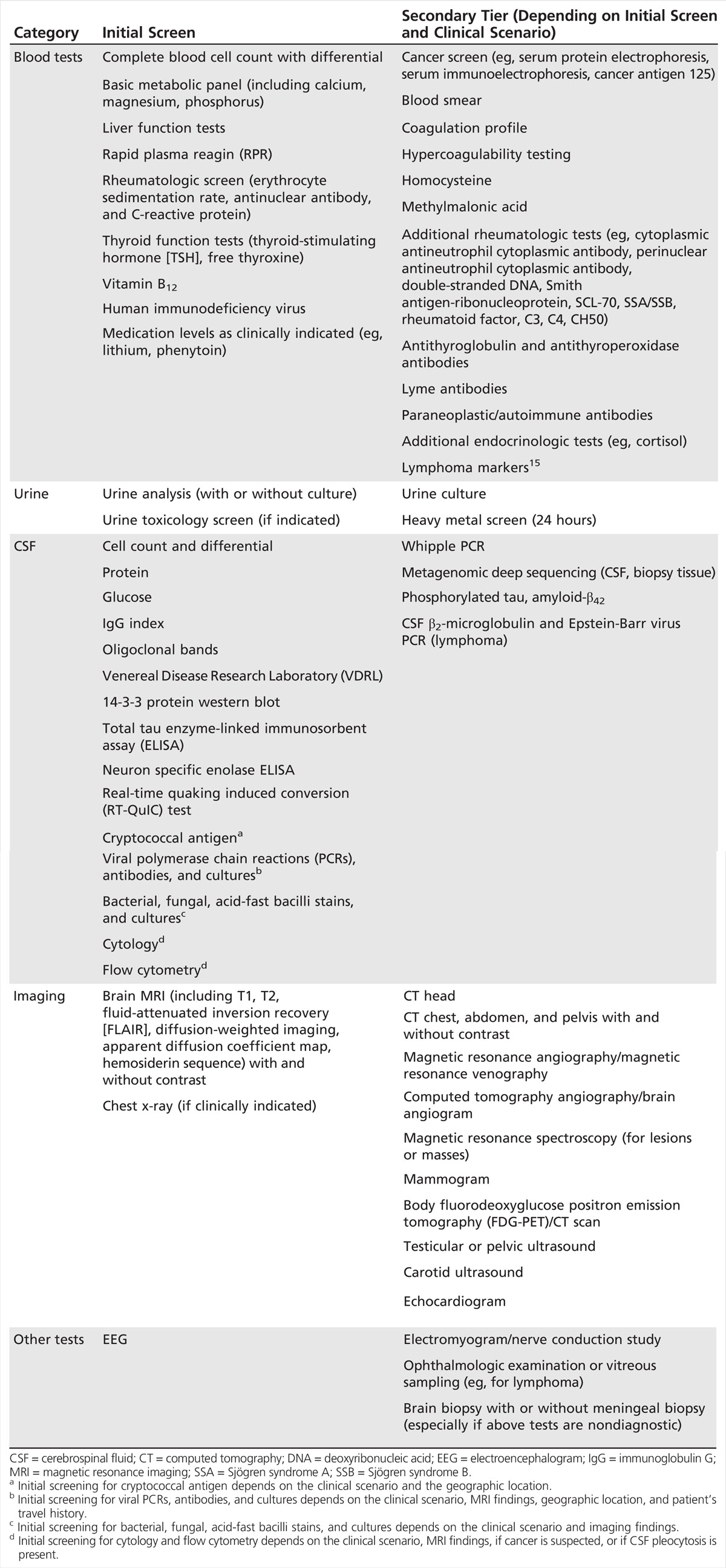
Ruling out a simple metabolic perturbation or infection is particularly important, especially in more susceptible persons, such as elderly patients or those with a preexisting mild dementia or cognitive impairment, as these patients sometimes experience a rapid decline in these circumstances. Simple blood tests and urine analysis, to rule out common toxic-metabolic causes, should be done. If seizures (including nonconvulsive status epilepticus) are possible, an EEG should be performed. Brain imaging usually is done prior to CSF analysis as it might direct what CSF tests are sent, as well as rule out a space-occupying lesion. Certain treatable or reversible conditions, such as autoimmune diseases, HIV, or other infections, depending on the geographic location, should always be considered. HIV can occur in anyone; there should be a low threshold for testing for treatable conditions, even if the likelihood is low. Although American Academy of Neurology (AAN) guidelines currently do not recommend rapid plasma reagin (RPR) as a screening test for dementia, at the UCSF center the author prefers to send this test, as it is a relatively inexpensive test for a treatable dementia. CSF pleocytosis or elevated CSF IgG index or oligoclonal bands might indicate an autoimmune or inflammatory process, such as paraneoplastic or other neuroimmunologic conditions. Although oligoclonal bands or elevated IgG index can occur in Jakob-Creutzfeldt disease,2 their presence probably should prompt an autoimmune and paraneoplastic evaluation. In the author’s UCSF Jakob-Creutzfeldt disease–RPD cohort, several patients with RPD were identified who had either an elevated IgG index or oligoclonal bands and the presence of novel antineuronal antibodies in their serum and/or CSF. Although some patients had cancer, in many, no cancers were identified despite thorough evaluation.5,16,17 At the author’s center, sending the CSF biomarkers 14-3-3, total tau (t-tau), and neuron-specific enolase is always recommended, not necessarily as diagnostic tests for prion disease, because they are not,2 but rather as markers of rapid neuronal injury. If these biomarkers are elevated, they help confirm the history of a rapidly progressive neurologic condition. If prion disease is in the differential, the CSF real-time quaking induced conversion (RT-QuIC) test that detects prion seeding activity should be performed.2 If AD is a consideration, consider sending CSF for phosphorylated tau (p-tau) and amyloid-β42 (Aβ42). As Aβ42 sticks very strongly to regular plastic (lowering the measured value of the enzyme-linked immunosorbent assay [ELISA]), only polypropylene tubes should be used for collection; the CSF for that test should not touch regular plastic tubes or the manometer found in lumbar puncture trays. For a patient with an undiagnosed RPD after the initial workup, a body CT scan with and without contrast should be performed for malignancy, sarcoid, or other etiology. If a body CT is not initially feasible, start with a chest x-ray. If paraneoplastic antibodies are positive, depending on the specific antibody identified, an aggressive cancer workup including fluorodeoxyglucose positron emission tomography (FDG-PET)/CT and various ultrasounds might be indicated.
Additional, or second-tier, tests that may be considered, depending on the results of the initial screen or the clinical scenario, are shown in Table 7-3. These include additional blood work, urine testing, CSF analysis, brain and body imaging, and other tests. With the exception of lymphomas, RPDs that present with space-occupying brain masses are easily identified by CT or MRI scan, and the details of most of these disorders are omitted from this chapter.
CONSIDERATION OF RAPIDLY PROGRESSIVE DEMENTIAS BY ETIOLOGIC CATEGORY
Several RPDs come under more than one etiologic category; paraneoplastic antibody-mediated encephalopathies, for example, are both autoimmune and neoplastic. In addition, not all conditions mentioned present as dementia, but may have other rapidly progressive neurologic signs, such as ataxia or chorea. One useful mnemonic device for the differential evaluation of RPDs is the acronym VITAMINS, which stands for vascular, infectious, toxic-metabolic, autoimmune, malignancy, iatrogenic, neurodegenerative, and systemic (as well as seizures and sarcoid) etiologies (Table 7-4). The following sections discuss several nonprion RPDs by their diagnostic category or etiology, in the order of the mnemonic VITAMINS. A diagnostic algorithm for RPDs is shown in Figure 7-2.
Table 7-4.
Partial Differential Diagnosis for Rapidly Progressive Dementias by Etiologic Category

Figure 7-2.
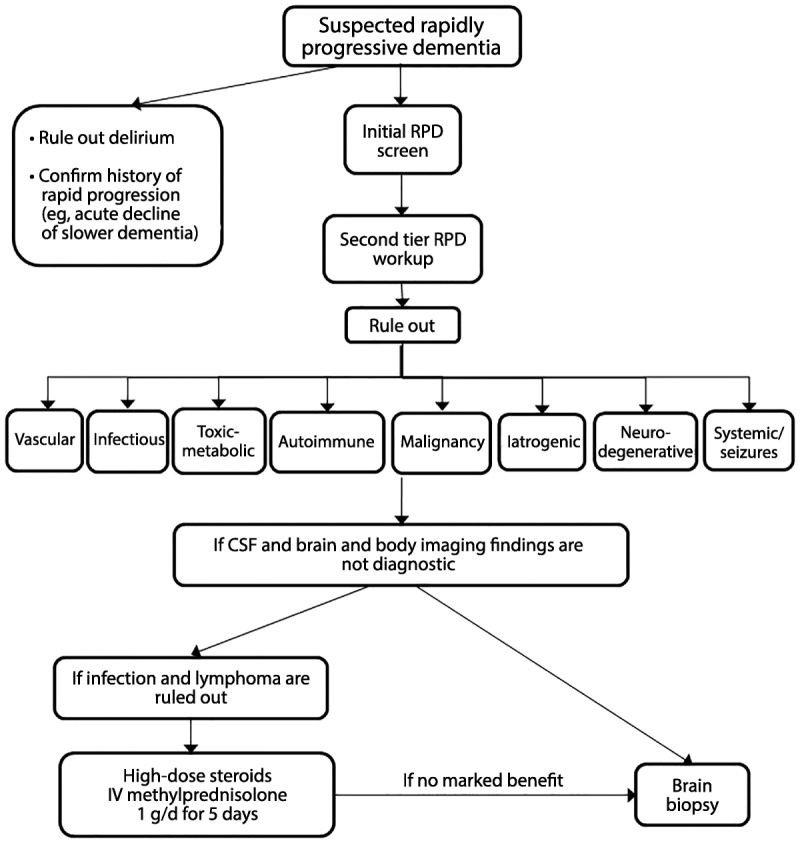
Algorithm for evaluating rapidly progressive dementia. Refer to the text and Table 7-3 for details about the workup for each diagnostic category.
CSF = cerebrospinal fluid; IV = intravenous; RPD = rapidly progressive dementia.
Vascular
As noted in Table 7-2 and Table 7-4, numerous vascular conditions can cause RPDs, including strokes or multiple infarcts, cerebral amyloid angiopathy, dural arteriovenous fistulas (DAVFs), and hypertensive encephalopathy.18–22 CNS vasculitis and intravascular lymphoma might also be considered under vascular etiologies, although they also might be considered under autoimmune and malignancy, respectively. Any vascular disruption of the thalamus, especially if bilateral involvement, such as through venous thrombosis, can result in an RPD. Even small focal strokes, particularly in the thalamus, can cause acute dementia.23,24 Diffusion-weighted imaging (DWI) and gradient echo (hemosiderin-sensitive sequence) MRI as well as vascular imaging such as magnetic resonance angiography (MRA), magnetic resonance venography (MRV), CT angiography, and conventional angiography can help diagnose vascular causes of RPD. At least 20 cases of DAVFs causing dementia, most presenting as RPDs, have been reported in the literature. Most patients presented with headache, progressive confusion, and memory loss, which progressed rapidly over the course of 2 weeks to 12 months. Some patients developed seizures, gait instability, or some focal neurologic deficit such as aphasia, facial palsy, or hemiparesis. In some cases, a bruit was heard over the skull. In all 20 cases, diagnosis was made by angiogram, and treatments such as surgery alone (one patient), embolization and surgery (two patients), or embolization alone (17 patients) improved cognitive function profoundly in all cases except one, who only showed minimal improvement and died of brain herniation after embolization. Mean age is 60 (range 55 to 77) years. Duration from onset to RPD ranges from about 2 months to just over 12 months.19–22,25–28 Common MRI findings are diffuse high-signal intensities in the cerebral or cerebellar white matter along with enlarged vessels over the hemispheric surface on T2-weighted images (Figure 7-3). Basal ganglia hyperintensity has been, albeit rarely, reported.19 Considering the reversibility of clinical symptoms, it is critical to consider DAVF in a patient presenting with RPD and MRI showing diffuse white matter lesions.
Figure 7-3.
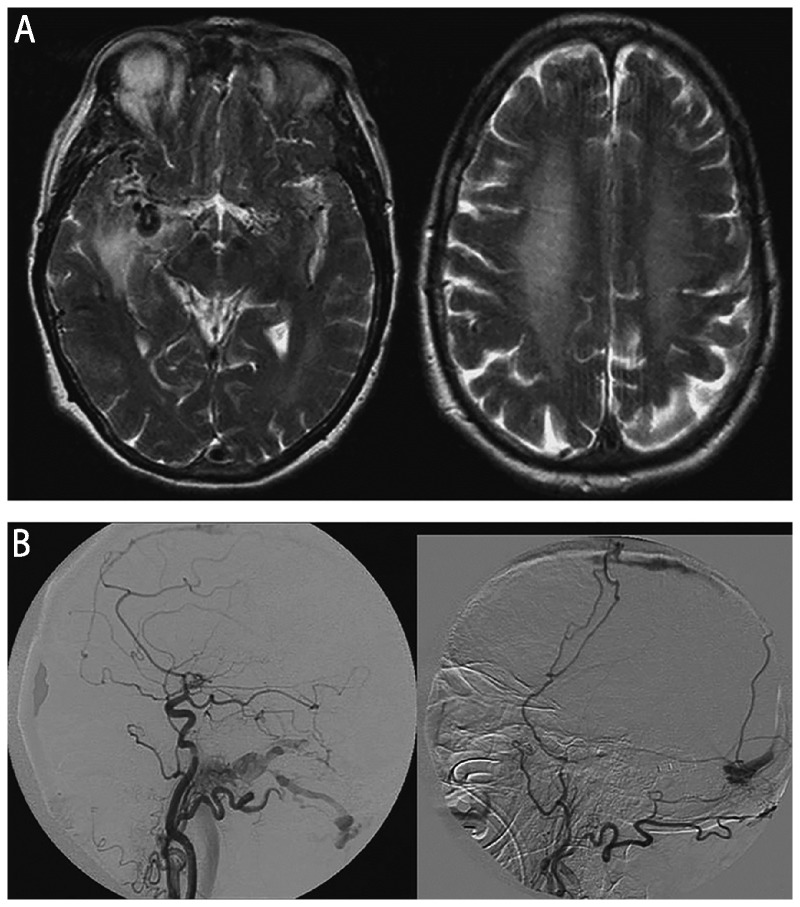
Imaging of a dural arteriovenous fistula (DAVF). A 70-year-old woman developed amnesia, aphasia, incoherent speech, and progressive gait disturbance over 2 weeks, progressing over the next 3 weeks to develop myoclonus with a startle reflex, flaccid tetraparesis with brisk deep tendon reflexes, extensor plantar responses, pronounced primitive reflexes, and akinetic mutism. Initial diagnosis was Jakob-Creutzfeldt disease until brain imaging revealed a DAVF. A, Axial T2-weighted MRI shows multiple dilated vessels in the temporal regions, predominantly in the right temporal region with venous ectasia and hyperintensity of the white matter including the centrum semiovale. B, Digital subtraction angiogram (right common carotid injection [left panel]) and left external carotid injection [right panel]), lateral views, shows multiple DAVFs of the superior sagittal sinus, torcula, and the lateral sinuses.Reprinted with permission from Mendonça N, et al, Neurologist.21journals.lww.com/theneurologist/pages/articleviewer.aspx?year=2012&issue=05000&article=00005&type=Abstract.© 2012 Lippincott Williams & Wilkins, Inc.
Venous sinus thrombosis often manifests with headaches, papilledema, visual loss, seizures, focal neurologic deficits, altered mental status, or coma depending on the location of the thrombus. Brain MRI shows cerebral edema and venous infarction, and CT venogram (CTV) or MRV can detect the thrombus. If the deep veins are thrombosed, there might be unilateral or symmetric basal ganglia involvement.29 Cerebral amyloid angiopathy involving small and medium-sized arteries in the brain may result in RPD.30 Hemosiderin sequences often show multiple large or small hemorrhages, often in areas in which there are T2-weighted white matter hyperintensities (Figure 7-431),32 particularly when there is cerebral amyloid angiopathy–related inflammation, which might be somewhat responsive to steroids.
Figure 7-4.
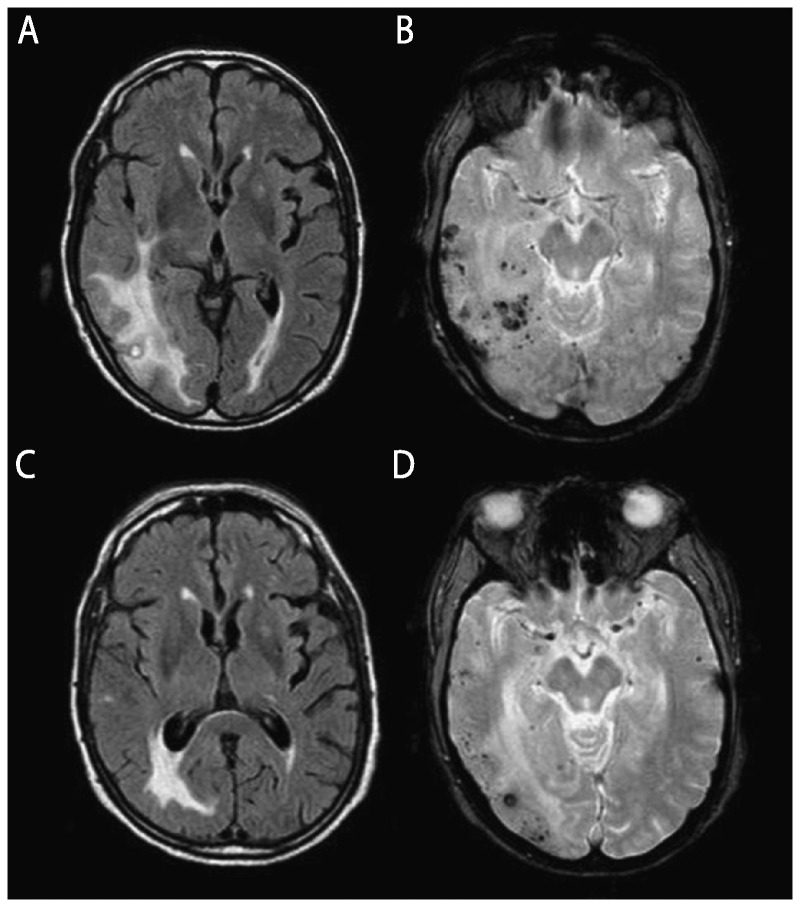
MRI findings of cerebral amyloid angiopathy–related inflammation. An elderly man presented with relatively acute onset left hemiparesis, left homonymous hemianopia, dysarthria, spatial and temporal disorientation, sensory aphasia, and psychomotor slowness. Cerebral amyloid angiopathy–related inflammation was suspected and the finding of APOE genotype ɛ4/ɛ4 supported the diagnosis. Anti–amyloid-β (Aβ) autoantibody concentration in CSF was elevated at 55.9 ng/mL. Physical therapy and corticosteroid therapy with dexamethasone 24 mg/d were started and the patient showed clinical improvement. Initial axial fluid-attenuated inversion recovery (FLAIR) MRI shows bilateral hyperintense lesions (A) and gradient recalled echo (GRE) image shows cortical and subcortical microhemorrhages (B). After 1 month of steroid therapy, FLAIR MRI (C) and GRE (D) sequence show reduction of both cerebral edema and microhemorrhages.
Modified with permission from Crosta F, et al, Case Rep Neurol Med.31www.hindawi.com/journals/crinm/2015/483020/. © 2015 Francesca Crosta et al.
Cerebroretinal microangiopathy with calcifications and cysts is a very rare condition that can manifest as RPD, more commonly in children. In one adult case, a 62-year-old man presented with a 2-month history of rapidly progressive cognitive decline, ataxia, spasticity, and seizures. CT showed multiple calcifications in the cerebral hemispheres, brainstem, and cerebellum and enhancement in the brain calcification areas on MRI. He died 3 years after onset without a definite diagnosis. Autopsy showed an adult-onset case of cerebroretinal microangiopathy with calcifications and cysts.33 Another rare vascular cause of RPD is subacute diencephalic angioencephalopathy, which is considered a severe form of posterior reversible encephalopathy syndrome (PRES). Approximately seven cases of subacute diencephalic angioencephalopathy have been reported to date and are characterized by acute cognitive changes with rapid progression to death, bilateral thalamic involvement, and profound vasculopathy with noninflammatory parenchymal necrosis. MRI typically shows nonenhancing confluent subcortical fluid-attenuated inversion recovery (FLAIR)/T2 hyperintensities in the occipital and temporal lobes and pons as well as the thalamus (Figure 7-5).34
Figure 7-5.
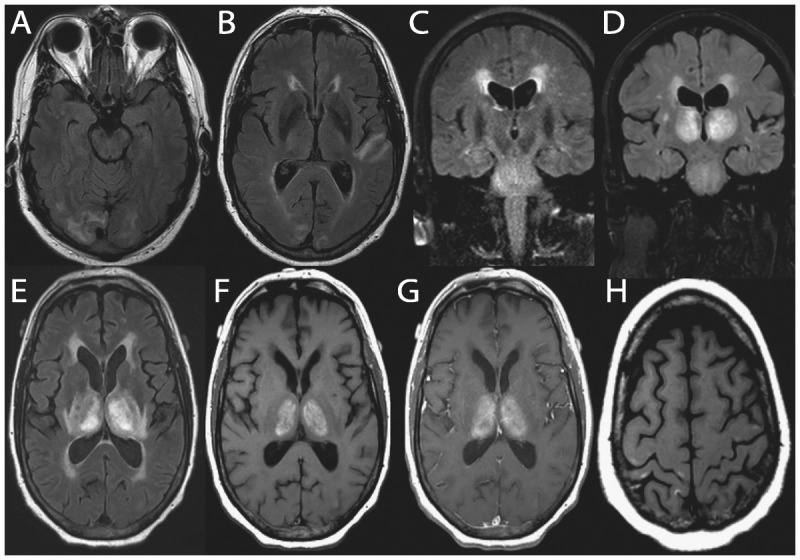
Brain MRI of a 68-year-old man with recent hyperintensive encephalopathy leading to subacute diencephalic angioencephalopathy. His first symptoms were hypernasal dysarthria and palatal weakness that resolved over 3 months after treatment of hypertension. Thirteen months after initial onset, he developed headaches, confusion, and speech and language problems due to hyperintensive encephalopathy, which again resolved with treatment. About 15 months after initial onset, he began a downward, but fluctuating course (with treatments), over 8 weeks, of a rapidly progressive neurologic decline leading to his death. Brain autopsy revealed findings consistent with subacute diencephalic angioencephalopathy. Nonenhancing, confluent subcortical axial fluid-attenuated inversion recovery (FLAIR) hyperintensities in the occipital lobes (A) and left temporal lobe (B) at the time of initial onset. Confluent nonenhancing abnormal coronal T2-weighted signal symmetrically in the pons, as well as confluent periventricular FLAIR signal, at 13 months after onset (C). One month later, imaging demonstrated bilaterally symmetric T1 and T2/FLAIR hyperintense abnormal signal in the thalami (D, coronal FLAIR; E, axial FLAIR). Thalamic abnormalities enhanced minimally following gadolinium administration on axial T1-weighted images (F, unenhanced; G, enhanced). Small T1-weighted (H) and T2-weighted hyperintense cortical foci were also seen.
Reprinted with permission from Graffeo CS, et al, J Clin Neurosci.34 www.jocn-journal.com/article/S0967-5868(15)00354-9/abstract. © 2015 Elsevier Ltd.
Infectious
Most infectious causes of encephalopathy have certain suggestive features, such as fever, pleocytosis, meningeal signs (if a meningoencephalitis), and have acute onset (eg, over days). CSF and imaging, however, may be normal. When viral encephalitis is suspected, depending on the clinical scenario, treatable conditions, such as herpes simplex virus, should be ruled out. With herpes simplex virus, serial CSF PCR may be needed. It is important to rule out HIV35,36 and, when clinically indicated, acquired immune deficiency syndrome (AIDS)-related CNS conditions such as toxoplasmosis, primary CNS lymphoma, and progressive multifocal leukoencephalopathy. Depending on the geographic location, Lyme disease or other local infectious agents, such as Balamuthia mandrillaris in California, most of which are treatable, should be considered.37 Fungal infections, such as aspergillosis in the CNS, may also cause RPD, although most commonly in immunocompromised patients. Although Whipple disease classically presents with gastrointestinal symptoms, lymphadenopathy, fever, and arthralgia, neurologic involvement occurs in 5% to 45% of cases (depending on the study) and can be the presenting symptom. Symptoms of CNS Whipple disease often include cognitive and psychiatric dysfunction, hemiparesis, seizures, and ataxia, and may present as an RPD. Although Whipple disease is rare, it is curable with antibiotics; when indicated, physicians should test for the bacterium Tropheryma whippelii by PCR from the blood, CSF, jejunal biopsy, or brain biopsy. Diagnosis may also be made by duodenal biopsy revealing periodic acid–Schiff positive macrophages in the lamina propria containing non–acid-fast Gram-positive bacilli.38 If the etiology of a probable infectious encephalopathy or encephalitis is not identified, both acute and convalescent serum and CSF should be saved to help with later identification of the infectious agent. In the United States, the Centers for Disease Control and Prevention, including several state health department encephalitis programs they fund, has divisions to help diagnose various encephalitides. These sites can help analyze acute and convalescent serum and CSF, which should be kept for analysis in all undiagnosed but suspected infectious cases. Historically, for most cases of encephalitis, the etiology unfortunately is never found. Among more than 300 referrals to the California Encephalitis Project over a 2.5-year period, 62% of cases remained unexplained,39 although some cases recently have been found to be due to anti–N-methyl-D-aspartate (NMDA) receptor antibodies.40 Although many suspected infectious dementias or encephalopathies have previously been unidentified, the advent of metagenomic deep sequencing in clinical practice will likely change this. Metagenomic deep sequencing involves the study of genetic material recovered from a sample, such as bodily fluids or tissue, and is a powerful diagnostic tool with the potential for rapid and unbiased pathogen identification. It recently has been used to detect nonhuman DNA and RNA in human CSF, leading to the identification of uncommon but treatable infections, including neuroleptospirosis and B. mandrillaris. At this time, this testing is primarily being done only through research, but soon should be clinically available.41 Several other infections to consider are shown in Table 7-4 and in the December 2015 issue of Continuum.2
Toxic-Metabolic
Identification of toxic causes of encephalopathy requires a thorough medication and exposure history, including assessing work and home environments. Elevated lithium levels, often iatrogenic, can cause encephalopathy. Inorganic lead poisoning in adults typically leads to peripheral neuropathy, whereas in children, cognitive impairment and encephalopathy can occur. Organic lead, a gasoline additive, is much more toxic than inorganic lead, and even minimal exposure may cause psychiatric and behavioral symptoms, including agitation, disturbed sleep patterns, and hallucinations. Inorganic and organic mercury, similar to lead toxicity, can lead to very toxic states, such as the Mad Hatter syndrome, caused by inorganic mercury exposure in industrial settings and marked by tremor and psychological disturbance. Organic mercury exposure, possibly due to grain contamination or industrial exposure, usually causes a triad of concentric visual field loss, paresthesia, and cerebellar ataxia, without tremor. Although commonly tested for in heavy metal screens, acute arsenic toxicity does not cause RPD but, rather, causes early gastrointestinal and circulatory problems, followed weeks later by a length-dependent sensorimotor polyneuropathy and desquamation of palms and soles.42 A 24-hour urine (not a spot urine) and possibly a hair or nail analysis should be performed for heavy metal testing. Bismuth toxicity, such as from excess use to treat gastrointestinal illnesses (bismuth subsalicylate [BSS] or Pepto-Bismol in the United States or colloidal bismuth [CBS] or De-Nol in Europe), can cause RPD, even mimicking Jakob-Creutzfeldt disease. Symptoms include cognitive dysfunction, tremor, ataxia, dysarthria, and myoclonus. A careful history and bismuth level testing can confirm the diagnosis. Symptoms are often reversible if caught early.43
There are far too many metabolic disorders that can cause RPD to be discussed in this article, from hypercalcemia44 to poikilothermia45 A few important reversible categories, however, are vitamin deficiencies and endocrinologic dysfunction. Niacin (vitamin B3) deficiency, or pellagra (“rough skin”), is often described as “the three Ds”: dermatitis, diarrhea, and dementia. Neurologic deficits can include peripheral neuropathy, myelopathy, and subacute cognitive deficits. Other nutritional deficiencies often occur in conjunction with pellagra. Niacin deficiency occurs most commonly in nutritionally deprived patients (eg, alcoholics, malnourished, impoverished), and in association with systemic disorders, including diabetes mellitus, neoplasms, chronic infections, cirrhosis, chronic gastrointestinal or diarrheal illnesses, and thyrotoxicosis. In industrialized nations, pellagra is most often seen in alcoholics and in patients taking isoniazid. Clinical diagnosis is usually made on suspicion, ruling out other etiologies, and then treating patients empirically (niacin 40 mg/d to 250 mg/d), usually resulting in improvement of neurologic symptoms.46,47
Thiamine (vitamin B1) deficiency can cause Wernicke encephalopathy, which classically presents as an acute/subacute dementia with ophthalmoparesis (with vertical or horizontal nystagmus), ataxia, and memory loss. Often, however, not all features are present. Pathologically, there may be hemorrhagic necrosis of the mammillary bodies or dorsomedial nucleus of the thalamus that may be detected on MRI. Restricted diffusion may be present on DWI and apparent diffusion coefficient (ADC) maps,48 and some imaging features can overlap with those seen in Jakob-Creutzfeldt disease (Case 7-1 and Figure 7-6). Any patient in whom Wernicke encephalopathy is a consideration should be urgently treated with empiric thiamine replacement. All patients with a dementia should be screened for vitamin B12 deficiency, as the clinical deficits are potentially reversible.
Figure 7-6.
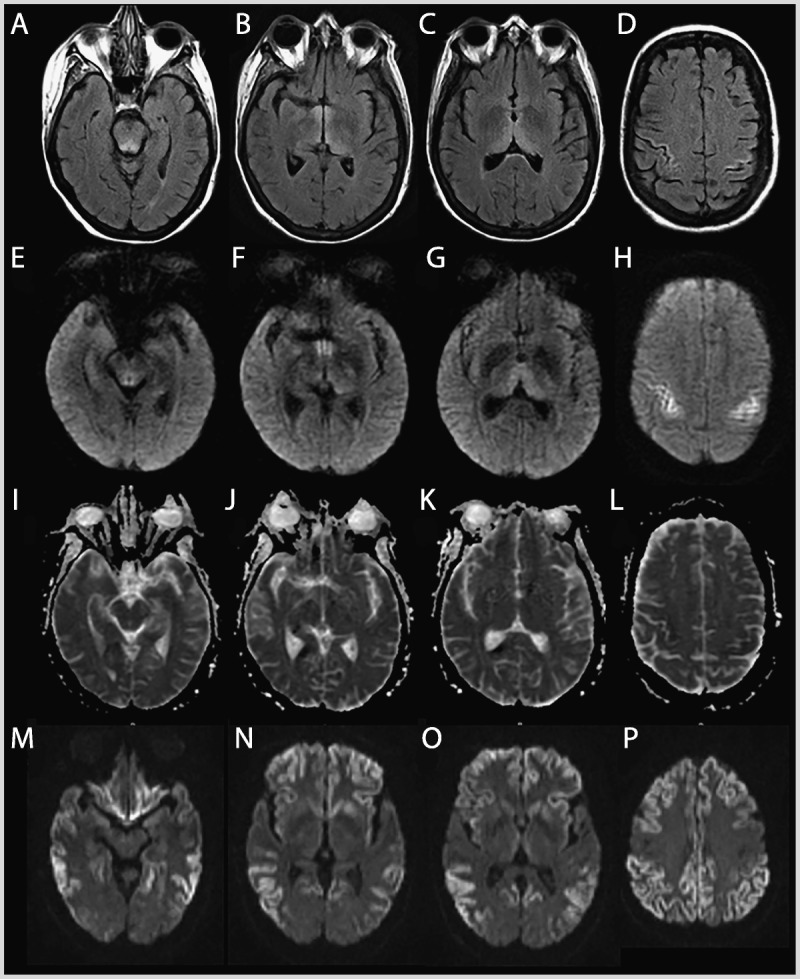
MRI of the patient in Case 7-1. Wernicke encephalopathy (compared to a sporadic Jakob-Creutzfeldt disease case). Fluid-attenuated inversion recovery (FLAIR) (A–D), diffusion-weighted imaging (DWI) (E–H), and apparent diffusion coefficient (ADC) map (I–L) sequences showing FLAIR and DWI hyperintense signal changes involving the periaqueductal gray and midbrain tectum, medial thalami, and perirolandic cortex in the patient with Wernicke encephalopathy. There is relative sparing of the mammillary bodies across all sequences (B,F,J). The ADC sequences (I–L) primarily show subtle hypointensity in the perirolandic cortex (L), corresponding to hyperintensities on FLAIR (D) and DWI (H). This pattern preferentially involving the perirolandic cortex is the opposite of what we typically see in sporadic Jakob-Creutzfeldt disease (M–P; DWI sequences), in which there is generally sparing of the perirolandic region, particularly the primary motor cortex.
As thyroid dysfunction is well known to cause cognitive impairment, all patients with RPD should have a basic thyroid function screen. Depending on the clinical scenario, other endocrine dysfunction should also be considered. It is important to include basic electrolytes (including serum calcium, magnesium, and phosphorus levels), in any dementia screen (Table 7-3). Some other metabolic conditions that might mimic sporadic Jakob-Creutzfeldt disease are adult-onset seizure disorders, due to severe hypoglycemia and hyperglycemia (Case 7-250 and Figure 7-7) or other causes, and extrapontine myelinolysis due to inappropriate correction of hyponatremia (Case 7-3 and Figure 7-8).50 These conditions can also have DWI and ADC MRI findings that overlap with those seen in Jakob-Creutzfeldt disease, including basal ganglia hyperintensity and cortical ribboning.
Figure 7-7.
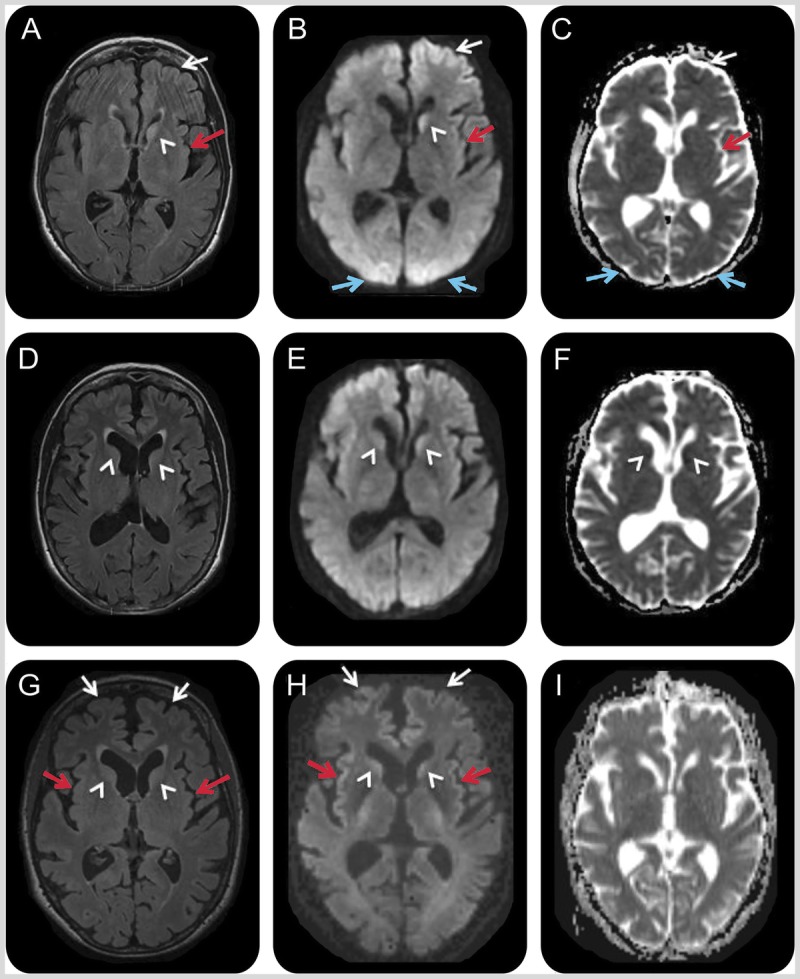
MRI of the patient in Case 7-2. A 66-year-old woman with hypoglycemic encephalopathy. Fluid-attenuated inversion recovery (FLAIR) (A, D, G), diffusion-weighted images (DWI) (B, E, H), and apparent diffusion coefficient (ADC) map (C, F, I) sequences 2 days (A–C), 3 weeks (D–F), and 1 month (G–I) after onset. Initial MRI (A–C) showed left frontal (white arrows), left insular (red arrows), bilateral medial occipital (blue arrows), and left caudate (white arrowhead) FLAIR/DWI hyperintensity with restricted diffusion, which is subtle but definitely appreciable. Repeat MRI about 3 weeks later (D–F) showed possible reduced FLAIR/DWI hyperintensity in the left caudate head and medial occipital regions, and possible increased right caudate FLAIR hyperintensity and restricted diffusion (D–F; white arrowheads). A third MRI 1 week later, 1 month after onset (G–I), revealed more intense FLAIR/DWI insular (G, H; red arrows) and frontal cortical hyperintensities (G, H; white arrows) and possible restricted diffusion and FLAIR hyperintensity still present in the caudate heads (G, H; arrowheads). The resolution of occipital cortical ribboning in such a short time argued against a diagnosis of sporadic Jakob-Creutzfeldt disease.
Reprinted with permission from Rosenbloom MH, et al, Neurol Clin Pract.50 cp.neurology.org/content/5/2/108.full. © 2015 American Academy of Neurology.
Figure 7-8.
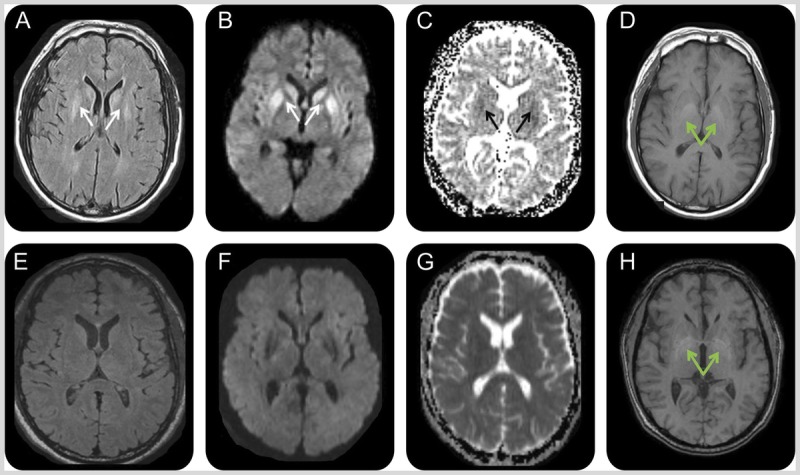
MRI of the patient in Case 7-3. A 50-year-old man with extrapontine myelinolysis. Initial MRI 2 months after onset (A–D) showed symmetric bilateral striatal fluid-attenuated inversion recovery (FLAIR) (A)/diffusion-weighted imaging (DWI) (B) hyperintensities (A, B; white arrows) with corresponding hypointensities on the apparent diffusion coefficient (ADC) map suggesting restricted diffusion (C; black arrows). Bilateral globus pallidus hyperintensities were present on T1-weighted images (D; green arrows). MRI 1 month later, 3 months after onset (E–H), showed resolution of the prior FLAIR (E), DWI (F), and ADC (G) map abnormalities but no change in the globus pallidus T1 hyperintensities (H; green arrows).
Reprinted with permission from Rosenbloom MH, et al, Neurol Clin Pract.50 cp.neurology.org/content/5/2/108.full. © 2015 American Academy of Neurology.
Case 7-1
A 61-year-old woman was evaluated at an outside hospital several times over the course of 8 months for insidious onset of psychiatric symptoms (depressed mood and severe anxiety with panic attacks) followed by gastrointestinal symptoms (eg, abdominal pain, nausea, vomiting) and anorexia of unclear etiology. She had a history of obesity and had lost more than 40% of her initial body weight over this time period, became progressively confused, and was unable to live independently. Results from broad urinary and serologic investigations were nondiagnostic, and her EEG reportedly showed diffuse slowing. Notably, her CSF analysis showed elevated total tau and 14-3-3 proteins, and brain MRI showed fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted imaging (DWI) hyperintensities in the deep nuclei and cortical ribboning along the rolandic cortex, interpreted as consistent with Jakob-Creutzfeldt disease. Given her rapidly progressive dementia and CSF and MRI findings, she was diagnosed with sporadic Jakob-Creutzfeldt disease, and further workup was not pursued. Upon review of her medical records, we assessed that her clinical syndrome and brain MRI findings (involvement of periaqueductal gray matter, midbrain tectum, medial thalamus, and perirolandic cortex) (Figure 7-6) were more consistent with thiamine deficiency and not with Jakob-Creutzfeldt disease. Unfortunately, the patient died prior to treatment for Wernicke encephalopathy could be initiated. Brain autopsy, performed through the US National Prion Disease Pathology Surveillance Center, confirmed thiamine deficiency without evidence of prion disease. The patient’s final neuropathologic diagnosis was Wernicke encephalopathy.
Comment. There is considerable clinical and radiographic overlap between Wernicke encephalopathy and Jakob-Creutzfeldt disease; FLAIR and DWI hyperintensities are observed commonly in the mammillary bodies, midbrain tectum, and medial thalami in Wernicke encephalopathy, which somewhat overlap with the imaging findings of Jakob-Creutzfeldt disease. Focal cortical involvement of the motor cortex, as shown in this case, however, was reported in Wernicke encephalopathy49 but this area is usually spared in Jakob-Creutzfeldt disease.6 Although Wernicke encephalopathy is largely known to occur in alcoholic patients with the classical triad of cognitive impairment, incoordination, and oculomotor abnormalities, now it more commonly occurs in nonalcoholic patients, resulting from malnutrition or malabsorption, which appears to have been the case with this patient. Occurrence of all three signs is also rare. Wernicke encephalopathy always should be considered in patients with rapidly progressive dementia and significant weight loss or malnutrition.
Case 7-2
A 66-year-old right-handed woman with a history of poorly controlled insulin-dependent diabetes mellitus, hypothyroidism, and primary biliary cirrhosis was found unconscious. She had not been compliant with her insulin treatment, and her blood glucose level was 99 mg/dL, which was taken by a paramedic, although it was not clear if this was after she was given glucose. For the first 3 days of admission her blood sugar was in the range of 96 mg/dL to 366 mg/dL, which decreased to 25 mg/dL and 42 mg/dL on the fourth and fifth days of admission. She regained her consciousness after a day or two without focal neurologic deficits, but was confused (eg, using her telephone as a television remote control). Blood tests were unremarkable. CSF analysis showed normal white blood cell count, red blood cell count, protein, and glucose, but positive 14-3-3 protein, elevated neuron-specific enolase of 78 ng/mL (more than 35 ng/mL consistent with Jakob-Creutzfeldt disease according to Mayo Medical Laboratories),51 very elevated total tau of 17,585 pg/mL (greater than 1200 ng/mL consistent with Jakob-Creutzfeldt disease according to Athena Diagnostics, Inc),52 and very elevated phosphorylated tau of 88.4 pg/mL (more than 61 pg/mL considered elevated according to Athena Diagnostics, Inc), and normal amyloidβ42 of 828.8 pg/mL (according to Athena Diagnostics Inc). Brain MRI fluid-attenuated inversion recovery (FLAIR), diffusion-weighted imaging (DWI), and apparent attenuation diffusion coefficient (ADC) map 2 days after the onset demonstrated cortical ribboning and left caudate hyperintensity with restricted diffusion (Figure 7-7A, 7-7B, and 7-7C). Her cognitive functions gradually improved but she had intermittent confusion and disorientation. A second MRI 3 weeks after the onset showed less diffusion restriction in the left caudate but possible new reduced diffusion in the right caudate head and anterior putamen (Figure 7-7D, 7-7D, 7-7F). She was referred to our center for a Jakob-Creutzfeldt disease treatment trial. Notably, 1 month after the onset, at our center, she had continued to improve significantly in cognitive and behavioral domains, although had mild cognitive impairment. A third MRI showed more intense DWI cortical ribboning and greater right caudate head involvement (Figure 7-7G, 7-7H, and7-7I).
Given her profound clinical improvement, she was diagnosed with hypoglycemia-related encephalopathy with MRI abnormalities that might be secondary to seizure or hypoglycemia. She was discharged home, but several weeks later had another episode of hypoglycemia leading to coma and death. Brain autopsy revealed hypoxic/hypoglycemic nerve loss in multiple brain regions (cortices, CA1/CA4 hippocampus, deep nuclei, cerebellar Purkinje cells, and dentate nucleus) without evidence of prion disease.
Comment. Sustained hypoglycemia can cause various symptoms such as confusion, seizures, weakness, and coma, and MRI FLAIR/DWI hyperintensities with restricted diffusion in the cortices, basal ganglia, and hippocampus.50 Seizures are also known to result in reduced diffusion in the cortex and deep nuclei, which is reversible.50 Although the patient’s CSF biomarkers and overall MRI findings were suggestive of prion disease, significant clinical improvement with less diffusion restriction in the left caudate within a few weeks was not consistent with Jakob-Creutzfeldt disease. A thorough clinical history, as well as an extensive metabolic workup, and serial MRIs can help differentiate a reversible metabolic encephalopathy from prion disease. A detailed description of this case is published.50
Modified with permission from Rosenbloom MH, et al, Neurol Clin Pract.50 cp.neurology.org/content/5/2/108.full. © 2015 American Academy of Neurology.
Case 7-3
A 50-year-old right-handed man with a history of alcoholism (current), hypertension, hypercholesterolemia, and hepatitis C developed nausea, vomiting, and diarrhea for 3 days. He reduced his alcohol consumption and increased his water intake for hydration. One week later, he had two episodes of generalized tonic-clonic seizures and was treated for encephalopathy and hyponatremia at a local intensive care unit. His brain CT was unremarkable but he had behavioral symptoms (emotional blunting, violent outbursts, delusions, and hallucinations), impaired episodic memory, speech disturbance (slurred, halting), executive problems, gait imbalance, and myoclonus of the hands and trunk. His first brain MRI at approximately 2 months after the onset of symptoms showed restricted diffusion with hyperintensities on T2, fluid-attenuated inversion recovery (FLAIR), and diffusion-weighted imaging (DWI) in the bilateral striata with corresponding hypointensity on apparent diffusion coefficient (ADC) map, and T1 hyperintensities in bilateral globus pallidi (Figure 7-8A, 7-8B, 7-8C, and 7-8D). Because of his clinical symptoms and MRI findings, he was diagnosed with sporadic Jakob-Creutzfeldt disease and referred to our center. At our center, 3 months after the onset, we noted that although he had deficits (mild cognitive and motor deficits), he had improved profoundly. A repeat brain MRI showed resolution of the diffusion and T2 striatal abnormalities (Figure 7-8E, 7-8F, 7-8G, and 7-8H).
Extensive laboratory workup for rapidly progressive dementia was negative, but a careful review of his outside medical records determined that at his initial hospitalization his first sodium level was 106 mEq/L, which decreased to 102 mEq/L within 3.5 hours and then was corrected to 130 mEq/L in less than 36 hours. Given the MRI findings and the history of rapidly corrected hyponatremia, he was diagnosed with extrapontine myelinolysis.
Comment. Extrapontine myelinolysis results from rapid metabolic shifts, such as rapid correction of hyponatremia, and mostly affects the basal ganglia, internal capsule, white matter, and cerebral cortices, sparing the pons.44 Three radiologic features suggested against Jakob-Creutzfeldt disease in this case: (1) such quick resolution of the restricted diffusion without volume loss; (2) the striatal involvement was uniformly bright, whereas in Jakob-Creutzfeldt disease there is usually an anterior to posterior decreasing gradient; and (3) isolated striatal involvement without any cortical ribboning is uncommon in Jakob-Creutzfeldt disease (approximately 2% of cases).1 Symptoms such as dysarthria, bradykinesia, dystonia, and akinetic mutism mimic sporadic Jakob-Creutzfeldt disease, but clinical and radiologic improvement are different from those seen in sporadic Jakob-Creutzfeldt disease. A detailed description of this case is published.50
Modified with permission from Rosenbloom MH, et al, Neurol Clin Pract.50 cp.neurology.org/content/5/2/108.full. © 2015 American Academy of Neurology.
Autoimmune
As seen in our and other RPD cohorts (Table 7-1 and Table 7-2), several autoimmune conditions present as RPDs. These conditions, particularly antibody-mediated causes of CNS dysfunction, including RPD, are discussed in detail in the article “Autoimmune Encephalopathies and Dementias” by Andrew McKeon, MD,9 in this issue of Continuum, so they will be discussed here only briefly. For most RPDs, we generally recommend a basic autoimmune screen. There is no consensus on what this should include, but one possible initial and a secondary screen are shown in Table 7-3. Unfortunately, most cases of CNS vasculitis have normal serologic findings.36 MRI may show contrast enhancement, infarcts, and involvement of gray or white matter.5,43 When MRI and the clinical picture suggest vasculitis, it is important to note that other conditions, such as intravascular lymphoma, can have the appearance of vasculitis on brain angiogram, and may temporarily respond to steroids. When performing a brain biopsy for potential vasculitis, it is important to take an appropriately sized piece of tissue (typically 1 cm3), including meninges, ideally from an area that is abnormal on MRI.36 If a paraneoplastic or antibody-mediated dementia syndrome is suspected in a patient who has none of the known or commercially available antibodies, the clinician may wish to send blood or CSF to a research laboratory that specializes in identifying novel antibodies.53
Neurodegenerative/Neoplastic
As shown in Table 7-1 and Table 7-2, nonprion neurodegenerative diseases are often the most common forms of RPD. Although many nonprion neurodegenerative diseases can truly present as RPD, at the author’s center, such patients are often found to have had a slow course over several years that has been unnoticed or undiagnosed until a rapid decline occurs. Many patients referred to our center with an RPD diagnosis did not have an RPD, but rather a slowly progressive neurodegenerative dementia. In the author’s experience, often, those closest to the patient, particularly when with the patient daily, miss the first signs of illness, and do not bring the patient to a doctor until the symptoms became obvious or there is a sudden steep decline. These declines are sometimes part of the natural disease course and, other times, are due to an infection (eg, urinary tract infection, pneumonia) or another metabolic perturbation. Determination of non-RPD dementia diagnoses is typically made by obtaining history from family members who did not see the patient regularly (eg, a child who comes to visit for the holidays), friends, or colleagues, who had noticed cognitive changes well before those who saw the patient on a daily basis.5 As noted in Table 7-1, Table 7-2, and Table 7-4, some nonprion neurodegenerative dementias, however, do rarely progress rapidly, with dementia occurring within 1 year from first symptom onset or from onset of first symptom to death in 1 to 3 years.54,55 These include corticobasal degeneration, FTD, FTD with motor neuron disease, and rare cases of AD and DLB.5,7,43 These neurodegenerative conditions are discussed elsewhere in this volume.
A patient with FTD with the C9ORF72 mutation recently was reported to manifest with an RPD.56 The initial symptoms were profound behavioral and personality changes (blunt affect, begging for money, collecting cigarette butts, and social withdrawal) followed by delusions, disorganized and aggressive behavior, progressive cognitive decline and motor neuron disease, leading to death within 1 year.
Neoplastic
Neoplastic causes of RPD can be quite difficult to diagnose, particularly when they do not present as isolated mass lesions on MRI. Primary CNS lymphoma and intravascular lymphoma often present as an RPD and even may resemble Jakob-Creutzfeldt disease clinically. MRI can be helpful in these diagnoses. Primary CNS lymphoma presents as a solitary or multiple mass lesions in contact with the subarachnoid space and is uniformly contrast-enhancing in immunocompetent patients. Multifocal lesions and peripheral enhancement, however, are characteristic MRI findings in immunosuppressed patients.15 In lymphoma, certain markers such as serum lactate dehydrogenase, β2-microglobulin, and CA-125 may be elevated, although some of these markers have somewhat low sensitivity and specificity. CSF β2-microglobulin appears to be somewhat correlated with CNS lymphoma rather than with systemic lymphoma in small numbers of patients. CSF lactate dehydrogenase 5 (LD5) (anaerobic lactate dehydrogenase [LDH] isoenzyme) proportions greater than or equal to 2.8% of total LDH had a high sensitivity (about 93%) and a low specificity for CNS lymphoma.15 In intravascular lymphoma, brain angiogram shows a pattern indistinguishable from vasculitis, and, thus, pathology or CSF cytology may be the necessary and preferred diagnostic methods. When considering CNS lymphoma, repeated large-volume CSF analyses for cytology and flow cytometry should be done, although they are often negative.36 Avoid giving steroids if a biopsy is being considered, as this will result in necrosis of the lymphoma cells, rendering pathologic diagnosis difficult to impossible (Figure 7-2). When a neoplastic condition is in the differential, a thorough general medical examination, a chest/abdomen/pelvis CT (with and without contrast), possibly with a full body PET scan, should be considered, depending on the clinical context.15
Systemic/Structural
There are too many systemic conditions that can present as RPDs to discuss in detail here, but several to consider are listed in Table 7-4.5,43 In a patient with an unexplained multisystem disorder, consider mitochondrial disease, particularly if there are gray and white matter MRI abnormalities or clinical features consistent with mitochondrial disease. Systems frequently affected are the peripheral nervous system (myopathy, polyneuropathy, lactic acidosis), brain (leukoencephalopathy, calcifications, strokes, seizures, upper motor neuron findings, ataxia, extrapyramidal symptoms, unexplained fatigue), endocrine (short stature, pubertal abnormalities), cardiac (conduction defects, cardiomyopathy, unexplained heart failure), visual (early cataracts, retinopathy, optic atrophy), auditory (deafness, vertigo), gastrointestinal (dysphagia, nausea/vomiting, diarrhea, liver abnormalities, pancreatic insufficiency), kidney (renal failure, cysts), and bone marrow (sideroblastic anemia).57 Mitochondrial dementia may rarely present acutely.58 A strong lactate signal on magnetic resonance spectroscopy and genetic analysis of mitochondrial mutations are useful for differentiating mitochondriopathy from Jakob-Creutzfeldt disease.59 An unusual pattern of neurologic disease in the family may also be a clue to mitochondrial disease. An MRI of a patient with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes syndrome (MELAS) presenting as an RPD with some imaging features overlapping with Jakob-Creutzfeldt disease is shown in Figure 7-9.59
Figure 7-9.
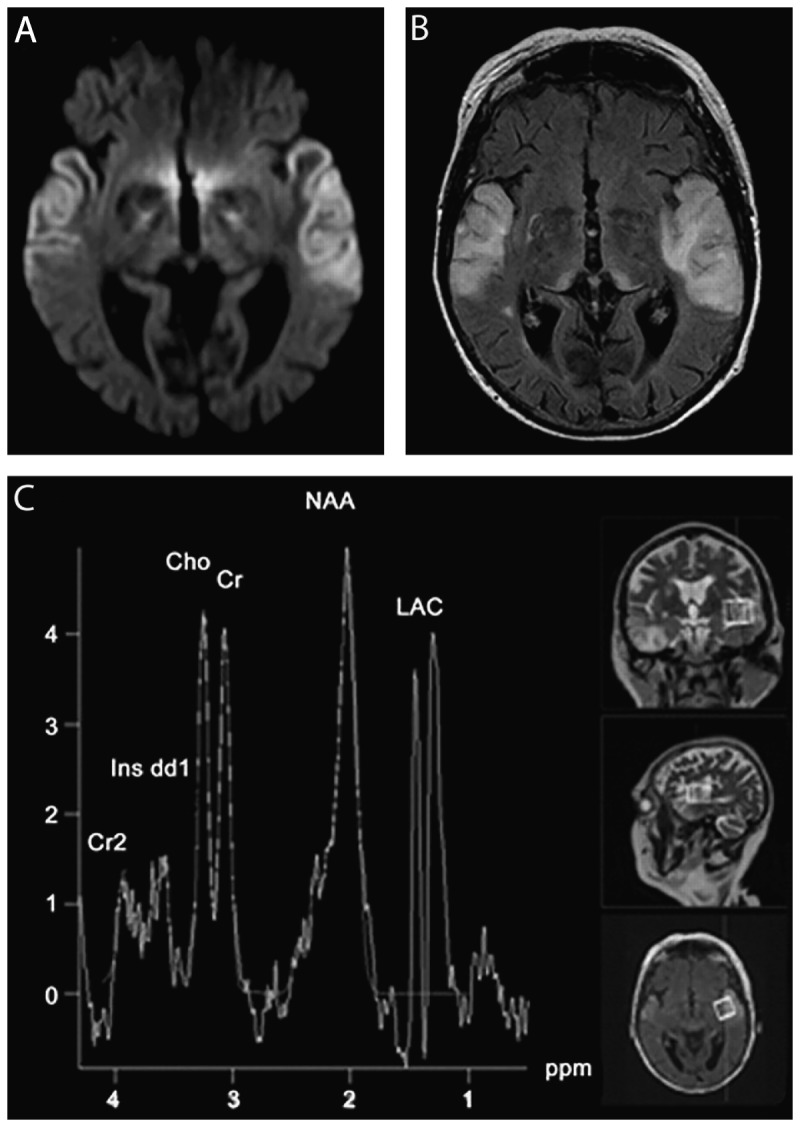
MRI and spectroscopy in a case of mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes syndrome (MELAS). A 59-year-old woman developed confusion, progressive aphasia, mutism, and fluctuations of alertness over 2 weeks. Diffusion-weighted imaging (DWI) MRI revealed abnormalities overlapping with Jakob-Creutzfeldt disease (A), although the fluid-attenuated inversion recovery (FLAIR) MRI (B) with white and gray matter hyperintensity was not consistent with Jakob-Creutzfeldt disease. CSF showed normal cell counts, negative polymerase chain reaction (PCR) for herpes simplex virus, elevated lactate (4.6 mmol/L), and increased levels of 14-3-3 and tau protein (1300 pg/L), both concerning for Jakob-Creutzfeldt disease. There were no periodic sharp-wave complexes on EEG recordings. Magnetic resonance spectroscopy revealed a lactate signal indicative of mitochondriopathy and genetic analysis confirmed the MELAS A3243G mutation. The DWI (A) displays bitemporal neocortical hyperintense signals. The FLAIR (B) 2 days after the initial MRI scan reveals newly emerging symmetric lesions in the pulvinar thalami. Magnetic resonance spectroscopy (C) displays a strong lactate signal.
Cho = choline; Cr = creatine; Cr2 = phosphocreatine; Ins dd1 = myoinositol; LAC = lactate; NAA = N-acetylaspartate; ppm = parts per million.Reprinted with permission from Weiss D, et al, Neurology.59 www.neurology.org/content/77/9/914.full. © 2011 American Academy of Neurology.
CONCLUSION
The evaluation of a patient with an RPD can be challenging and time consuming. Therefore, it is important to have a structured approach to the diagnostic evaluation. A few important factors perhaps have led to the improved ability to differentiate and diagnose RPDs over the past few years. Diagnosis of many forms of human prion diseases and distinction from other RPDs has become easier and more accurate because of the more common use of diffusion MRI (DWI and ADC maps) for diagnosis of Jakob-Creutzfeldt disease as well as improved methods for detection of prions (not just biomarkers) in the CSF using protein misfolding cyclic amplification assay and RT-QuIC.2 The identification of novel antibody-mediated dementias, particularly those with cell surface antigens, has expanded dramatically over the past decade. These conditions have led to a new field within neurology that is particularly exciting because of the reversibility of many of these conditions.
KEY POINTS
In most larger rapidly progressive dementia series, the most common causes of rapidly progressive dementia are nonprion neurodegenerative diseases, prion diseases, and autoimmune/antibody-mediated encephalopathies.
Sending the CSF biomarkers 14-3-3, total tau, and neuron-specific enolase is always recommended, not necessarily as diagnostic tests for prion disease, because they are not, but rather as markers of rapid neuronal injury. If these biomarkers are elevated, they help confirm the history of a rapidly progressive neurologic condition.
The use of a mnemonic device, such as VITAMINS (vascular, infectious, toxic-metabolic, autoimmune, malignancy, iatrogenic, neurodegenerative, and systemic), when evaluating a patient with rapidly progressive dementia can help the clinician consider many diverse etiologies for such conditions.
Any patient in whom Wernicke encephalopathy is a consideration should be urgently treated with empiric thiamine replacement.
In sporadic Jakob-Creutzfeldt disease, there is usually relative sparing of the perirolandic cortex on fluid-attenuated inversion recovery and diffusion-weighted imaging MRI, particularly in comparison to adjacent cortex.
ACKNOWLEDGMENTS
The author would like to thank the National Institutes of Health/National Institute on Aging (R01 AG031189; K23 AG0219889; P01 AG021601); National Institutes of Health/National Center for Research Resources, University of California, San Francisco, Clinical and Translational Science Institute (UL1 RR024131); the Michael J. Homer Family Fund; and Quest Diagnostics for support of his research in rapidly progressive dementia and related disorders. He also thanks Mee-Ohk Kim, MD, PhD, for help in preparation of this manuscript.
REFERENCES
- 1. Grossman M, Irwin DJ. The mental status examination in patients with suspected dementia. Continuum (Minneap Minn) 2016; 22(2 Dementia): 385– 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Geschwind MD. Prion diseases. Continuum (Minneap Minn) 2015; 21(6 Neuroinfectious Disease): 1612– 1638. doi:10.1212/CON.0000000000000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gibbs CJ., Jr Spongiform encephalopathies—slow, latent, and temperate virus infections—in retrospect. In: Prusiner SB, Collinge J, Powell J, et al. eds. Prion diseases of humans and animals. London, UK: Ellis Horwood, 1992: 53– 62. [Google Scholar]
- 4. Katscher F. It’s Jakob’s disease, not Creutzfeldt’s. Nature 1998; 393(6680): 11 doi:10.1038/29862. [DOI] [PubMed] [Google Scholar]
- 5. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol 2008; 64(1): 97– 108. doi:10.1002/ana.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology 2011; 76(20): 1711– 1719. doi:10.1212/WNL.0b013e31821a4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Poser S, Mollenhauer B, Kraubeta A, et al. How to improve the clinical diagnosis of Creutzfeldt-Jakob disease. Brain 1999; 122(pt 12): 2345– 2351. doi:10.1093/brain/122.12.2345. [DOI] [PubMed] [Google Scholar]
- 8. Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol 2011; 70(3): 437– 444. doi:10.1002/ana.22454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McKeon A. Autoimmune encephalopathies and dementias. Continuum (Minneap Minn) 2016; 22(2 Dementia): 538– 558. [DOI] [PubMed] [Google Scholar]
- 10. Renaud DL. Adult-onset leukoencephalopathies. Continuum (Minneap Minn) 2016; 22(2 Dementia): 559– 578. [DOI] [PubMed] [Google Scholar]
- 11. Papageorgiou SG, Kontaxis T, Bonakis A, et al. Rapidly progressive dementia: causes found in a Greek tertiary referral center in Athens. Alzheimer Dis Assoc Disord 2009; 23(4): 337– 346. doi:10.1097/WAD.0b013e31819e099b. [DOI] [PubMed] [Google Scholar]
- 12. Sala I, Marquié M, Sánchez-Saudinós MB, et al. Rapidly progressive dementia: experience in a tertiary care medical center. Alzheimer Dis Assoc Disord 2012; 26(3): 267– 271. doi:10.1097/WAD.0b013e3182368ed4. [DOI] [PubMed] [Google Scholar]
- 13. Lindau M, Almkvist O, Kushi J, et al. First symptoms—frontotemporal dementia versus Alzheimer’s disease. Dement Geriatr Cogn Disord 2000; 11(5): 286– 293. doi:10.1159/000017251. [DOI] [PubMed] [Google Scholar]
- 14. Claassen DO, Josephs KA, Ahlskog JE, et al. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology 2010; 75(6): 494– 499. doi:10.1212/WNL.0b013e3181ec7fac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scott BJ, Douglas VC, Tihan T, et al. A systematic approach to the diagnosis of suspected central nervous system lymphoma. JAMA Neurol 2013; 70(3): 311– 319. doi:10.1001/jamaneurol.2013.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ng AS, Kramer J, Centurion A, et al. Clinico-pathological correlation in adenylate kinase 5 autoimmune limbic encephalitis. J Neuroimmunol 2015; 287: 31– 35. doi:10.1016/j.jneuroim.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Höftberger R, van Sonderen A, Leypoldt F, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology 2015; 84(24): 2403– 2412. doi:10.1212/WNL.0000000000001682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harkness KA, Coles A, Pohl U, et al. Rapidly reversible dementia in cerebral amyloid inflammatory vasculopathy. Eur J Neurol 2004; 11(1): 59– 62. doi:10.1046/j.1351-5101.2003.00707.x. [DOI] [PubMed] [Google Scholar]
- 19. Geraldes R, Albuquerque L, Ferro JM, et al. Rapidly progressive cognitive impairment, ataxia, and myoclonus: an unusual presentation of a dural arteriovenous fistula. J Stroke Cerebrovasc Dis 2012; 21(7): 619.e3– 619.e5. doi:10.1016/j.jstrokecerebrovasdis.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 20. Labeyrie MA, Lenck S, Saint-Maurice JP, et al. Dural arteriovenous fistulas presenting with reversible dementia are associated with a specific venous drainage. Eur J Neurol 2014; 21(3): 545– 547. doi:10.1111/ene.12300. [DOI] [PubMed] [Google Scholar]
- 21. Mendonça N, Santos G, Duro D, et al. Multiple dural arteriovenous fistulas presenting as rapidly progressive dementia. Neurologist 2012; 18(3): 130– 132. doi:10.1097/NRL.0b013e318251e695. [DOI] [PubMed] [Google Scholar]
- 22. Pasi M, Nappini S, Salvadori E, et al. Rapidly progressive cognitive impairment in a patient with high flow dural arteriovenous fistulas, cerebral sinus thrombosis and protein S deficiency. J Clin Neurosci 2014; 21(9): 1654– 1656. doi:10.1016/j.jocn.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 23. Carrera E, Bogousslavsky J. The thalamus and behavior: effects of anatomically distinct strokes. Neurology 2006; 66(12): 1817– 1823. doi:10. 1212/01. wnl. 0000219679.95223.4c. [DOI] [PubMed] [Google Scholar]
- 24. Auchus AP, Chen CP, Sodagar SN, et al. Single stroke dementia: insights from 12 cases in Singapore. J Neurol Sci 2002; 203–204: 85– 89. doi:10.1016/S0022-510X(02)00272-1. [DOI] [PubMed] [Google Scholar]
- 25. Zeidman SM, Monsein LH, Arosarena O, et al. Reversibility of white matter changes and dementia after treatment of dural fistulas. AJNR Am J Neuroradiol 1995; 16(5): 1080– 1083. [PMC free article] [PubMed] [Google Scholar]
- 26. Hurst RW, Bagley LJ, Galetta S, et al. Dementia resulting from dural arteriovenous fistulas: the pathologic findings of venous hypertensive encephalopathy. AJNR Am J Neuroradiol 1998; 19(7): 1267– 1273. [PMC free article] [PubMed] [Google Scholar]
- 27. Abe K, Okuda O, Ohishi H, et al. Multiple dural arteriovenous fistulas causing rapid progressive dementia successfully treated by endovascular surgery: case report. Neurol Med Chir (Tokyo) 2014; 54(2): 145– 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Imazeki R, Amari K, Sekiguchi T, et al. Rapidly progressive dementia caused by a superior sagittal sinus dural arteriovenous fistula: a case report. Tokai J Exp Clin Med 2015; 40(1): 22– 26. doi:10.2176/nmc.nmc-2012-0080. [PubMed] [Google Scholar]
- 29. Wasay M, Azeemuddin M. Neuroimaging of cerebral venous thrombosis. J Neuroimaging 2005; 15(2): 118– 128. doi:10.1111/j.1552-6569.2005.tb00296.x. [DOI] [PubMed] [Google Scholar]
- 30. Sarazin M, Amarenco P, Mikol J, et al. Reversible leukoencephalopathy in cerebral amyloid angiopathy presenting as subacute dementia. Eur J Neurol 2002; 9(4): 353– 358. doi:10.1046/j.1468-1331.2002.00393.x. [DOI] [PubMed] [Google Scholar]
- 31. Crosta F, Orlandi B, De Santis F, et al. Cerebral amyloid angiopathy-related inflammation: report of a case with very difficult therapeutic management. Case Rep Neurol Med 2015; 2015: 483020 doi:10.1155/2015/483020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke 2015; 17(1): 17– 30. doi:10.5853/jos.2015.17.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cachia D, Smith T, Paydarfar D, Pomorska G. A case of early-onset rapidly progressive dementia. JAMA Neurol 2014; 71(11): 1445– 1449. doi:10.1001/jamaneurol.2014.836. [DOI] [PubMed] [Google Scholar]
- 34. Graffeo CS, Dawson ET, Murphy ME, et al. Expanding the spectrum of subacute diencephalic angioencephalopathy. J Clin Neurosci 2016; 23: 8– 13. doi:10.1016/j.jocn.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 35. Bouwman FH, Skolasky RL, Hes D, et al. Variable progression of HIV-associated dementia. Neurology 1998; 50(6): 1814– 1820. doi:10.1212/WNL.50.6.1814. [DOI] [PubMed] [Google Scholar]
- 36. Josephson SA, Papanastassiou AM, Berger MS, et al. The diagnostic utility of brain biopsy procedures in patients with rapidly deteriorating neurological conditions or dementia. J Neurosurg 2007; 106(1): 72– 75. [DOI] [PubMed] [Google Scholar]
- 37. Schuster FL, Honarmand S, Visvesvara GS, Glaser CA. Detection of antibodies against free-living amoebae Balamuthia mandrillaris and Acanthamoeba species in a population of patients with encephalitis. Clin Infect Dis 2006; 42(9): 1260– 1265. doi:10.1086/503037. [DOI] [PubMed] [Google Scholar]
- 38. Hurth K, Tarawneh R, Ghoshal N, et al. Whipple’s disease masquerades as dementia with Lewy bodies. Alzheimer Dis Assoc Disord 2015; 29(1): 85– 89. doi:10.1097/WAD.0b013e3182a715da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Glaser CA, Gilliam S, Schnurr D, et al. In search of encephalitis etiologies: diagnostic challenges in the California Encephalitis Project, 1998-2000. Clin Infect Dis 2003; 36(6): 731– 742. doi:10.1086/367841. [DOI] [PubMed] [Google Scholar]
- 40. Gable MS, Sheriff H, et al. The frequency of autoimmune N-methyl-d-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California encephalitis project. Clin Infect Dis 2012; 54(7): 899– 904. doi:10.1093/cid/cir1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilson MR, Shanbhag NM, Reid MJ, et al. Diagnosing Balamuthia mandrillaris encephalitis with metagenomic deep sequencing. Ann Neurol 2015; 78(5): 722– 730. doi:10.1002/ana.24499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bolla K. Exogenous acquired metabolic disorders of the nervous system: toxins and illicit drugs. In: Goetz C, Pappaert, editors. Clinical neurology. 3rd ed New York: Elsevier, 1999: 865– 896. [Google Scholar]
- 43. Geschwind MD, Haman A, Miller BL. Rapidly progressive dementia. Neurol Clin 2007; 25(3): 783– 807, vii. doi:10.1016/j.ncl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rösche J, Sieveking C, Kampf C, Benecke R. Creutzfeldt-Jakob-Like syndrome due to hypercalcemic encephalopathy. Clin EEG Neurosci 2015; 46(4): 327– 330. doi:10.1177/1550059414529764. [DOI] [PubMed] [Google Scholar]
- 45. Bouwman NA, Verhagen WI, Meulstee J. EEG abnormalities in poikilothermia suggesting Creutzfeldt-Jakob disease. Clin EEG Neurosci 2009; 40(3): 196– 199. doi:10.1177/155005940904000313. [DOI] [PubMed] [Google Scholar]
- 46. Kertesz SG. Pellagra in 2 homeless men. Mayo Clin Proc 2001; 76(3): 315– 318. doi:10.4065/76.3.315. [DOI] [PubMed] [Google Scholar]
- 47. Pitsavas S, Andreou C, Bascialla F, et al. Pellagra encephalopathy following B-complex vitamin treatment without niacin. Int J Psychiatry Med 2004; 34(1): 91– 95. doi:10.2190/29XV-1GG1-U17K-RGJH. [DOI] [PubMed] [Google Scholar]
- 48. Halavaara J, Brander A, Lyytinen J, et al. Wernicke’s encephalopathy: is diffusion-weighted MRI useful? Neuroradiology 2003; 45(8): 519– 523. doi:10.1007/s00234-003-1043-8. [DOI] [PubMed] [Google Scholar]
- 49. Kinoshita Y, Inoue Y, Tsuru E, et al. Unusual MR findings of Wernicke encephalopathy with cortical involvement. No To Shinkei 2001; 53(1): 65– 68. [PubMed] [Google Scholar]
- 50. Rosenbloom MH, Tartaglia MC, Forner SA, et al. Metabolic disorders with clinical and radiologic features of sporadic Creutzfeldt-Jakob disease. Neurol Clin Pract 2015; 5(2): 108– 115. doi:10.1212/CPJ.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mayo Medical Laboratories. Neuron-specific enolase (NSE), spinal fluid. neurology.testcatalog.org/show/NSESF. Published 2015. Accessed February 15, 2016.
- 52.Athena Diagnostics. ADmark® phospho-tau/total-tau/Aβ42 CSF analysis and interpretation (symptomatic). www.athenadiagnostics.com/view-full-catalog/a/admark-reg;-phospho-tau-total-tau-ab42-csf-analysi#section1. Accessed February 15, 2016.
- 53. Lancaster E, Dalmau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012; 8(7): 380– 390. doi:10.1038/nrneurol.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Josephs KA, Ahlskog JE, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol 2009; 66(2): 201– 207. doi:10.1001/archneurol.2008.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schmidt C, Haïk S, Satoh K, et al. Rapidly progressive Alzheimer’s disease: a multicenter update. J Alzheimers Dis 2012; 30(4): 751– 756. doi:10.3233/JAD-2012-120007. [DOI] [PubMed] [Google Scholar]
- 56. Grau-Rivera O, Gelpi E, Carballido-López E, et al. Rapidly progressive dementia with psychotic onset in a patient with the C9ORF72 mutation. Clin Neuropathol 2015; 34(5): 294– 297. doi:10.5414/NP300853. [DOI] [PubMed] [Google Scholar]
- 57. Finsterer J. Mitochondriopathies. Eur J Neurol 2004; 11(3): 163– 186. doi:10.1046/j.1351-5101.2003.00728.x. [DOI] [PubMed] [Google Scholar]
- 58. Carroll MB. MELAS masquerading as a systemic vasculitis. J Clin Rheumatol 2007; 13(6): 334– 337. doi:10.1097/RHU.0b013e31815c2516. [DOI] [PubMed] [Google Scholar]
- 59. Weiss D, Brockmann K, Nägele T, et al. Rapid emergence of temporal and pulvinar lesions in MELAS mimicking Creutzfeldt-Jakob disease. Neurology 2011; 77(9): 914 doi:10.1212/WNL.0b013e31822c6275. [DOI] [PubMed] [Google Scholar]