

Abstract
Therapeutic gene transfer holds the promise of providing lasting therapies and even cures for diseases that were previously untreatable or for which only temporary or suboptimal treatments were available. For some time, clinical gene therapy was characterized by some impressive but rare examples of successes and also several setbacks. However, effective and long-lasting treatments are now being reported from gene therapy trials at an increasing pace. Positive outcomes have been documented for a wide range of genetic diseases (including hematological, immunological, ocular, and neurodegenerative and metabolic disorders) and several types of cancer. Examples include restoration of vision in blind patients, eradication of blood cancers for which all other treatments had failed, correction of hemoglobinopathies and coagulation factor deficiencies, and restoration of the immune system in children born with primary immune deficiency. To date, about 2,000 clinical trials for various diseases have occurred or are in progress, and many more are in the pipeline. Multiple clinical studies reported successful treatments of pediatric patients. Design of gene therapy vectors and their clinical development are advancing rapidly. This article reviews some of the major successes in clinical gene therapy of recent years.
Gene therapy seeks to treat a disease by transferring one or more therapeutic nucleic acids to a patient’s cells or by correcting a defective gene, for example by gene editing. Hence, this technology has the potential to cure diseases that are treatable but not curable with conventional medications, and to provide treatments for diseases previously classified as untreatable. As with any new medical technology, translation of this concept initially led to a mixture of encouraging and disappointing results in clinical trials, and also some major setbacks. However, fueled by successful treatment of ocular diseases and primary immune deficiencies, the “comeback of gene therapy” was highlighted as one of the major scientific breakthroughs of the year by Science magazine in 2009 (refs. 1,2). Advances in the development of gene therapy vector systems, optimized for in vivo and ex vivo gene transfer, and increasing clinical experience with these technologies were major factors that have finally allowed medicine to capitalize on the potential of gene transfer for the treatment of human disease. As the field advanced gene therapy beyond correction of genetic disorders, the spectrum of applications vastly increased. In fact, eradication of blood cancers using chimeric antigen receptor (CAR)-modified T cells prompted Science magazine to select cancer immunotherapy as the biggest scientific breakthrough of 2013 (ref. 3).
Effective strategies for clinical gene therapy are based on either in vivo gene delivery to postmitotic target cells or tissues or ex vivo gene delivery into autologous cells followed by adoptive transfer back into the patient (Figure 1). Among the various viral based vector systems, adeno-associated virus (AAV) vectors have demonstrated the greatest clinical success for in vivo gene delivery (Figure 2). A wide array of serotypes and capsid variants enables the targeting of a variety of tissues and cell types. Clinical application of ex vivo gene therapy has primarily focused on gene delivery to autologous hematopoietic stem cells (HSC) to treat hematological and other disorders, or into differentiated lineages such as T lymphocytes for cancer immunotherapy. Retroviral vectors (γ-retroviral or lentivirus derived) are capable of integrating their therapeutic genetic payload into the target cells’ genome and have proven effective for hematopoietic cells. Early adverse events with γ-retroviral vectors have promoted a shift to the use of vectors based on lentivirus (Figure 2), which show a better preclinical safety profile and more efficient gene delivery to nondividing cells.4,5
Figure 1.
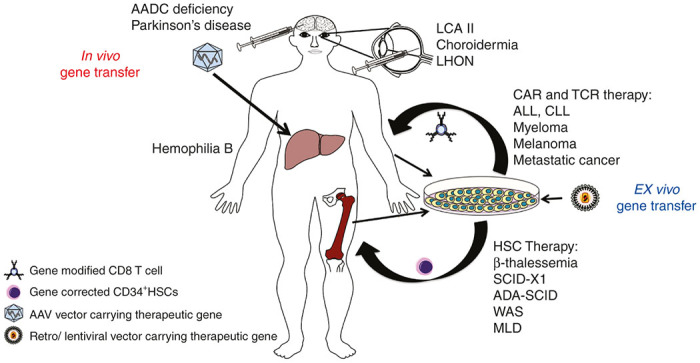
In vivo versus ex vivo gene therapies for the treatment of genetic diseases and cancer. In vivo gene therapy involves direct introduction of vector (carrying the therapeutic gene) into the patient (either into or near the target organ). This strategy has achieved success in the treatment of eye diseases, neurological disorders, and hemophilia In ex vivo gene therapy, a patient’s cells (e.g., hematopoietic cells) are taken out of the body and then transduced by a vector in culture to incorporate the therapeutic gene. Finally, the gene-modified cells are transplanted back to the patient. Various inherited metabolic and immunological disorders and different types of cancers have been successfully treated with ex vivo gene therapy. AADC, aromatic L-amino acid decarboxylase; ADA-SCID, adenosine deaminase severe combined immunodeficiency; ALL, acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; LCA II, Leber’s congenital amaurosis II; LHON, Leber’s hereditary optic neuropathy; MLD, metachromatic leukodystrophy; SCID-X1, X-linked severe combined immunodeficiency; WAS, Wiskott-aldrich syndrome.
Figure 2.
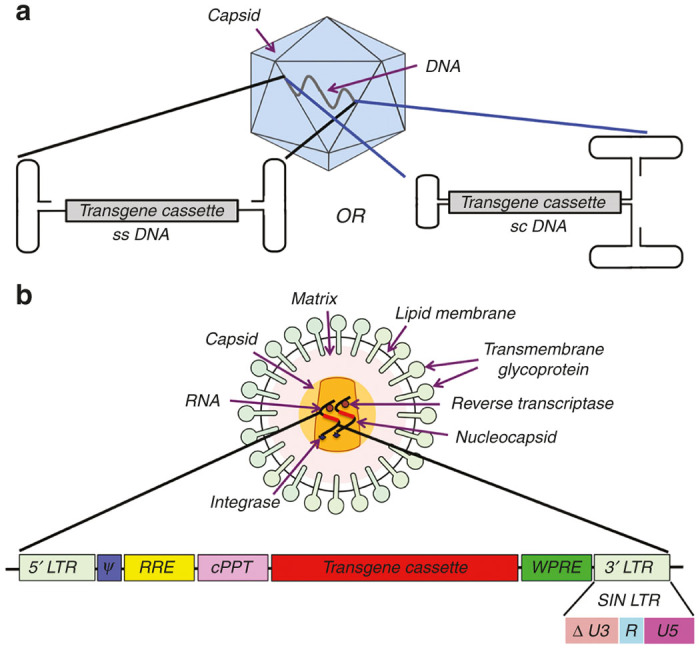
Schematic illustration of two viral vectors widely used in clinical gene therapy. (a) Adeno-associated viral (AAV) vectors are prevalently used for in vivo gene therapy. Given the many serotypes and capsid variants that have been developed, these vectors can target a wide variety of tissues but are limited by their transgene carrying capacity (~5 kb for single-stranded, ssAAV, and 2.5–3 kb for self-complementary, scAAV). (b) Lentiviral vectors (LV) can carry up to 8 kb of transgene and are used in many ex vivo gene therapy protocols, in particular for HSC gene transfer. LV can be pseudotyped with envelopes from different viruses and thereby adapted to a broad range of targets. cPPT, central polypurine tract; LTR, long terminal repeat; Ψ: Packaging signal; RRE, Rev responsive elements; SIN LTR, self-inactivating LTR (with partial deletion in U3 region of 3’LTR); WPRE, Woodchuck hepatitis viral post-transcriptional regulatory element.
Approval of the first-gene therapy product Glybera, an AAV vector for treatment of lipoprotein lipase deficiency, by the European Medicines Agency was an important first step in gene-based drug development.6 Recent breakthroughs in clinical gene therapy trials have now emerged in a variety of monogenic diseases including primary immune deficiencies, hemoglobinopathies, hemophilia B, neurological diseases, ocular diseases, and cancer immunotherapies (excluding oncolytic cancer therapy, which is reviewed elsewhere).7 This review aims to highlight recent successes in gene therapy clinical trials.
Trials and Tribulations on the Path of Treating Primary Immunodeficiencies
Primary immunodeficiencies (PIDs) are rare but life-threatening genetic diseases that severely compromise the integrity and functions of the immune system. Children born with these diseases are often referred to as “bubble boys” or “bubble girls”, as they have to live in a germ free environment because their immune system is unable to fight off microbes that are harmless to immune competent individuals. PIDs targeted by gene therapy include X-linked severe combined immunodeficiency (SCID-X1), adenosine deaminase–deficient severe combined immunodeficiency (ADA-SCID), chronic granulomatous disease, and Wiskott-Aldrich syndrome (WAS). These children typically suffer from recurrent infections, failure to thrive, and death in the first few years after birth (unless they undergo successful bone marrow transplantation). Patients with PID mostly rely on the availability of human leukocyte antigens (HLA)-matched donors for HSC transplantation. With only a small proportion of patients (<20%) finding compatible donors, alternate strategies to treat PIDs are highly desirable.8 Growing success of various gene therapy protocols involving autologous HSCs opened up new treatment avenues for patients without a need for an HLA-matched donor while avoiding a need for immune suppression and the complication of graft versus host disease.9–14
Early experience with gene therapy for SCID-X1, an immunodeficiency disorder characterized by the absence of T cells, impaired B-cell function, lack of natural killer (NK) cell development and γ-chain (γc) dependent cytokines, verified the concept that gene-corrected cells had a selective advantage and could therefore effectively reconstitute immune competence in treated patients. However, these trials also experienced a major setback, as use of murine γ-retroviral vector for ex vivo gene transfer led to the development of leukemia in 5 of the 20 patients treated, thus raising safety concerns regarding the use of γ-retroviral vectors.15–18 Use of self-inactivating (SIN) viral vectors, devoid of long terminal repeats promoter/enhancer function, in recent gene therapy protocols has reduced the risk for insertional mutagenesis and clonal dominance.19–21
A multicenter phase 1/2 clinical trial (#NCT01410019, Paris; #NCT01175239, London; and #NCT01129544, United States) of SCID-X1 employed a SIN γ-retroviral vector to deliver a corrected copy of the interleukin-2 receptor γ chain (IL2RG) gene to autologous HSCs of nine patients.22 Infusion of IL2RG gene transduced autologous HSCs into SCID-X1 patients restored the T-cell population in most patients, who were subsequently able to resolve pre-existing infections. During follow up of 1 to 3 years post-gene therapy, no adverse event related to insertional mutagenesis was reported. Although it is still early for assessment of long-term safety, these results clearly demonstrate the efficacy of these vectors in treating PIDs. Recent data also document that these gene therapy protocols result in faster immunological reconstitution after transplant than conventional haploidentical HSC transplant.23
Murine γ-retroviral vectors have also been employed in gene therapy trials of ADA-SCID, a fatal primary immunodeficiency with impaired T-, B-, and NK-cell development, which puts patients at risk for severe opportunistic infections.11,12,14,24 Patients with ADA-SCID have mutations in a gene-encoding adenosine deaminase (ADA), an enzyme responsible for clearance of toxic purine metabolites from the body. Initial gene therapy trials for ADA-SCID in the early 1990’s utilized γ-retroviral vectors carrying a corrected copy of ADA gene.25,26 These trials were encouraging in that normalization of T lymphocyte counts in some of the treated patients was observed. However, overall results strongly reflected the need to improve engraftment and frequency of gene-corrected HSCs. Moreover, in these trials, the direct effect of gene transfer could not be ascertained as treated patients simultaneously received enzyme replacement therapy. In subsequent clinical trials, patients were preconditioned with a nonmyeloablative regimen, and enzyme replacement therapy was discontinued before infusing autologous HSCs transduced with γ-retroviral vector carrying a functional copy of ADA.12,24,27,28 These measures could entirely reverse the disease phenotype. Follow-up studies in these patients confirmed gene correction in multiple cell lineages, leading to expression of normal ADA levels and restoration of immune competence. It is further encouraging that more than 40 ADA-SCID patients have been treated with these vectors without any signs of vector-related genotoxicity.
More recently, Otsu et al.29 reported clinical findings of γ-retroviral vector mediated gene therapy for ADA-SCID in two patients. The study found that although systemic detoxification and immune recovery was only partial, both patients have not required enzyme replacement therapy for more than 5 years, with observations ongoing. Again, no adverse event related to vector integration was observed. However, due to safety concerns with γ-retroviral vectors, SIN lentiviral vectors (with cellular promoter and codon optimized ADA cDNA) are also being explored for ADA-SCID in clinical studies (NCT01380990, NCT01852071).30,31 These studies employed a mild non-myeloablative conditioning with busulfan (5 mg/kg) before infusing gene-corrected autologous HSCs. Five patients have been treated so far, and significant improvements in total T-cell count and overall immune recovery have been observed at about 1 year of follow-up. These promising results emphasize that though γ-retroviral vectors appeared to be safe in ADA-SCID gene therapy, LVs represents a viable alternate for the future.
WAS is another PID, for which retroviral vector has been employed in clinical studies. During 2006–2009, 10 WAS patients were treated with autologous HSCs transduced retrovirally to carry a corrected copy of WASp gene (following mild myeloablation).32,33 Gene-corrected HSCs engrafted well and showed proliferative and selective advantage over noncorrected cells. Therapeutics levels of WAS protein correlated with clinical benefits (partial to complete resolution of bleeding, eczema, immunodeficiency, and autoimmunity) in these patients. However, following 1–5 years of gene therapy, seven of these patients were reported to develop leukemia, of which two patients died. These clinical outcomes further emphasize the need to address potential risks and safety concerns associated with these vectors.
Based on the preclinical studies, SIN-LVs were developed for subsequent clinical trial for treatment of WAS (#NCT01515462). Results of this study support the safety and efficacy of these vectors.10 In this trial, three children with WAS were treated with bone marrow derived autologous CD34+ HSCs, which were transduced ex vivo with LV-w1.6W vector carrying a corrected copy of the WASp gene. Gene-corrected cells were observed in bone marrow and peripheral blood of all three patients. Stable levels of WASp protein were observed. Most importantly, these patients showed considerable improvement in their clinical symptoms (such as eczema and recurrent infections) and experienced reduced disease severity as early as 6 months after gene therapy. In contrast to γ-retroviral gene therapy for WAS, no insertional mutagenesis or clonal dominance was observed in any of these patients after 20 to 32 months of follow-up.34 Similar positive outcomes have recently been reported for additional patients, who now no longer require a germ-free environment or frequent hospitalization due to bleeding or infection.35,36
Chronic granulomatous disease is a rare genetic disorder caused by mutation in the gp91phox subunit of nicotinamide adenine dinucleotide phosphate oxidase, leading to inability of phagocytic cells (mainly neutrophils and macrophages) to produce reactive oxygen species and therefore to clear bacterial and fungal infections efficiently. Initial clinical trials on gene therapy for chronic granulomatous disease proved relatively unsuccessful as it only provided transient clinical benefits to patients, mainly due to low engraftment of gene-corrected cells.37 Moreover, use of γ-retroviral vectors resulted in clonal dominance of gene-corrected cells leading to the development of myelodysplastic syndrome and monosomy 7 in some of the patients.31,37,38 Current research focuses on utilizing SIN LVs to avoid such insertional mutations and approaches such as the use of myeloid-specific promoter are being adapted to achieve better reconstitution of gene-corrected cells.
Progress Toward Treating Hemoglobin Disorders: β-Thalassemia and Sickle Cell Disease
Worldwide, the monogenic hemoglobin disorders sickle cell anemia and β-thalassemia are major causes of morbidity and early mortality. There are no ideal long-term treatments. Available therapies, while aiming to improve the quality and duration of life, are not curative and have side effects from long-term use. While bone marrow transplantation can be curative, a matched donor is required. Thus, these hematological diseases should be ideal targets for therapeutic gene transfer to HSC. However, since these diseases provide no selective survival advantage to the gene-corrected HSC, it has been a challenge to generate a sufficient number of gene-corrected HSC expressing appropriate levels of the corrected globin protein to correct the defect in erythrocytes. The common blood disorder β-thalassemia results from the loss of functional β-globin, an essential component of hemoglobin in erythrocytes. Current treatment for β-thalassemia patients is frequent blood transfusions and chelation therapy to prevent the accumulation of iron. The first success in correcting β-thalassemia was reported by Cavazzana-Calvo et al.9 where they transduced autologous CD34+ HSCs with a SIN-LV encoding a functional copy of the β-globin gene followed by myeloablative conditioning prior to reinfusion of the gene-corrected HSCs. Since treatment, the subject has displayed stable hemoglobin levels and has been transfusion free for almost 2 years. Recently, early results were reported from 17 patients enrolled in phase 1/2 studies, in which LV transfer of an engineered β-globin gene to HSC was performed. Treatment was particularly successful in patients with some level of endogenous β-globin such as those with βE form. Here, expression of >2 g/dl largely eliminated the need for transfusions and iron chelation therapy, thus vastly improving the patients’ quality of life (even allowing these children to regularly attend school).39,40 A more difficult situation occurs in patients with β0/β0 genotype, as they lack endogenous expression entirely. Thus far, gene therapy more typically has resulted in only partial correction of the disease. These ongoing phase 1/2 trials for β-thalassemia (#NCT01639690, #NCT01745120, #NCT02151526, and #NCT02453477) will provide important long-term follow-up data for the safety and efficacy of gene-corrected HSC and the stability of β-globin expression. With clinical studies being conducted in United States, Australia, Thailand, France, and Italy, a much more comprehensive assessment of the potential of gene therapy for globin disorders should be possible in the near future.
In patients with sickle cell disease, mutations in β-globin generate an abnormal form of hemoglobin, called hemoglobin S or sickle hemoglobin. As a result, red blood cells appear “sickle-shaped”, lack flexibility and stick to vessel walls. In addition to pain, blockage of blood vessels can occur that slows or stops the flow of blood, so that oxygen may not reach nearby tissues. LV-mediated HSC gene transfer of a β-globin sequence with a missense mutation that results in “anti-sickling” properties (βA-T87Q) has now been successfully carried out in a 13-year-old patient with sickle cell disease who did not respond to treatment with hydroxyuria (which aims to increase fetal globin gene expression).40,41 This subject has achieved 47% of β-globin expression derived from the therapeutic transgene and no longer requires red blood cell transfusions (>1-year follow-up). This outcome marks the beginning of a wider use of gene therapy in the treatment of globin disorders.
Toward a Cure for the Coagulation Disorder Hemophilia B
Hemophilia is a hematological disorder caused by mutations in the X-linked gene encoding coagulation factor VIII (hemophilia A) or IX (hemophilia B) and occurs in 1 in 5,000 or 1 in 30,000 male births worldwide, respectively. In its severe form (<1% coagulation activity), the resulting failure of the blood to clot causes spontaneous bleeds into joints and soft tissues, and can be life threatening. Hemophilia patients are currently treated with intravenous infusion of either recombinant or plasma derived FVIII or FIX proteins, which are infused up to two to three times per week in order to prevent serious internal bleeds. This life-long treatment is burdensome and expensive and, due to the costs of the protein drugs, is typically not available in third world countries. In contrast, gene therapy has the potential to be curative, lasting for many years after a single round of gene transfer (>10 years in canine models). Since there is no need for regulated gene expression, levels as low as 5% of normal have a significant impact on bleeding frequencies (FVIII and FIX are secreted in an inactive form with normal plasma levels of 200 and 5,000 ng/ml, respectively). FVIII is primarily synthesized in a subset of endothelial cells, including liver sinusoidal endothelial cells, while hepatocytes are the site of FIX synthesis. Nonetheless, design of a gene therapy for hemophilia offers many choices for target cells and tissues because a number of cell types are capable of synthesizing biologically active FVIII or FIX upon gene transfer.
In preclinical studies using both small and large animals with hemophilia, in vivo gene transfer to the liver using AAV vectors emerged as one of the most efficient and promising protocols. Hemophilia B is considered an ideal first target for this approach because FIX is more easily expressed at high levels than is the case for FVIII. Initial clinical trials by High and colleagues provided proof-of-principle that AAV2 gene transfer via the hepatic artery can correct hemophilia B (FIX deficiency).42–44 A patient in the high-dose cohort of 2 × 1012 vg/kg had expression of FIX for over 2 months, with activity levels peaking at ~12%. However, a cytotoxic T-cell response to the viral capsid prevented sustained therapeutic FIX expression, resulting in mild, vector-dose-dependent liver toxicity manifested as self-limited, asymptomatic elevation of transaminases.43–45 Further studies revealed a memory CD8+ T cells against AAV capsid in humans (who are naturally infected with AAV) that likely eliminated transduced hepatocytes.46 In addition, there is considerable prevalence of neutralizing antibodies against AAV (in particular against serotype 2) in the human population, which blocks gene transfer to the liver above a certain titer. While AAV vectors alone do not provoke strong immune responses unlike other viruses such as adenovirus, these results highlight that the immune system remains a hurdle for in vivo gene transfer.
Nathwani et al. initiated a second clinical trial using a self-complementary genome, a codon-optimized F9 sequence, a different AAV serotype (AAV8), with a reduced frequency of neutralizing antibodies, and a transient immune suppression (IS) regimen with prednisolone if patients presented with a loss in circulating FIX or mild transaminitis.47,48 The switch to AAV8 also allowed for a less-invasive peripheral vein administration, and the lower incidence of neutralizing antibodies made the therapy available to more patients. Extensive testing in nonhuman primates supported the safety and efficacy of this vector in human clinical trials (#NCT00979238).49 Ten hemophilia B patients lacking neutralizing antibodies to AAV8 were enrolled in escalating dose groups. A dose-dependent persistent expression of FIX (1 to 6% of normal levels) was observed in all participants after a single intravenous injection, allowing either the discontinuation of prophylactic FIX protein infusions or a significant reduction in frequency.50 However, capsid-specific antibodies arose in all participants, which would likely block future readministration of vector.51 Relatively high vector doses were required to lift the patients from severe disease (<1% of normal coagulation activity) to mild disease (>5% of normal). A dose of 2 × 1012 vg/kg resulted in a rise of liver enzyme levels (aspartate aminotransferase and alanine aminotransferase) in four out of six participants. These patients were started on prednisolone at 60 mg/day, which was tapered and stopped over a period of ~8 weeks. While this regimen prevented the complete loss of circulating FIX protein, as seen in the initial AAV2 clinical trial, some patients had a reduction in circulating FIX levels from the early peak levels. However, it should be noted that all subjects experienced sustained multi-year expression of FIX.52,53
A new phase 1/2 clinical trial was recently started by Monahan et al. in which they gained approval to use a naturally occurring hyperactive FIX variant (R338L, FIX-Padua) in a self-complementary AAV8 vector (#NCT01687608).54–58 In preclinical studies using animal models of hemophilia B, long-term expression with improved catalytic activity of FIX variant (FIXR338L) at lower vector doses (scAAV8-FIXR338L) were observed.59 Three dose cohorts (of up to 3 × 1012 vg/kg) were incorporated. Early treatment data show that one patient receiving a mid-range dose of 1 × 1012 vg/kg has achieved sustained levels of 20–25% of normal, which is considered curative.60,61 However, subjects treated with the highest vector dose lost expression, showing transaminitis and IFN-γ producing T cells in response to viral capsid antigen. In contrast to the trial by Nathwani et al., immune suppression with prednisolone could not rescue expression. Other patients of the mid-range dose group also achieved only transient therapeutic levels, though the reason for their loss of expression is unclear as no liver toxicity or T-cell responses were observed. On the positive side, with over 20 hemophilia B patients treated with different AAV-F9 vectors there has been no indication of patients developing an immune response against the FIX protein. Recent data from ongoing clinical trials suggest that stable therapeutic and even curative levels of FIX protein activity are now obtainable in patients. Going forward, several critical questions need to be answered. For example, why did one trial using scAAV8 consistently yield sustained levels >5% at the highest dose while a second trial with a similar vector and dose did not? Can this be explained by differences in the design (e.g., presence of immune stimulatory CpG motifs) or production of the vectors or by other factors? Why was immune suppression with steroid drugs successful in one trial but not the other? Could other immune suppression regimens and/or advancements in vector engineering provide superior results?
Advances in design and molecular evolution of AAV capsids and testing in humanized mouse models will hopefully result in vectors with better performance in the human liver, which will be critical in adapting this approach to hemophilia A, as expression and efficient secretion of the larger FVIII molecule is more challenging.62–66 To what extent vectors with higher transduction efficiency allow for a reduction in vector dose in humans (which would reduce capsid antigen presentation) remains to be seen, as it is possible that a threshold in the form of a minimally required number of particles exists for efficient transduction. Similar efforts are ongoing to design protocols that overcome pre-existing immunity in humans and that limit capsid antigen presentation.67–69 Superior immune suppression protocols are being developed in parallel in case vector development by itself is insufficient to solve the immunological hurdles.49,70,71 With sufficient levels of hepatocyte transduction and transgene expression, AAV hepatic gene transfer mediated immunological tolerance induction may ultimately be used as a dual therapy to eliminate established inhibitory antibodies to coagulation factors and other therapeutic proteins and provide therapeutic protein expression.72–75
Inherited Neurological Disorders
Neurological disorders are among the most difficult diseases to treat with conventional pharmacological drugs because of the complexity of the central nervous system (CNS) and the existence of physical barriers such as the blood brain barrier. Gene therapy can potentially overcome these limitations but also faces substantial hurdles to delivery of the vector, targeting specific cells types within the CNS, and having to achieve adequate levels of gene expression within a therapeutic window. Nonetheless, successful gene therapies have now been reported for various genetic diseases of the CNS such as adrenoleukodystrophy (ALD), metachromatic leukodystrophy, and aromatic L-amino acid decarboxylase (AADC) deficiency. Both integrating (LV) and nonintegrating (AAV) vectors have been successfully used in these gene therapy trails.
Successful use of LV in neurological disorders was first reported in ALD, a genetic disorder of CNS in which mutations in ABCD1 gene (encoding ALD enzyme) results in accumulation of very long chain fatty acids causing demyelination of CNS and the adrenal cortex. In this clinical study, two ALD patients (after a complete myeloablative conditioning) were treated with LV-mediated gene-corrected autologous HSCs.76 More than 3 years of follow-up studies in these patients showed persistent therapeutic levels of ALD protein with no further demyelination of CNS and stabilization of disease. Moreover, no major safety concern was reported in any of these patients.77 However, larger cohorts with longer follow-up periods are needed to strengthen the safety and efficacy profile of these vectors to promote their use in gene therapy of ALD and other neurological disorders.
Late infantile metachromatic leukodystrophy is a fatal genetic disorder, in which first sign of symptoms appear in the second year of life and patients die within the first decade of their life. These patients lack arylsulfatase A (ARSA), an enzyme whose deficiency leads to accumulation of sulfatide (a glycolipid with sulfate group) in myelin-producing cells, causing demyelination of the nervous system leading to severe motor and cognitive damage. Unfortunately, bone marrow transplant or HSC transplant are not effective treatments for this disease because replacement of resident tissue macrophages and microglia by the transplanted hematopoietic cell progeny does not keep pace with the rapidly progressing disease. While bone marrow transplant, if given early enough, may have some stabilizing effect on neurocognitive function, it typically fails to halt loss of motor function. Biffi et al.13 (#NCT01560182) hypothesized that overexpression of ARSA in gene-modified hematopoietic cells might overcome these limitations of bone marrow transplant by delivering a level of ARSA that would correct neighboring cells and thus halt demyelination. The authors employed SIN-LV-mediated gene transfer in autologous CD34+ HSCs of three presymptomatic patients. A dose-adjusted treatment of busulfan prior to gene transfer resulted in engraftment of bone marrow and peripheral blood with high frequencies of gene-corrected cells. In this approach, microglia derived from gene-corrected HSC serve to deliver ARSA to the CNS. After 2 years of gene transfer, these children continued to produce therapeutic levels of functional ARSA enzyme and showed normal motor and cognitive development for their ages. Moreover, these patients are well past their expected age of disease manifestation. Insertional mutagenesis was not observed in these patients, although long-term results in these patients are still pending.
AADC deficiency is another devastating neuronal genetic disorder for which gene therapy is being explored, as existing drug therapy provides little to no benefit to the patients. AADC deficiency impairs the synthesis and secretion of neurotransmitters such as dopamine and serotonin leading to developmental delay, oculogyric crises, dystonia, truncal hypotonia, sweating, severe movement disorders, tongue protrusion, jaw spasms, and neurological impairment in infants. In a recent clinical trial (#NCT01395641) of AADC deficiency, Hwu et al. directly injected AAV2 vector carrying AADC gene into the bilateral putamen of four 4- to 6-year-old patients.78,79 All subjects were reported to gain weight after 3 to 6 months post-gene transfer and had better head control and emotional stability. Importantly, their Alberta Infant Motor Scale, Peabody Developmental Motor Scale, and Comprehensive Developmental Inventory for Infants and Toddlers scores were better after 15 to 24 months of gene therapy, indicating improvement in their motor and cognitive functions. The only adverse events observed were transient increases in dyskinesia in two patients and frequent episodes of apnea in one patient, which declined to normal by 10 months after gene transfer. These results demonstrate the potential of AAV-mediated gene therapy delivered by intracerebral infusion. However, long-term efficacy of treatment remains to be documented. For example, a phase 1 clinical trial (#NCT00229736) of Parkinson’s disease showed initial improvement post-AAV-mediated gene transfer but failed to achieve a lasting effect.80 Nonetheless, multi-year transgene expression in the human brain was documented in this as well as in a trial on Canavan disease.80,81 In evaluation of these approaches, one has to keep in mind that design of phase 1 clinical safety studies for neurodegenerative disease is difficult. In older patients with advanced disease, it may not be possible to sufficiently restore the damage to achieve a therapeutic effect or to target gene transfer to the ideal part of the CNS. However, there are also limitations to the gene delivery technology, resulting in transduction of too few neurons or other targeted cell types in the CNS. Early AAV2-based vectors could not spread from the injection site because of binding to extracellular matrix components. Spread from the injection site is much improved with use of alternative serotypes and capsid engineering. Nonetheless, the route of administration also needs to be optimized to achieve delivery to wider areas of the CNS. For example, infusion of the vector into the cisterna magna (for delivery into cerebrospinal fluid) or use of vectors that can cross the blood brain barrier is being explored to replace conventional intracranial injections.82,83 Finally, promising results on correction of motor neurons in infant children have now been reported for a clinical trial on intravenous AAV9 delivery for spinal muscular atrophy type 1 (#NCT02122952; 2016 annual meeting of the American Society of Gene and Cell Therapy).
Inherited Retinal Diseases
AAV vectors are highly effective in ocular gene transfer and have therefore been extensively used in gene therapy protocols of various retinal diseases, including inherited forms of blindness for which no treatment existed. Leber’s congenital amaurosis type 2 (LCA2) is the first retinal hereditary disease that showed impressive clinical success with this type of gene therapy.84–87 In LCA2 patients, mutations in RPE65 gene prevent the expression of retinal pigment epithelium 65 kilodalton protein (RPE65), thereby impairing the process of visual photo-transduction and thus severely limiting vision in these patients. Three clinical studies carried out independently by different centers demonstrated that a single subretinal injection of AAV2 vector carrying the therapeutic gene (RPE65) improved vision in treated regions of the retina, resulting in improved vision that was stable for at least 3 years.84–86,88–92 One protocol was successfully expanded to a trial in pediatric patients, demonstrating that early intervention vastly improved the potential for restoring vision.93 Patients showed a 2 log or more unit increase in pupillary light responses, and an 8-year-old child gained light sensitivity to nearly that of age-matched normal-sighted individuals. This approach has now been further advanced to a phase 3 clinical trial.94
Recent long-term evaluation of patients from two of the aforementioned clinical trials (#NCT00481546 and #NCT00643747) suggested a decline in retinal sensitivity, visual acuity, and functional gain over time, which however has not been observed in the third study.95,96 Differences in vector design, final formulation, immunomodulatory regimens used (transient around vector administration), and surgical approach, may all contribute to the observed differences. For safety reasons, patients enrolled in these early trials had received gene transfer to only one eye. Although safety and efficacy data from these clinical trials supported subsequent treatment of the contralateral eye, there was concern that induction of immunity to the viral capsid or possibly the transgene product would not only limit therapy from a second injection, but may also affect the functionality gained by the first treatment. On the other hand, since the eye is considered immune privileged and relatively low vector doses are used, it is hypothesized that repeated gene transfer is possible without toxicity. To address these points, Bennett et al.97 designed a protocol (#NCT01208389) to inject the contralateral eye of three patients with the identical AAV2 vector encoding the RPE65 gene. Within 3 months of the second gene transfer, patients showed gradually improved sensitivity to dim light, activation of the visual cortex on fMRI, and navigational skills using the recently injected eye without any adverse effect to their previously injected eye. These functional gains were more pronounced in younger patients suggesting that older patients with highly degenerated retinas might benefit less. The extent of retinal degeneration in LCA2 patients, as in many inherited retinal dystrophies, advances with age. Preclinical studies in murine and canine models have shown that gene augmentation could slow down the process of degeneration provided that the therapeutic intervention starts at an early, predegenerative stage of the disease.98–100 More recently, Cideciyan et al.101 found that though there was markedly improved visual functionality in LCA2 patients treated with gene therapy, this did not halt progression of retinal degeneration. However, these results may reflect suboptimal vector dose/gene transfer at a stage of retinal degeneration that could not be salvaged. Therefore, future clinical studies will be designed to better address both visual functionality and slowing down/halting the process of retinal degeneration. Approaches to halt other genetic causes of retinal degeneration are also being developed and show promise.102
Success with the LCA2 gene therapy has generated interest in developing gene therapy for other retinal diseases. MacLaren et al.103 reported successful initial results of a gene therapy trial (#NCT01461213) for choroidermia. This retinal genetic disease is due to a nonfunctional copy of the CHM gene, resulting in slow and progressive degeneration of the patient’s photoreceptors, choroid and retinal pigmented epithelium, and leading to complete blindness by middle age. A subretinal administration of the AAV2 vector carrying the CHM gene substantially improved vision in two patients and increased retinal sensitivity in four more patients after 6 months of gene therapy. Working toward a gene therapy for Leber hereditary optic neuropathy, Koilkonda et al.104 demonstrated in a murine model of this disease that a single intravitreal injection of scAAV2 (with triple Y-F mutations in AAV2 capsid) carrying a wild-type human ND4 gene (a mitochondrial gene affecting complex I of electron transport chain) was able to arrest further deterioration of the optic nerve. Further, a significantly high rate of complex-I-dependent ATP synthesis was observed in eyes rescued with ND4, suggesting correction of the impaired electron transport chain. Two gene therapy trials (#NCT01267422 and NCT02161380) utilizing AAV as a vector for Leber hereditary optic neuropathy gene therapy are currently enrolling patients.
Chimeric Antigen Receptor-Based Immunotherapy
Successful gene therapy is not limited to genetic diseases. Cancer immunotherapy based on genetic modification of autologous T cells has received much attention as it is highly effective at eradicating B-cell leukemias and lymphomas that are resistant to standard therapies in cancer patients. Autologous CD8+ T cells are engineered to recognize and kill cells bearing tumor-specific antigens through a CAR that combines the specificity of a monoclonal antibody with the proliferative and cytotoxic abilities of an activated CD8+ T cell. Antigen receptor and costimulatory molecule signaling is complexed with antibody-based antigen recognition, bypassing the need for HLA restriction, which is often downregulated in transformed cells, or the requirement for antigen presenting cells. CAR-modified autologous CD8+ T cells are generated by ex vivo gene transfer with LV, expanded and can be banked for repeated transfusions under well-established cGMP-compliant manufacturing processes.105 Three generations of CARs have been developed with different combinations of signaling domains, with second- and third-generation CARs showing the greatest efficacy.106,107 CARs have been successfully employed in clinical trials of modified T cells in patients with relapsed and refractory B-cell leukemias, B-cell lymphoma, chronic lymphocytic leukemia (CLL), and acute lymphoblastic leukemia (ALL).
In one of the three seminal studies published recently, Brentjens et al.108, at the Memorial Sloan Kettering Cancer Center reported on the safety of infusing a second-generation CAR that coupled the T-cell receptor ζ chain with the costimulatory CD28 molecule. This 19-28z CAR was transduced into autologous T cells of eight patients with CLL (#NCT00466531) and one patient with ALL (#NCT01044069), with or without prior conditioning therapy. Patients had poor genetic prognostic markers, including deletions in the p53 gene and exhibited advanced disease as evidenced by bulky lymphadenopathy. All patients tolerated the infusion well and did not develop cytokine release syndrome. Of the four CLL patients treated with cyclophosphamide before T-cell infusion, three patients exhibited either stable disease or a marked reduction of peripheral lymphadenopathy, whereas B-cell aplasia was observed in B-ALL patients. Rapid trafficking of 19-28z+ T cells to sites of CD19+ tumor and in vivo persistence of transplanted cells was observed. In another clinical study (#NCT01029366) at the University of Pennsylvania, CD19 targeting CAR T cells that contained a costimulatory domain from CD137 (4-1BB) and the T-cell receptor ζ chain (CTL019) showed potent non-cross-resistant clinical activity after infusion in three of three patients treated with advanced and refractory CLL.109,110 High levels of expansion of CTL019 CAR T cells (>1,000-fold) were observed in all three patients, who continued to express functional CARs at high levels for at least 6 months. Moreover, a portion of these cells persisted as memory CAR+ T cells and retained anti-CD19 effector functionality. Of the three patients treated, there were two complete responses and one partial response lasting greater than 8 months after CTL019 infusion. In a third study, Kochenderfer et al.111 at the NIH reported a phase 1 clinical trial of CLL (#NCT00924326), in which patients received chemotherapy followed by an infusion of autologous anti CD19-CAR transduced T cells and a follow-up course of intravenous IL-2. Six of the eight treated patients showed remissions of their malignancies, and four of eight patients had long-term elimination of CD19+ B-lineage cells. Anti-CD19 CAR-transduced T cells could be detected in the blood of patients for up to 181 days after infusion. Elevations in serum levels of IFN-γ and TNF-α correlated with significant toxicity observed in the patients. The same group also reported on a phase 1/2 clinical trial (#NCT01087294) using allogenic donor hematopoietic stem cells, with 3 out of 10 patients achieving remission. Donor-derived anti-CD19 CAR T cells were detected in the blood of 8 of the 10 patients. Associated toxicities were transient, and none of the patients receiving donor derived T cells experienced graft versus host disease.112
Given these encouraging responses in patients with CLL, researchers extended their findings to the more aggressive B-ALL, which also expresses the CD19 antigen. Long-term survival of adult patients with relapsed B-ALL is dependent upon achieving a complete remission induced through chemotherapy followed by allo-HSC therapy. Lee et al.113 recently reported a phase 1 dose-escalation trial (#NCT01593696) that had enrolled 21 patients with relapsed or refractory B-ALL. The maximum tolerated dose was established, and a complete response was observed in 14 of the 21 patients (representing 66.7% complete response rate). Cytokine release syndrome was observed in 3 of the 21 patients but was controlled by treatment. All other toxicities observed were also reversible. Although CD19 CAR T-cell expansion was observed, long time persistence was not seen past day 68. In another clinical study (#NCT01044069), Brentjens et al.114 also infused autologous 19-28z+ CAR T cells into five relapsed adult B-ALL subjects with persistent morphological disease or minimal residual disease after salvage chemotherapy. Rapid tumor eradication and complete remissions were observed in all patients, with one relapse 90 days into therapy. While the infused CAR T cells were otherwise well tolerated, significantly elevated cytokine levels required lympholytic steroid therapy. Patients were subsequently able to undergo allo-HSC therapy 1–4 months after 19-28z+ CAR T-cell infusion. In a larger cohort, an overall complete response of 88% was observed, which in some cases occurred at 2 weeks or sooner after treatment began, allowing most treated patients to transition to allo-HSC therapy.115 Maude et al.116 used CTL019 CAR T cells to treat 30 patients with relapsed and refractory ALL (#NCT01626495 and #NCT01029366). Complete remission was observed in 90% patients after the infusion of LV transduced autologous CTL019 cells. CTL019 cells proliferated in vivo and were detectable in the blood, bone marrow and cerebrospinal fluid of patients for up to 11 months. Here, all patients developed cytokine release syndrome, which was severe in eight patients but was resolved by treatment with the anti-IL-6 receptor antibody tocilizumab. In an unexpected outcome from a previous study by this group (with CTL019 T-cell infusion into two patients with ALL), a relapse occurred in one patient with blast cells that no longer expressed CD19. These findings highlight the need to develop CARs that recognize other tumor-associated antigens in B-cell leukemias and lymphomas.117
While the data summarized above demonstrate significant remission of malignancy by CAR T-cell therapy in CLL and ALL patients, serious but manageable adverse events, including B-cell aplasia, tumor lysis syndrome and cytokine release syndrome, have also been reported. Approaches providing a controlled inflammatory response to tumor lysis such as determining optimal costimulatory signaling domains or suicide gene switches are being explored to enhance safety. Further identification of tumor-specific antigens and their use in CAR T-cell therapy would avoid the killing of nonmalignant cells, thereby providing better specificity. CAR T-cell doses and conditioning regimens have definitive influences on the outcome of therapy and thus need to also be further evaluated. Use of CARs in other cell types may provide an alternate strategy for patients, in which the use of T cells is not feasible. For example, CAR NK cells have been shown to be cytotoxic to B-cell leukemia, and transducing CARs into FoxP3+ regulatory CD4+ T cells (Treg) could provide specific immunosuppression for the treatment of autoimmune diseases.118–120 Similarly, transfer of T-cell receptor (TCR) genes into Treg or fusion proteins into B cells could be used clinically for immune tolerance induction.121,122 CAR gene transfer to T and NK cells also shows potential for generating immunity to human immunodeficiency virus.123 Finally, CAR T-cell therapies are now being developed for solid tumors and other malignancies.
TCR Gene-Modified T Cells for Cancer Immunotherapy
Gene transfer of cloned TCRs isolated from tumor infiltrating T cells represents another approach for T-cell-based cancer immunotherapy, especially for tumor antigens not expressed on the cell surface. In TCR gene therapy, the patient’s T cells are again engineered ex vivo to redirect their specificity toward a particular tumor antigen. These tumor-specific engineered T cells are then reinfused back to the patient, where they recognize tumor antigen in the context of HLAs in the tumor microenvironment. Various clinical trials have employed genetically modified TCRs to treat a wide variety of cancers (synovial cell sarcoma, neuroblastoma, melanoma, and colorectal cancer) with long-term tumor regression.124–129 However, due to high sensitivity, these engineered T cells can target normal cells expressing low levels of target antigen, which has lead to serious “on target-off tumor” toxicities in treated patients.125,127,130 Cancer-testes antigens (CTAs), a group of tumor-associated antigens, provide an excellent alternative target for TCR gene therapy, since their expression is limited to male germ cells in the testis and in some cases in ovary, trophoblasts, or placenta. Moreover, various types of cancers (including lung, breast, ovary, bladder, and melanoma) share many CTAs, with their expression frequency ranging from 30–80%. Therefore, one type of CTA-specific TCR gene therapy could be used against several types of cancers. Several recent clinical studies (#NCT01350401; #NCT01352286; #NCT01273181) utilized affinity enhanced TCRs to target MAGE-A3 (one of the CTAs highly expressed in different types of cancers) in melanoma, myeloma, and metastatic cancer patients.131,132 Five of the nine metastatic patients treated by Morgan et al.132 responded positively to the treatment and underwent tumor regression. However, three of the nine treated patients developed serious neurotoxicity related to the treatment, resulting in two fatalities. Post-autopsy assays on the deceased patients’ brains suggested that expression of MAGE-A12 in human brain tissue might have contributed to the neurotoxicity. Similarly, two melanoma patients treated by Linette et al.131, died due to cardiac shock following MAGE-A3-specific T-cell infusion. Post-autopsy assays suggested that nonspecific recognition of an unrelated peptide derived from the striated muscle-specific protein titin led to severe myocardial damage in these patients. Though tumor regression in five patients treated by Morgan et al., demonstrate efficacy of the approach, overall results from these two studies raise serious safety concerns for use of MAGE-A family members as the target for TCR gene therapy. Moreover, inability to predict such adverse events in preclinical studies of these targets clearly underscores the need for superior means to identify potential off-targets of engineered TCRs.
Encouragingly, a very recent clinical study (#NCT01352286) by Rapoport et al.133 reported the safety and efficacy of NY-ESOc259, a human-derived affinity-enhanced TCR that recognizes a peptide shared by two CTAs (NY-ESO-1- and LAGE-1), in 20 patients with multiple myeloma. Infusion of the engineered T cells did not cause adverse events. Moreover, the infused cells showed target-specific antitumor activity with significant proliferation and persistence, which was well correlated with progression free survival in 16 of the 20 treated patients up to 20 months. These results suggest that targeting multiple antigens increases the chance to achieve complete remission. Such advances would probably be able to target a larger cohort of patients with improved outcomes. Although outside the scope of this review, it should be pointed out that cancer immunotherapy of solid tumors using genetically engineered oncolytic viruses has had similar success.134
Future Outlook: Precision Gene Therapy Through Gene Editing
Common characteristics shared by the success of ongoing clinical gene therapy trials are that: (i) disease is caused by the absence of a functional protein and conversely the presence of a mutated protein does not interfere with the therapy; (ii) regulated expression is not necessary due to suboptimal expression levels and the specific disease; and (iii) phenotypic correction with gene therapy can be achieved either directly through gene delivery to postmitotic tissues or indirectly through genetic modification of stem cells. Thus for the broader application of gene therapy to treat genetic-based diseases, the field needs to advance beyond gene addition strategies. One such path is the incorporation of genome-editing technology to either correct endogenous disease causing genes or to specifically target the integration of a therapeutic gene into a defined genetic locus. Such tools include zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeat (CRISPR)–associated systems (CRISPR–Cas). These engineered endonucleases can be programmed to specifically target and alter a DNA sequence by introducing a double-strand break and can therefore be employed to correct a disease-causing mutation with great efficiency, representing a sophisticated tool for precision medicine. Much of initial work has focused on establishing proof of principle and developing reagents and animal models for these gene-editing tools. ZFNs are the first to be investigated in clinical trial (#NCT00842634). Tebas et al.135 infused 12 patients with autologous CD4+ T cells, in which the CCR5 gene, a coreceptor of human immunodeficiency virus, was inactivated by ZFNs. The study reports a significant increase in CD4+ T cells postinfusion and long-term persistence of CCR5-modified CD4+ T cells in peripheral blood and mucosal tissue. In parallel, TALENs and CRISPR-Cas system are undergoing preclinical development as potentially more versatile gene-editing tools. For instance, the CRISPR-Cas system was employed to study the effects of gene modifications in postmitotic neurons in the mouse brain or to correct a hereditary disease, Tyrosinemia, in a mouse model.136,137 ZFN-mediated gene editing and a system for targeting without using nucleases have shown promise in correction of the F9 gene in hepatocytes of hemophilia B mice.138,139 However, the current efficiency of gene editing may be subtherapeutic for certain diseases, where edited cells have no proliferative or survival advantage, and off-target double strand breaks may induce genotoxicity. Therefore, these gene-editing tools need further refinement before they can be safely and effectively used in the clinic. In addition, a guideline should be established on the ethical use of gene-editing tools such as for the editing of embryos to correct germ line mutations.140
Conclusions
Clinical gene therapy has matured over the past decade, so that the field can now point to several impressive successes. These include a wide variety of diseases and modes of gene transfer. LV and AAV vectors have primarily been used in these trials, while other vector systems are expected to also further advance in their clinical applications. Lessons learned from successes as well as from problems and impediments encountered in recent trials will drive innovation of clinical gene therapy approaches. Next-generation protocols are already being developed, which will also help expand the spectrum of diseases that can be treated by gene therapy. For some of the most difficult targets such as muscular dystrophies and several of the lysosomal storage and neurological disorders, rapid success is less likely. Nonetheless, continuous progress is being made toward future treatments. Ultimately, gene therapy will become more precise with the incorporation of gene-editing tools, as has recently demonstrated in genome editing of HSC.141,142
Dr. High is a co-founder, employee, and equity holder in Spark Therapeutics. She also holds issued patents related to AAV gene therapy. Dr. Herzog holds issued patents related to AAV gene therapy and has been receiving royalty payments from Spark Therapeutics.
References
- Naldini, L (2009). Medicine. A comeback for gene therapy. Science 326: 805–806. [DOI] [PubMed] [Google Scholar]
- Herzog, RW, Cao, O and Srivastava, A (2010). Two decades of clinical gene therapy–success is finally mounting. Discov Med 9: 105–111. [PMC free article] [PubMed] [Google Scholar]
- Couzin-Frankel, J (2013). Breakthrough of the year 2013. Cancer immunotherapy. Science 342: 1432–1433. [DOI] [PubMed] [Google Scholar]
- Wang, X, Shin, SC, Chiang, AF, Khan, I, Pan, D, Rawlings, DJ et al. (2015). Intraosseous delivery of lentiviral vectors targeting factor VIII expression in platelets corrects murine hemophilia A. Mol Ther 23: 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, BD (2015). A shot in the bone corrects a genetic disease. Mol Ther 23: 614–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylä-Herttuala, S (2012). Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol Ther 20: 1831–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, HL, Kohlhapp, FJ and Zloza, A (2015). Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 14: 642–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith, LM, Cowan, MJ, Notarangelo, LD, Kohn, DB, Puck, JM, Pai, SY et al. workshop participants. (2014). Primary Immune Deficiency Treatment Consortium (PIDTC) report. J Allergy Clin Immunol 133: 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo, M, Payen, E, Negre, O, Wang, G, Hehir, K, Fusil, F et al. (2010). Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 467: 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Biasco, L, Scaramuzza, S, Ferrua, F, Cicalese, MP, Baricordi, C et al. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341: 1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Cattaneo, F, Galimberti, S, Benninghoff, U, Cassani, B, Callegaro, L et al. (2009). Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med 360: 447–458. [DOI] [PubMed] [Google Scholar]
- Aiuti, A, Slavin, S, Aker, M, Ficara, F, Deola, S, Mortellaro, A et al. (2002). Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 296: 2410–2413. [DOI] [PubMed] [Google Scholar]
- Biffi, A, Montini, E, Lorioli, L, Cesani, M, Fumagalli, F, Plati, T et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341: 1233158. [DOI] [PubMed] [Google Scholar]
- Gaspar, HB, Cooray, S, Gilmour, KC, Parsley, KL, Zhang, F, Adams, S et al. (2011). Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med 3: 97ra80. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina, S, Garrigue, A, Wang, GP, Soulier, J, Lim, A, Morillon, E et al. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 118: 3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina, S, von Kalle, C, Schmidt, M, Le Deist, F, Wulffraat, N, McIntyre, E et al. (2003). A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 348: 255–256. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina, S, Von Kalle, C, Schmidt, M, McCormack, MP, Wulffraat, N, Leboulch, P et al. (2003). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302: 415–419. [DOI] [PubMed] [Google Scholar]
- Howe, SJ, Mansour, MR, Schwarzwaelder, K, Bartholomae, C, Hubank, M, Kempski, H et al. (2008). Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest 118: 3143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini, E, Cesana, D, Schmidt, M, Sanvito, F, Ponzoni, M, Bartholomae, C et al. (2006). Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol 24: 687–696. [DOI] [PubMed] [Google Scholar]
- Modlich, U, Navarro, S, Zychlinski, D, Maetzig, T, Knoess, S, Brugman, MH et al. (2009). Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther 17: 1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W, Russ, JL and Eiden, MV (2012). Evaluation of residual promoter activity in γ-retroviral self-inactivating (SIN) vectors. Mol Ther 20: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina, S, Pai, SY, Gaspar, HB, Armant, M, Berry, CC, Blanche, S et al. (2014). A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med 371: 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzot, F, Moshous, D, Creidy, R, Neven, B, Frange, P, Cros, G et al. (2015). Faster T-cell development following gene therapy compared with haploidentical HSCT in the treatment of SCID-X1. Blood 125: 3563–3569. [DOI] [PubMed] [Google Scholar]
- Candotti, F, Shaw, KL, Muul, L, Carbonaro, D, Sokolic, R, Choi, C et al. (2012). Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood 120: 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn, DB, Hershfield, MS, Carbonaro, D, Shigeoka, A, Brooks, J, Smogorzewska, EM et al. (1998). T lymphocytes with a normal ADA gene accumulate after transplantation of transduced autologous umbilical cord blood CD34+ cells in ADA-deficient SCID neonates. Nat Med 4: 775–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn, DB, Weinberg, KI, Nolta, JA, Heiss, LN, Lenarsky, C, Crooks, GM et al. (1995). Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med 1: 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Cassani, B, Andolfi, G, Mirolo, M, Biasco, L, Recchia, A et al. (2007). Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J Clin Invest 117: 2233–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar, HB (2012). Gene therapy for ADA-SCID: defining the factors for successful outcome. Blood 120: 3628–3629. [DOI] [PubMed] [Google Scholar]
- Otsu, M, Yamada, M, Nakajima, S, Kida, M, Maeyama, Y, Hatano, N et al. (2015). Outcomes in two Japanese adenosine deaminase-deficiency patients treated by stem cell gene therapy with no cytoreductive conditioning. J Clin Immunol 35: 384–398. [DOI] [PubMed] [Google Scholar]
- Gaspar, BB, Rivat, C, Himoudi, N, Gilmour, K, Booth, C and Xu-Bayford, J (2014). Immunological and metabolic correction after lentiviral vector mediated haematopoietic stem cell gene therapy for ADA deficiency. J Clin Immunol 34: Suppl 2:S167–S168. [Google Scholar]
- Cicalese, MP and Aiuti, A (2015). Clinical applications of gene therapy for primary immunodeficiencies. Hum Gene Ther 26: 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boztug, K, Schmidt, M, Schwarzer, A, Banerjee, PP, Díez, IA, Dewey, RA et al. (2010). Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med 363: 1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, CJ, Boztug, K, Paruzynski, A, Witzel, M, Schwarzer, A, Rothe, M et al. (2014). Gene therapy for Wiskott-Aldrich syndrome–long-term efficacy and genotoxicity. Sci Transl Med 6: 227ra33. [DOI] [PubMed] [Google Scholar]
- Qasim, W and Gennery, AR (2014). Gene therapy for primary immunodeficiencies: current status and future prospects. Drugs 74: 963–969. [DOI] [PubMed] [Google Scholar]
- Ferrua, FM, Galimberti, S, Scaramuzza, S, Giannelli, S, Pajno, R, Dionisio, F, et al. (2015). Safety and clinical benefit of lentiviral hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Blood 126: 259. [Google Scholar]
- Chu, JI, Myriam Armant, L, Male, F, Dansereau, CH, MacKinnon, B, Burke, CJ, et al. (2015). Gene therapy using a self-inactivating lentiviral vector improves clinical and laboratory manifestations of Wiskott-Aldrich syndrome. Blood 126: 260. [Google Scholar]
- Grez, M, Reichenbach, J, Schwäble, J, Seger, R, Dinauer, MC and Thrasher, AJ (2011). Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther 19: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, S, Ott, MG, Schultze-Strasser, S, Jauch, A, Burwinkel, B, Kinner, A et al. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 16: 198–204. [DOI] [PubMed] [Google Scholar]
- Walters, MC, Suradej Hongeng, J, Kwiatkowski, J, Schiller, GJ, Kletzel, M, Ho, PJ, et al. (2015). Update of results from the Northstar Study (HGB-204): a phase 1/2 study of gene therapy for beta-thalassemia major via transplantation of autologous hematopoietic stem cells transduced ex-vivo with a lentiviral beta AT87Q-globin vector (LentiGlobin BB305 Drug Product). Blood 126: 201. [Google Scholar]
- Cavazzana, M, Emmanuel Payen, J-A, Suarez, F, Beuzard, Y, Touzot, F, Cavallesco, R, et al. (2015). Outcomes of gene therapy for severe sickle disease and beta-thalassemia major via transplantation of autologous hematopoietic stem cells transduced ex vivo with a lentiviral beta AT87Q-globin vector. Blood 126: 202. [Google Scholar]
- Malik, P (2016). Gene therapy for hemoglobinopathies: tremendous successes and remaining caveats. Mol Ther 24: 668–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay, MA, Manno, CS, Ragni, MV, Larson, PJ, Couto, LB, McClelland, A et al. (2000). Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet 24: 257–261. [DOI] [PubMed] [Google Scholar]
- Manno, CS, Chew, AJ, Hutchison, S, Larson, PJ, Herzog, RW, Arruda, VR et al. (2003). AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 101: 2963–2972. [DOI] [PubMed] [Google Scholar]
- Manno, CS, Pierce, GF, Arruda, VR, Glader, B, Ragni, M, Rasko, JJ et al. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 12: 342–347. [DOI] [PubMed] [Google Scholar]
- Mingozzi, F, Maus, MV, Hui, DJ, Sabatino, DE, Murphy, SL, Rasko, JE et al. (2007). CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med 13: 419–422. [DOI] [PubMed] [Google Scholar]
- Hui, DJ, Podsakoff, GM, Pein, GC, Ivanciu, L, Camire, RM, Ertl, H, et al. High Etiena Basner-Tschakarjan (2015). AAV capsid CD8+ T cell epitopes are highly conserved across AAV serotypes. Mol Ther Methods Clin Dev 2: 15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Gray, JT, McIntosh, J, Ng, CY, Zhou, J, Spence, Y et al. (2007). Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood 109: 1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Gray, JT, Ng, CY, Zhou, J, Spence, Y, Waddington, SN et al. (2006). Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 107: 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Rosales, C, McIntosh, J, Rastegarlari, G, Nathwani, D, Raj, D et al. (2011). Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther 19: 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Reiss, UM, Tuddenham, EG, Rosales, C, Chowdary, P, McIntosh, J et al. (2014). Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, GL and Herzog, RW (2015). Gene therapy for hemophilia. Front Biosci (Landmark Ed) 20: 556–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VandenDriessche, T and Chuah, MK (2015). Moving forward toward a cure for hemophilia B. Mol Ther 23: 809–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri, N, Grove, NC, Zuo, Y, Edwards, R, Palmer, D, Cerullo, V et al. (2009). Bioengineered factor IX molecules with increased catalytic activity improve the therapeutic index of gene therapy vectors for hemophilia B. Hum Gene Ther 20: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantore, A, Nair, N, Della Valle, P, Di Matteo, M, Màtrai, J, Sanvito, F et al. (2012). Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood 120: 4517–4520. [DOI] [PubMed] [Google Scholar]
- Finn, JD, Nichols, TC, Svoronos, N, Merricks, EP, Bellenger, DA, Zhou, S et al. (2012). The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood 120: 4521–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettrumpf, J, Herzog, RW, Schlachterman, A, Kaufhold, A, Stafford, DW and Arruda, VR (2005). Factor IX variants improve gene therapy efficacy for hemophilia B. Blood 105: 2316–2323. [DOI] [PubMed] [Google Scholar]
- Suwanmanee, T, Hu, G, Gui, T, Bartholomae, CC, Kutschera, I, von Kalle, C et al. (2014). Integration-deficient lentiviral vectors expressing codon-optimized R338L human FIX restore normal hemostasis in Hemophilia B mice. Mol Ther 22: 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan, PE, Sun, J, Gui, T, Hu, G, Hannah, WB, Wichlan, DG et al. (2015). Employing a gain-of-function factor IX variant R338L to advance the efficacy and safety of hemophilia B human gene therapy: preclinical evaluation supporting an ongoing adeno-associated virus clinical trial. Hum Gene Ther 26: 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan, P, Powell, JS, Konkle, BA, Josephson, NC, Escobar, M, McPhee, SJ, et al. (2015). Update on a phase 1/2 open-label trial of BAX335, an adeno-associated virus 8 (AAV8) vector-based gene therapy program for hemophilia B. J Thromb Haemost 13: 87, 13 87. [Google Scholar]
- Herzog, RW (2015). Hemophilia gene therapy: caught between a cure and an immune response. Mol Ther 23: 1411–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsic, D, Govindasamy, L, Currlin, S, Markusic, DM, Tseng, YS, Herzog, RW et al. (2014). Vector design Tour de Force: integrating combinatorial and rational approaches to derive novel adeno-associated virus variants. Mol Ther 22: 1900–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig-Choisat, B, Wang, L, Legras, X, Saha, PK, Chen, L, Bell, P et al. (2015). Development and rescue of human familial hypercholesterolaemia in a xenograft mouse model. Nat Commun 6: 7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisowski, L, Dane, AP, Chu, K, Zhang, Y, Cunningham, SC, Wilson, EM et al. (2014). Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 506: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer, DV (2014). AAV shuffles to the liver: commentary on Lisowski et al. Mol Ther Methods Clin Dev 1: 14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotterman, MA and Schaffer, DV (2014). Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15: 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F, Anguela, XM, Pavani, G, Chen, Y, Davidson, RJ, Hui, DJ et al. (2013). Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med 5: 194ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, BE and Herzog, RW (2013). Covert warfare against the immune system: decoy capsids, stealth genomes, and suppressors. Mol Ther 21: 1648–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino, AT, Basner-Tschakarjan, E, Markusic, DM, Finn, JD, Hinderer, C, Zhou, S et al. (2013). Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood 121: 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti, M, Elder, M, Falk, D, Lawson, L, Smith, B, Nayak, S et al. (2014). B-Cell Depletion is Protective Against Anti-AAV Capsid Immune Response: A Human Subject Case Study. Mol Ther Methods Clin Dev 1: 14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar, D, Biswas, M, Liao, G, Seay, HR, Perrin, GQ, Markusic, DM et al. (2014). Ex vivo expanded autologous polyclonal regulatory T cells suppress inhibitor formation in hemophilia. Mol Ther Methods Clin Dev 1: 14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack, BK, Herzog, RW, Terhorst, C and Markusic, DM (2014). Development of gene transfer for induction of antigen-specific tolerance. Mol Ther Methods Clin Dev 1: 14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markusic, DM, Hoffman, BE, Perrin, GQ, Nayak, S, Wang, X, LoDuca, PA et al. (2013). Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol Med 5: 1698–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crudele, JM, Finn, JD, Siner, JI, Martin, NB, Niemeyer, GP, Zhou, S et al. (2015). AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood 125: 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, JD, Ozelo, MC, Sabatino, DE, Franck, HW, Merricks, EP, Crudele, JM et al. (2010). Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood 116: 5842–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier, N, Hacein-Bey-Abina, S, Bartholomae, CC, Veres, G, Schmidt, M, Kutschera, I et al. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326: 818–823. [DOI] [PubMed] [Google Scholar]
- Cartier, N, Hacein-Bey-Abina, S, Bartholomae, CC, Bougnères, P, Schmidt, M, Kalle, CV et al. (2012). Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol 507: 187–198. [DOI] [PubMed] [Google Scholar]
- Hwu, WL, Muramatsu, S, Tseng, SH, Tzen, KY, Lee, NC, Chien, YH et al. (2012). Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci Transl Med 4: 134ra61. [DOI] [PubMed] [Google Scholar]
- Lee, NC, Muramatsu, S, Chien, YH, Liu, WS, Wang, WH, Cheng, CH et al. (2015). Benefits of neuronal preferential systemic gene therapy for neurotransmitter deficiency. Mol Ther 23: 1572–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittermeyer, G, Christine, CW, Rosenbluth, KH, Baker, SL, Starr, P, Larson, P et al. (2012). Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther 23: 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone, P, Shera, D, McPhee, SW, Francis, JS, Kolodny, EH, Bilaniuk, LT et al. (2012). Long-term follow-up after gene therapy for canavan disease. Sci Transl Med 4: 165ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch, L, Salegio, EA, San Sebastian, W, Kells, AP, Bringas, JR, Forsayeth, J et al. (2013). Strong cortical and spinal cord transduction after AAV7 and AAV9 delivery into the cerebrospinal fluid of nonhuman primates. Hum Gene Ther 24: 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury, SR, Hudry, E, Maguire, CA, Sena-Esteves, M, Breakefield, XO and Grandi, P (2016). Viral vectors for therapy of neurologic diseases. Neuropharmacology (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Bainbridge, JW, Smith, AJ, Barker, SS, Robbie, S, Henderson, R, Balaggan, K et al. (2008). Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 358: 2231–2239. [DOI] [PubMed] [Google Scholar]
- Cideciyan, AV, Hauswirth, WW, Aleman, TS, Kaushal, S, Schwartz, SB, Boye, SL et al. (2009). Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther 20: 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire, AM, Simonelli, F, Pierce, EA, Pugh, EN Jr, Mingozzi, F, Bennicelli, J et al. (2008). Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 358: 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye, SE, Boye, SL, Lewin, AS and Hauswirth, WW (2013). A comprehensive review of retinal gene therapy. Mol Ther 21: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan, AV, Aleman, TS, Boye, SL, Schwartz, SB, Kaushal, S, Roman, AJ et al. (2008). Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA 105: 15112–15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth, WW, Aleman, TS, Kaushal, S, Cideciyan, AV, Schwartz, SB, Wang, L et al. (2008). Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther 19: 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, SG, Cideciyan, AV, Ratnakaram, R, Heon, E, Schwartz, SB, Roman, AJ et al. (2012). Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 130: 9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonelli, F, Maguire, AM, Testa, F, Pierce, EA, Mingozzi, F, Bennicelli, JL et al. (2010). Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther 18: 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa, F, Maguire, AM, Rossi, S, Pierce, EA, Melillo, P, Marshall, K et al. (2013). Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology 120: 1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire, AM, High, KA, Auricchio, A, Wright, JF, Pierce, EA, Testa, F et al. (2009). Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 374: 1597–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmer, J and Breazzano, S (2015). Investor Outlook: Significance of the Positive LCA2 Gene Therapy Phase III Results. Hum Gene Ther Clin Dev 26: 208–210. [DOI] [PubMed] [Google Scholar]
- Jacobson, SG, Cideciyan, AV, Roman, AJ, Sumaroka, A, Schwartz, SB, Heon, E et al. (2015). Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med 372: 1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge, JW, Mehat, MS, Sundaram, V, Robbie, SJ, Barker, SE, Ripamonti, C et al. (2015). Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med 372: 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, J, Ashtari, M, Wellman, J, Marshall, KA, Cyckowski, LL, Chung, DC et al. (2012). AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med 4: 120ra15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic, C, Crippa, SV, Pignat, V, Bemelmans, AP, Samardzija, M, Grimm, C et al. (2011). Gene therapy regenerates protein expression in cone photoreceptors in Rpe65(R91W/R91W) mice. PLoS One 6: e16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X, Li, W, Dai, X, Kong, F, Zheng, Q, Zhou, X et al. (2011). Gene therapy rescues cone structure and function in the 3-month-old rd12 mouse: a model for midcourse RPE65 leber congenital amaurosis. Invest Ophthalmol Vis Sci 52: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat, FM, Breuwer, AR, Bartoe, JT, Annear, MJ, Zhang, Z, Smith, AJ et al. (2013). RPE65 gene therapy slows cone loss in Rpe65-deficient dogs. Gene Ther 20: 545–555. [DOI] [PubMed] [Google Scholar]
- Cideciyan, AV, Jacobson, SG, Beltran, WA, Sumaroka, A, Swider, M, Iwabe, S et al. (2013). Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci USA 110: E517–E525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanab, AY, Mysona, BA, Matragoon, S and El-Remessy, AB (2015). Silencing p75(NTR) prevents proNGF-induced endothelial cell death and development of acellular capillaries in rat retina. Mol Ther Methods Clin Dev 2: 15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren, RE, Groppe, M, Barnard, AR, Cottriall, CL, Tolmachova, T, Seymour, L et al. (2014). Retinal gene therapy in patients with choroideremia: initial findings from a phase ½ clinical trial. Lancet 383: 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koilkonda, RD, Yu, H, Chou, TH, Feuer, WJ, Ruggeri, M, Porciatti, V et al. (2014). Safety and effects of the vector for the Leber hereditary optic neuropathy gene therapy clinical trial. JAMA Ophthalmol 132: 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee, AP (2015). Manufacturing genetically modified T cells for clinical trials. Cancer Gene Ther 22: 67–71. [DOI] [PubMed] [Google Scholar]
- Carpenito, C, Milone, MC, Hassan, R, Simonet, JC, Lakhal, M, Suhoski, MM et al. (2009). Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA 106: 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, BD, Suryadevara, CM, Gedeon, PC, Herndon, JE 2nd, Sanchez-Perez, L, Bigner, DD et al. (2014). Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J Clin Neurosci 21: 189–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, RJ, Rivière, I, Park, JH, Davila, ML, Wang, X, Stefanski, J et al. (2011). Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118: 4817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos, M, Levine, BL, Porter, DL, Katz, S, Grupp, SA, Bagg, A et al. (2011). T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, DL, Levine, BL, Kalos, M, Bagg, A and June, CH (2011). Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, JN, Dudley, ME, Feldman, SA, Wilson, WH, Spaner, DE, Maric, I et al. (2012). B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119: 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, JN, Dudley, ME, Carpenter, RO, Kassim, SH, Rose, JJ, Telford, WG et al. (2013). Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 122: 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, DW, Kochenderfer, JN, Stetler-Stevenson, M, Cui, YK, Delbrook, C, Feldman, SA et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, RJ, Davila, ML, Riviere, I, Park, J, Wang, X, Cowell, LG et al. (2013). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5: 177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila, ML, Riviere, I, Wang, X, Bartido, S, Park, J, Curran, K et al. (2014). Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6: 224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude, SL, Frey, N, Shaw, PA, Aplenc, R, Barrett, DM, Bunin, NJ et al. (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371: 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp, SA, Kalos, M, Barrett, D, Aplenc, R, Porter, DL, Rheingold, SR et al. (2013). Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blat, D, Zigmond, E, Alteber, Z, Waks, T and Eshhar, Z (2014). Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther 22: 1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson, M, Piras, E, Burman, J, Nilsson, B, Essand, M, Lu, B et al. (2012). CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation 9: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimasaki, N, Fujisaki, H, Cho, D, Masselli, M, Lockey, T, Eldridge, P et al. (2012). A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy 14: 830–840. [DOI] [PubMed] [Google Scholar]
- Kim, YC, Zhang, AH, Su, Y, Rieder, SA, Rossi, RJ, Ettinger, RA et al. (2015). Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood 125: 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Moghimi, B, Zolotukhin, I, Morel, LM, Cao, O and Herzog, RW (2014). Immune tolerance induction to factor IX through B cell gene transfer: TLR9 signaling delineates between tolerogenic and immunogenic B cells. Mol Ther 22: 1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen, A, Kamata, M, Rezek, V, Rick, J, Levin, B, Kasparian, S et al. (2015). HIV-specific immunity derived from chimeric antigen receptor-engineered stem cells. Mol Ther 23: 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval, L, Schmidt, H, Kaltoft, K, Fode, K, Jensen, JJ, Sorensen, SM et al. (2006). Adoptive transfer of allogeneic cytotoxic T lymphocytes equipped with a HLA-A2 restricted MART-1 T-cell receptor: a phase I trial in metastatic melanoma. Clin Cancer Res 12: 1229–1236. [DOI] [PubMed] [Google Scholar]
- Johnson, LA, Morgan, RA, Dudley, ME, Cassard, L, Yang, JC, Hughes, MS et al. (2009). Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114: 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Dudley, ME, Wunderlich, JR, Hughes, MS, Yang, JC, Sherry, RM et al. (2006). Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314: 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst, MR, Yang, JC, Langan, RC, Dudley, ME, Nathan, DA, Feldman, SA et al. (2011). T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 19: 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule, MA, Savoldo, B, Myers, GD, Rossig, C, Russell, HV, Dotti, G et al. (2008). Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14: 1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Morgan, RA, Feldman, SA, Yang, JC, Sherry, RM, Dudley, ME et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29: 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Yang, JC, Kitano, M, Dudley, ME, Laurencot, CM and Rosenberg, SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linette, GP, Stadtmauer, EA, Maus, MV, Rapoport, AP, Levine, BL, Emery, L et al. (2013). Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122: 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Chinnasamy, N, Abate-Daga, D, Gros, A, Robbins, PF, Zheng, Z et al. (2013). Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 36: 133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport, AP, Stadtmauer, EA, Binder-Scholl, GK, Goloubeva, O, Vogl, DT, Lacey, SF et al. (2015). NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21: 914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichty, BD, Breitbach, CJ, Stojdl, DF and Bell, JC (2014). Going viral with cancer immunotherapy. Nat Rev Cancer 14: 559–567. [DOI] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Tang, WW, Frank, I, Wang, SQ, Lee, G et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiech, L, Heidenreich, M, Banerjee, A, Habib, N, Li, Y, Trombetta, J et al. (2015). In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, H, Xue, W, Chen, S, Bogorad, RL, Benedetti, E, Grompe, M et al. (2014). Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32: 551–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzel, A, Paulk, NK, Shi, Y, Huang, Y, Chu, K, Zhang, F et al. (2015). Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature 517: 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H, Haurigot, V, Doyon, Y, Li, T, Wong, SY, Bhagwat, AS et al. (2011). In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson, R and Ylä-Herttuala, S (2015). Molecular therapy special issue on gene editing technologies and applications. Mol Ther 23: 1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J, Exline, CM, DeClercq, JJ, Llewellyn, GN, Hayward, SB, Li, PW et al. (2015). Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol 33: 1256–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather, BD, Romano Ibarra, GS, Sommer, K, Curinga, G, Hale, M, Khan, IF et al. (2015). Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med 7: 307ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]