

Abstract
Context:
Type 2 diabetes is characterized by a β-cell deficit and a progressive defect in β-cell function. It has been proposed that the deficit in β-cells may be due to β-cell degranulation and transdifferentiation to other endocrine cell types.
Objective:
The objective of the study was to establish the potential impact of β-cell dedifferentiation and transdifferentiation on β-cell deficit in type 2 diabetes and to consider the alternative that cells with an incomplete identity may be newly forming rather than dedifferentiated.
Design, Setting, and Participants:
Pancreata obtained at autopsy were evaluated from 14 nondiabetic and 13 type 2 diabetic individuals, from four fetal cases, and from 10 neonatal cases.
Results:
Whereas there was a slight increase in islet endocrine cells expressing no hormone in type 2 diabetes (0.11 ± 0.03 cells/islet vs 0.03 ± 0.01 cells/islet, P < .01), the impact on the β-cell deficit would be minimal. Furthermore, we established that the deficit in β-cells per islet cannot be accounted for by an increase in other endocrine cell types. The distribution of hormone negative endocrine cells in type 2 diabetes (most abundant in cells scattered in the exocrine pancreas) mirrors that in developing (embryo and neonatal) pancreas, implying that these may represent newly forming cells.
Conclusions:
Therefore, although we concur that in type 2 diabetes there are endocrine cells with altered cell identity, this process does not account for the deficit in β-cells in type 2 diabetes but may reflect, in part, attempted β-cell regeneration.
Type 2 diabetes is characterized by a progressive decline in β-cell function (1, 2). In studies of human pancreas obtained at autopsy or from brain-dead organ donors, there is a deficit in β-cells (3–6). This has been attributed to an imbalance between sufficient β-cell formation, pre- or postnatally, and increased β-cell loss through apoptosis or necrosis. Support for this model of the progressive decline in β-cell function in type 2 diabetes is the striking similarity between the loss of cell mass and function in neurodegenerative diseases such as Alzheimer's disease that share much in common with type 2 diabetes (7). In both the hippocampus in Alzheimer's disease and the islet in type 2 diabetes, the cells of interest express closely related amyloidogenic proteins (Alzheimer's β-protein and islet amyloid polypeptide) that misfold and form toxic membrane permeant oligomers and accumulate over time as extracellular amyloid. Moreover, the cell signaling changes in β-cells and hippocampal cells in type 2 diabetes and Alzheimer's disease are also shared, with mitochondrial dysfunction, endoplasmic reticulum stress, calpain hyperactivation, accumulation of polyubiquinated proteins, and defective autophagy/lysosomal pathways (7). Furthermore, both the pathological and functional changes in Alzheimer's disease and type 2 diabetes are recapitulated in models expressing human Alzheimer's β-protein and islet amyloid polypeptide, respectively (8, 9), accompanied by an increase in cell death (10).
Recently, based initially on genetically manipulated mouse models (11), it has been suggested that the underlying basis of the β-cell deficit in type 2 diabetes is β-cell degranulation and β-cell dedifferentiation and then transdifferentiation, rather than β-cell loss through apoptosis (11). Proponents of this hypothesis have suggested that the therapeutic approach to β-cell dysfunction in type 2 diabetes is best directed at the degranulation/dedifferentiation defects rather than preservation or expansion of β-cell mass (11). The purpose of the present studies was to test the hypothesis that the deficit in β-cells in type 2 diabetes can be accounted for by the degranulation of β-cells and/or the conversion of β-cells to other endocrine cell types. As a secondary question, we sought to compare human endocrine pancreas during late development and early childhood with that in type 2 diabetes, with consideration that some of the recently reported observations of changes in the endocrine identity in diabetes might be a consequence of attempted β-cell regeneration.
Research Design and Methods
Design and case selection
For the neonatal and adult subjects, sections of pancreas were obtained from the Mayo Clinic autopsy archives with institutional review board permission (institutional review board number 15-004992). For the adult subjects, two groups were identified: obese nondiabetic (14 subjects) and obese subjects with a documented history of type 2 diabetes (13 subjects). Obesity was defined as a body mass index (BMI) greater than 27 kg/m2. Potential cases were identified by retrospective analysis of the Mayo Clinic autopsy database. To be included, case requirements were a full autopsy within 24 hours of death, a general medical examination including at least one fasting blood glucose documented in the year prior to death, and stored pancreatic tissue of adequate size and quality. Exclusion criteria included any potential secondary cause of diabetes, exposure to chronic glucocorticoid treatment, and pancreatic tissue that had undergone autolysis or showed features of pancreatitis. Neonatal autopsy cases (n = 10) were selected to be as recently after delivery as possible while using the same inclusion and exclusion criteria for pancreatic tissue quality.
Sections of fetal pancreas (n = 4) were obtained from the Institute of Pathology and the Division of Clinical and Functional Anatomy, Medical University of Innsbruck (Innsbruck, Austria). They were obtained from miscarriage and legal abortions including parental consent and in compliance with the local governmental and institutional guidelines.
Case characteristics (Supplemental Tables 1–3)
The obese nondiabetic and obese type 2 diabetic groups were matched for age (61.4 ± 2.6 y vs 59.1 ± 3.5 y, obese type 2 diabetic [OT2D] vs obese nondiabetic [OND], P = NS). BMI was higher in the OT2D group (43.3 ± 2.0 kg/m2 vs 36.1 ± 1.2 kg/m2, OT2D vs OND, P < .005). When compared with the obese nondiabetic subjects, the obese diabetic subjects had higher fasting plasma glucose values despite being on treatment (150.9 ± 22.1 mg/dL vs 100.5 ± 3.8 mg/dL, OT2D vs OND, P < .05). Most individuals with type 2 diabetes had been treated with insulin (10 subjects), whereas one subject had received a sulfonylurea and two had been treated by diet alone.
To assure that differences in BMI between the nondiabetic and type 2 diabetic groups were not a factor in the results obtained, a subset of cases (11 obese nondiabetic and 10 obese type 2 diabetic), matched for both age (62.8 ± 2.6 y vs 60.1 ± 4.4 y, OT2D vs OND, P = NS) and BMI (40.4 ± 1.4 kg/m2 vs 37.5 ± 1.2 kg/m2, OT2D vs OND, P = NS), was examined (Supplemental Tables 4 and 5).
Sections of pancreas from 10 neonatal subjects were obtained, death having occurred between 8 hours and 12 weeks after delivery. Cause of death is noted in Supplemental Table 3. Sections of fetal pancreatic tissue were acquired from four fetuses, at 26, 30, 32, and 34 weeks of gestation.
Pancreatic tissue processing and staining
Paraffin tissue sections from each subject were stained for chromogranin A, insulin, glucagon, somatostatin, pancreatic polypeptide, and ghrelin. Standard immunohistochemistry protocol was used for fluorescent immunodetection of various proteins in pancreatic sections. One pancreas section obtained from the tail of the pancreas was analyzed for each subject. Briefly, slides were incubated at 4°C overnight with a cocktail of primary antibodies prepared in blocking solution (3% BSA in Tris buffered saline and Tween 20) at the following dilutions: rabbit antichromogranin A (1:200, NB120-15160; Novus Biologicals); mouse antiglucagon (1:1000, G2654-.2ML; Sigma-Aldrich); guinea pig antiinsulin (1:200, 7842; Abcam), rat antisomatostatin (1:300, MAB354; EMD Millipore), goat antipancreatic polypeptide (1:3000; Everest Biotech), rat antighrelin (1:50, MAB8200; R&D Systems). The primary antibodies were detected by a cocktail of appropriate secondary antibodies (Jackson ImmunoResearch) conjugated to Cy3 (1:200 for chromogranin A), fluorescein isothiocyanate (1:200 each to detect glucagon, somatostatin, pancreatic polypeptide, and ghrelin) or Cy5 (1:100 to detect insulin). Slides were counterstained to mark the nuclei using a mounting medium containing 4′,6′-diamino-2-phenylindole (DAPI; Vectashield; Vector Laboratories) and viewed using a Leica DM6000 microscope (Leica Microsystems), and images were acquired using the ×20 objective (×200 magnification) using a Hamamatsu Orca-ER camera (C4742-80-12AG; Indigo Scientific) and Openlab software (Improvision).
Morphometric analysis
Fifty islets per subject were imaged at ×20 magnification. An islet was defined as a grouping of four or more chromogranin-positive cells. A cluster was defined as a grouping of three or fewer chromogranin-positive cells. Islets were selected by starting at the top left corner of the pancreatic tissue section and working across the tissue from left to right and back again in a serpentine fashion, imaging all islets in this systematic excursion across the tissue section. Analysis was performed in a blinded fashion (by A.E.B.), and all chromogranin-positive, hormone-negative (CPHN) cells were confirmed by a second observer (S.D.). The endocrine cells contained within each islet were manually counted and the following data recorded: 1) the number of cells staining only for chromogranin; 2) the number of cells costaining for the endocrine hormone cocktail and chromogranin; and 3) the number of cells costaining for insulin and chromogranin.
The mean number of endocrine cells counted within the islets for the nondiabetic group was 2240 ± 294 cells per subject and for the type 2 diabetic group, 1696 ± 189 cells per subject. The mean number of cells counted in clusters for the nondiabetic group was 72 ± 9 cells per subject, and for the type 2 diabetic group, 88 ± 8 cells per subject. The mean number of CPHN cells per individual identified in islets from the nondiabetic subjects was 1.5 ± 0.5 cells per individual and from the type 2 diabetic group, 5.5 ± 1.5 cells per individual. The mean number of CPHN cells per individual counted in clusters for the nondiabetic group was 2.1 ± 0.3 per individual and for the type 2 diabetic group, 7.8 ± 1.0 cells per individual.
At ×200 magnification, using the Leica DM6000 with Hamamatsu Orca-ER camera and a 0.7x C-mount, each field of view was calculated to be 0.292 mm2. Within the fields imaged to obtain the 50 islets per subject, all clusters of endocrine cells (one, two, or three adjacent endocrine cells) were counted and recorded as outlined above.
Statistical analysis
Statistical analysis was performed using the Student's t test with GraphPad Prism 5.0 software (GraphPad Software). Data in graphs and tables are presented as means ± SEM. Findings were assumed statistically significant at P < .05.
Results
Nonhormone-expressing endocrine cells are abundant during endocrine pancreas formation, particularly in small scattered endocrine cell clusters
During late gestation, small clusters of pancreatic endocrine cells assemble into small islets, and this process continues in early infancy (Figure 1, A and B). By 6 months of age, most endocrine cells in the pancreas are assembled into islets (12), although infrequent small scattered foci of one to three endocrine cells are detectable throughout adult life (Figures 1C and 2, A and B).
Figure 1.
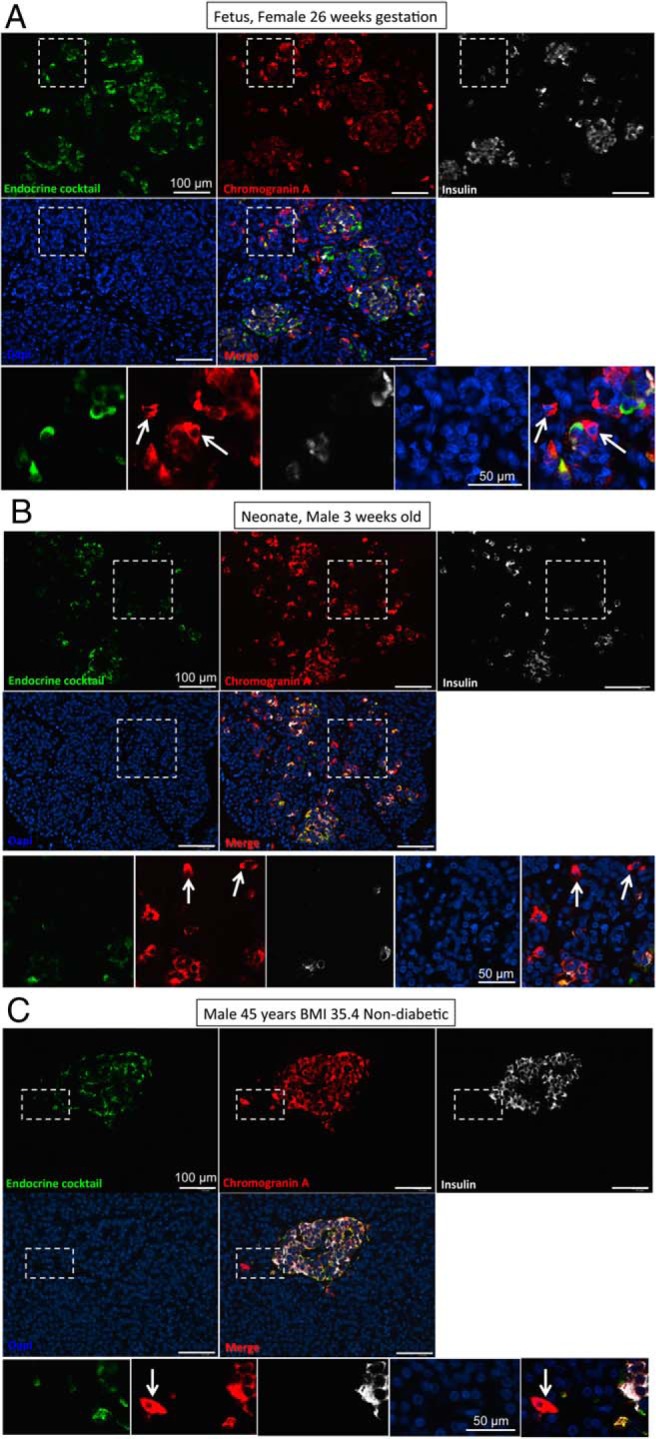
Representative images taken at low power (×10 objective) to show pancreas from a fetus at 26 weeks' gestation (A), a neonate 3 weeks after delivery (B), and a 45-year-old nondiabetic adult with a BMI of 35.4 kg/m2 (C). Pancreas sections in all cases were stained for chromogranin A (red), endocrine cocktail (glucagon, somatostatin, pancreatic polypeptide, and ghrelin) (green), insulin (white), and DAPI (blue), and the merged image is shown. Insets in all panels show cells staining for chromogranin A but none of the other endocrine hormones. These CPHN cells are frequently found in fetal pancreas, become less frequent in neonatal life, and are infrequently found in nondiabetic adult human pancreas. Scale bar, 100 μm (low power images), 50 μm in high power inset images).
Figure 2.

The number of endocrine cells present in scattered clusters of from one to three cells in the exocrine pancreas parenchyma (A, B). Scattered clusters of endocrine cells are most numerous in sections of fetal pancreas, and the frequency decreases after birth. By adulthood, endocrine clusters are relatively infrequent, although they are always present in pancreatic sections (52.66 ± 13.55 vs 9.41 ± 1.67 endocrine cells/mm2; fetus vs OND, ***, P < .0001; 37.80 ± 6.56 vs 9.41 ± 1.67 endocrine cells/mm2, neonate vs OND, ***, P < .0001). The percentage of endocrine cells that are CPHN in human fetus and neonate compared with nondiabetic adult subjects. The results are grouped according to their location, either within islets (C) or in small clusters of up to three endocrine cells (D). CPHN cells are found frequently in fetal and neonatal tissue, with the frequency rapidly declining after birth. Most CPHN cells are found in small clusters of between one and three endocrine cells scattered throughout the exocrine pancreas. Islets are as follows: 15.06% ± 6.79% vs 1.89% ± 0.79% vs 0.17% ± 0.09% CPHN cells in islets, fetus vs neonate vs OND; P < .01, fetus vs neonate; P < .001, fetus vs OND; P < .01, neonate vs OND. Clusters are as follows: 41.40% ± 10.94% vs 26.79% ± 2.31% vs 3.22% ± 0.37% CPHN cells in clusters, fetus vs neonate vs OND; P < .05, fetus vs neonate; fetus and neonate vs OND, P < .0001. Panels A, C, and D: black circles, fetus; black squares, neonate; black triangles, OND; panel B, white bar, fetus; striped bar, neonate; black bar, OND.
Within islets, the proportion of endocrine cells expressing no known islet hormone decreases from approximately 30% to approximately 5% during the last 10 weeks of gestation and then further declines from approximately 4% in early infancy to approximately 0.1% throughout adult life in nondiabetic individuals (Figure 2C).
The proportion of scattered pancreatic endocrine cells that express no known islet hormone follows a similar pattern but is consistently higher than in the defined islets over the corresponding period, falling from approximately 60% to approximately 20% during the last 10 weeks of gestation and then from approximately 20% to less than approximately 3% in adults (Figure 2D).
Therefore, during development of the human endocrine pancreas, nonhormone-expressing endocrine cells, particularly those scattered within the exocrine pancreas, appear to be the predecessors of differentiated endocrine cells that ultimately assemble into well-defined islets.
Nonhormone endocrine cells are more frequent in type 2 diabetes (Figure 3)
Figure 3.

Examples of CPHN cells in pancreas from an OND subject (A) and two OT2D subjects (B, C). Individual layers stained for endocrine cocktail (glucagon, somatostatin, pancreatic polypeptide, and ghrelin) (green), chromogranin A (red), insulin (white), and DAPI (blue) are shown along with the merged image. Arrows indicate CPHN cells. Scale bar, 50 μm.
It has been suggested that dedifferentiation of pancreatic β-cells transforming into hormone-negative β-cells might be the predominant cause for the β-cell deficit in type 2 diabetes rather than attrition of the cells through apoptosis (11, 13). We also identify occasional CPHN cells in islets in both type 2 diabetes and controls. Whereas these nonhormone-expressing endocrine cells are more frequent in the islets in type 2 diabetes than controls (0.11 ± 0.03 cells/islet vs 0.03 ± 0.01 cells/islet, OT2D vs OND, P < .01) (Figure 4, A and B), they are notably much less frequent than reported in genetically manipulated mouse models with diabetes (11). Because similar cells are characteristic of the developing pancreas in humans, and in particular in the small clusters of endocrine cells that precede mature islet development, we questioned whether these cells might represent attempted β-cell regeneration as opposed to dedifferentiation.
Figure 4.

The number of nonhormone-expressing endocrine cells (CPHN cells) in OND and OT2D. A and B, The number of nonhormone-expressing endocrine cells (CPHN cells) per islet cross-section is increased in the OT2D subjects (0.11 ± 0.03 vs 0.03 ± 0.01 cells/islet, OT2D vs OND, *, P < .01). C and D, The number of nonhormone-expressing endocrine cells (CPHN cells) present in scattered clusters of endocrine cells is increased in OT2D (1.54 ± 0.21 vs 0.30 ± 0.05 cells/mm2, OT2D vs OND, ***, P < .0001). E and F, The number of endocrine cells (both hormone expressing and nonhormone expressing) present in scattered clusters of endocrine cells is increased in OT2D (13.53 ± 1.31 vs 9.41 ± 1.67 cells/mm2 of pancreas, OT2D vs OND, ^, P < .05). Panels A, C, and E, black circles, OND subjects; black squares, OT2D subjects. Panels B, D, and F, white bars, OND subjects; black bars, OT2D subjects.
Scattered endocrine cells, with a disproportionate increase in nonhormone-expressing cells, are more frequent in pancreas of type 2 diabetes
To approach the possibility that hormone-negative cells in type 2 diabetes might indicate attempted regeneration, we first quantified the abundance of scattered foci of endocrine (chromogranin positive) cells in pancreas of individuals with type 2 diabetes and controls. We find an approximately 1.7-fold increase in the scattered foci of pancreatic endocrine cells in type 2 diabetes (13.5 ± 1.3 cells/mm2 vs 9.4 ± 1.7 cells/mm2 of pancreas, OT2D vs OND, P < .05) (Figure 4, E and F). Subset analysis likewise revealed an increase in scattered endocrine cells in type 2 diabetes (13.9 ± 1.6 cells/mm2 vs 8.8 ± 1.8 cells/mm2 of pancreas, OT2D vs OND, P < .05).
Moreover, scattered endocrine cells that are nonhormone expressing (chromogranin positive islet hormone negative) are approximately 6-fold increased in type 2 diabetes (1.54 ± 0.21 cells/mm2 vs 0.30 ± 0.05 cells/mm2 of pancreas, OT2D vs OND, P < .0001) (Figure 4, C and D). Subset analysis also revealed an increase in scattered endocrine cells that are nonhormone expressing (chromogranin positive islet hormone negative) in type 2 diabetes (1.43 ± 0.21 cells/mm2 vs 0.30 ± 0.05 cells/mm2 of pancreas, OT2D vs OND, P < .0001). The increase in scattered endocrine cells in type 2 diabetes, including a disproportionate number of nonhormone-expressing cells (∼12% vs ∼3%), is reminiscent of the developing human endocrine pancreas. This raises the possibility that the increase in hormone-negative cells in type 2 diabetes might represent attempted β-cell regeneration rather than necessarily being a consequence of β-cell dedifferentiation.
Age, BMI, and disease duration do not predict frequency of nonhormone-expressing endocrine cells
To investigate potential factors that might influence the frequency of nonhormone-expressing endocrine cells occurring in clusters of scattered endocrine cells, we evaluated the abundance of these cells in relation to the age, BMI, and documented duration of diabetes (Figure 5). None of these variables was related to the frequency of nonhormone-expressing endocrine cells. Notably, because BMI was not related to the frequency of hormone-negative-expressing endocrine cells, the greater abundance of these cells in type 2 diabetes compared with nondiabetic controls is not attributable to the greater obesity in the former. Whereas 13 type 2 diabetic subjects are included in the study, in only 10 of those subjects was there a documented duration of disease.
Figure 5.

The relationship between the scattered clusters of nonhormone-expressing endocrine cells (CPHN cells) in OND and OT2D subjects with age, BMI, and duration of disease. There was no relationship of the CPHN cells with age (A), BMI (B), or duration of disease (C). Panels A and B show the 13 OT2D subjects included in the study; panel C shows only 10 OT2D subjects because duration of disease was not known in three OT2D subjects. In all panels: open triangles, OND subjects; black circles, OT2D subjects.
The impact of nonhormone-expressing endocrine cells on the deficit in β-cell mass in type 2 diabetes is minimal
It has been proposed that the reported deficit in β-cell mass in type 2 diabetes may have been overestimated because the increase in hormone-expressing β-cells may be dedifferentiated or degranulated β-cells (11, 13). We addressed this by considering the impact that the hormone-negative endocrine cells could have on the deficit in β-cells in type 2 diabetes. The relative abundance of nonhormone-expressing endocrine cells per islet vs insulin-expressing endocrine cells per islet in type 2 diabetes and controls is shown in Figure 6A. If all the nonhormone-expressing endocrine cells per islet were added to β-cells per islet, the deficit in β-cells per islet in type 2 diabetes would decrease from 30.5% to 30.1%.
Figure 6.

The impact of nonhormone-expressing endocrine cells on the deficit in β-cell mass in type 2 diabetes is minimal. The number of cells/islet of the CPHN cell fraction is very small when compared with the number of β-cells/islet in the OND, and the deficit in β-cells in the OT2D subjects cannot be accounted for by the increase in CPHN cells in type 2 diabetes. A, There is an approximately 30% deficit in β-cells in the islets from the subjects with type 2 diabetes (18.23 ± 2.49 vs 25.23 ± 2.59 β-cells/islet, OT2D vs OND, ^, P < .05). The number of CPHN cells/islet is increased in type 2 diabetes (0.11 ± 0.03 vs 0.03 ± 0.01 CPHN cells/islet, OT2D vs OND, *, P < .01), but this increase does not account for the β-cell deficit seen in type 2 diabetes. B, Islet endocrine cells expressing islet hormones other than insulin are not increased in type 2 diabetes (14.83 ± 1.83 vs 18.67 ± 3.47 endocrine cocktail cells/islet, OT2D vs OND, P = NS). Therefore, the β-cell deficit in type 2 diabetes (18.23 ± 2.49 vs 25.23 ± 2.59 β-cells/islet, OT2D vs OND, ^, P < .05) cannot be explained by conversion of β-cells to other endocrine cells. White bars, OND subjects; black bars, OT2D subjects.
To further address the suggestion that nonhormone-expressing endocrine cells in type 2 diabetes might be degranulated β-cells, we compared these cells with the nascent nonhormone-expressing endocrine cells in neonates to discern whether there was a difference in vesicle density (granulation). As noted in Supplemental Figure 1, the granule density was comparable in neonates and in nondiabetic and individuals with type 2 diabetes. This further supports the possibility that the nonhormone-expressing cells in adults with type 2 diabetes are newly formed rather than degranulated β-cells. Taken together, therefore, degranulation of β-cells does not explain the deficit in β-cells in type 2 diabetes in humans.
Islet endocrine cells expressing islet hormones other than insulin are not increased in type 2 diabetes
Advocates of the dedifferentiation hypothesis have also proposed that the deficit in β-cells in type 2 diabetes may be due to transdifferentiation of β-cells to cells expressing other islet hormones such as glucagon (11, 13). To evaluate this, we quantified the endocrine cells expressing islet hormones other than insulin (glucagon, somatostatin, pancreatic polypeptide, and ghrelin) in comparison with cells expressing insulin in the same islets in type 2 diabetes vs controls. If the deficit in β-cells is due to transdifferentiation of β-cells to other endocrine cell types, then there should be a corresponding increase in endocrine cells expressing other islet hormones to match the deficit in β-cells. However, there is no increase in islet endocrine cells that do not express insulin in type 2 diabetes (Figure 6B). Collectively, although compelling evidence has been presented that there is a loss of cellular identity (as defined by hormone and transcriptional factor expression) in a subset of endocrine cells in type 2 diabetes (13–16), the deficit in β-cells in type 2 diabetes is not explained by either degranulated β-cells or conversion of β-cells to other endocrine cell types. Moreover, the deficit in β-cells in type 2 diabetes is accounted for by both a decrease in β-cells per islet and a decrease in islet density (4, 13). Studies that focus on the former only underestimate the β-cell deficit.
Discussion
Type 2 diabetes is characterized by a progressive decline in β-cell function in vulnerable individuals in the face of insulin resistance (1, 2). In cross-sectional studies of human pancreas obtained at autopsy or from brain-dead organ donors, there is a β-cell deficit in type 2 diabetes (3–6). In humans, pigs, and nonhuman primates, there is a wide variance in β-cell mass in nondiabetic individuals. However, once the β-cell mass approaches approximately 50% of the mean, an apparent threshold is reached beyond which there is a steep rise in fasting blood glucose (and therefore decline in β-cell function) for any further decrement in β-cells (17–20). Collectively these findings highlight the importance of a sufficiency of β-cells and that an insufficient β-cell mass, even if induced by a partial pancreatectomy (18, 21), leads to β-cell dysfunction.
Recently the basis of the loss of β-cell mass in type 2 diabetes has been an area of increased interest. We and others have reported that β-cells in type 2 diabetes are characterized by endoplasmic reticulum stress and an increase in β-cell apoptosis (22–24). In species vulnerable to type 2 diabetes, islet pathology is notable for the presence of islet amyloid deposits derived from islet amyloid polypeptide, a protein that forms toxic oligomers in β-cells in type 2 diabetes (25). Although it is not possible to perform prospective studies comparing islet pathology with evolving diabetes in humans, in a unique prospective study of a nonhuman primate (Macaca nigra), there was a progressive increase in islet amyloid and loss of islet cells in relationship to the transition from nondiabetic through borderline diabetes to diabetes (26). A recently published cross-sectional study of both humans and nonhuman primates with and without type 2 diabetes, reported an increased proportion of β-cells that display altered identity in type 2 diabetes and that these cells were more abundant in proximity to islet amyloid deposits (16). Loss of cell identity in that study was characterized by cells expressing both insulin and glucagon or by cells expressing the β-cell transcription factor Nkx6.1 and glucagon, both of which were increased in type 2 diabetes.
We previously reported a comparable increased proportion of β-cells (∼5%) coexpressing glucagon in type 2 diabetes (6). Perhaps more significant with regard to the impact on functional β-cell mass, the β-cell transcriptional factor musculoaponeurotic fibrosarcoma oncogene family A was detectable in less than 2% of β-cells in humans with type 2 diabetes compared with approximately 46% in nondiabetic controls, a change that has most often been attributed to glucotoxicity (15). The challenge with ascribing these changes in cell identity to cause or consequence in type 2 diabetes in humans is that all studies of the human pancreas are necessarily cross-sectional. Moreover, although partially differentiated endocrine cells may represent dedifferentiation, an alternative hypothesis is that they are derived from newly forming cells or regeneration (16, 27).
To further consider that possibility, we examined human pancreas obtained through the third trimester of human development and early infancy as a period of known new islet cell formation from endocrine precursors to probe for parallels with the findings in pancreas of adults with type 2 diabetes. As previously reported, newly forming islets in late gestation and early infancy appear to form via the coalescence of scattered foci of endocrine cells (28). Consistent with the assumption that scattered cells in the exocrine pancreas are the precursors of the developing islets, in the fetal pancreas, approximately 40% of these scattered endocrine cells marked as chromogranin A positive did not yet express a known islet hormone. These chromogranin A-positive, hormone-negative cells were also observed within the islets, amounting to approximately 15% of islet endocrine cells. By infancy, the corresponding values were approximately 20% of scattered endocrine cells and 4% of islet endocrine cells that expressed no known islet hormone. Collectively these findings support the concept that newly forming human islet cells arise from dispersed foci in pancreas, initially expressing chromogranin A but no known islet hormone and then coalescing to form islets while differentiating to an endocrine cell lineage.
Given that the distribution of hormone-negative, chromogranin A-positive cells in type 2 diabetes mirrors that present during islet formation during development, it is plausible that the pattern in type 2 diabetes signals ongoing attempted β-cell regeneration. Of note, whereas scattered hormone-negative endocrine cells are more abundant in type 2 diabetes than nondiabetics, the magnitude of these are still much less than in the developing pancreas, consistent with a relatively low, and apparently insufficient, rate of regeneration. Of interest, the increase in β-cell mass in pregnant humans is also characterized by an increase in small scattered foci of β-cells (29). Taken together, these data suggest that there may be a relatively low rate of attempted regeneration of β-cells in adults with type 2 diabetes. On the other hand, an alternative explanation for the predominantly scattered localization of these hormone-negative endocrine cells within pancreas could be that these cells are the product of β-cell dedifferentiation that have lost cell-cell contact and migrated (30). In the absence of lineage tracing in humans, however, it is unknown whether the hormone-negative endocrine cells, and those with a mixed identity, in type 2 diabetes arise from degranulation of previously mature endocrine cells or are incompletely mature newly formed endocrine cells.
Whereas our data question the concept that the measured deficit in β-cells in type 2 diabetes in humans can be primarily explained by degranulation or dedifferentiation, we agree that the partial loss of molecular identity of β-cells in diabetes likely plays a major role in β-cell dysfunction, most likely in both type 1 and 2 diabetes as well as secondary diabetes (for example, in pancreatitis) (11, 16). Moreover, we agree with the group of Accili (31) that reversal of β-cell deficit through the induction of endogenous β-cell formation without correcting the alterations in cell identity may be therapeutically unsuccessful. In a small study of brain-dead organ donors, we previously reported that although prior glucagon-like peptide-1-based therapy in type 2 diabetes was associated with a complete reversal of the deficit in β-cell mass, the individuals still had diabetes (6). Of interest, glucagon-like peptide-1-based therapy in humans was also associated with a marked increase in the percentage of cells coexpressing insulin and glucagon, further suggesting that these cells with a mixed identity may be newly formed rather than formed by β-cell dedifferentiation (6).
In conclusion, based on the present studies, the deficit in β-cells in type 2 diabetes is not attributable alone to degranulation or dedifferentiation of β-cells. As previously discussed, given the cross-sectional nature of human studies, it is not possible to know whether the deficit in β-cells precedes disease onset and contributes to diabetes risk or is only a consequence of loss of β-cells (32). Given the closely shared molecular and pathological characteristics of the islet in humans with type 2 diabetes and the relevant neurological tissue in neurodegenerative diseases, the concept that β-cell dysfunction and attrition occurs through mechanisms related to protein misfolding is not inconsistent with studies reporting changes in β-cell transcriptional networks and cellular identity. Moreover, type 2 diabetes is a heterogeneous disease, and it is plausible that there are other groups of patients (perhaps lean patients, as opposed to the obese patients reported here) in which β-cell dedifferentiation and/or transdifferentiation play a more prominent role toward β-cell deficit. Moreover, given that the remaining β-cells in type 2 diabetes, as in the remaining neurons in neurodegenerative diseases, are dysfunctional and have an altered transcriptional identity (14–16), we agree with the proposition that efforts to reverse those functional changes, perhaps by addressing the altered molecular identity (11), is a worthy therapeutic target.
Acknowledgments
We thank Bonnie Lui for editorial assistance.
Author contributions included the following: A.E.B. and S.D. performed the studies, undertook the microscopy with assistance from J.H., M.C., and K.Z., and performed the morphological analysis. A.E.B., S.D., H.F., J.J.M., R.A.R., and P.C.B. researched the data, wrote the manuscript, reviewed the manuscript, edited the manuscript, and contributed to the discussion.
This work was supported by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases Grant DK077967 and Larry Hillblom Foundation Grant 2014-D-001-NET (to P.C.B.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BMI
- body mass index
- CPHN
- chromogranin positive hormone negative
- DAPI
- 4′,6′-diamino-2-phenylindole
- OND
- obese nondiabetic
- OT2D
- obese type 2 diabetic.
References
- 1. Buchanan TA, Xiang AH, Peters RK, et al. Preservation of pancreatic β-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk Hispanic women. Diabetes. 2002;51:2796–2803. [DOI] [PubMed] [Google Scholar]
- 2. Xiang AH, Takayanagi M, Black MH, et al. Longitudinal changes in insulin sensitivity and β cell function between women with and without a history of gestational diabetes mellitus. Diabetologia. 2013;56:2753–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. 1985;4:110–125. [DOI] [PubMed] [Google Scholar]
- 4. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-Cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. [DOI] [PubMed] [Google Scholar]
- 5. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(suppl 4):32–42. [DOI] [PubMed] [Google Scholar]
- 6. Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes. 2013;62:2595–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mukherjee A, Morales-Scheihing D, Butler PC, Soto C. Type 2 diabetes as a protein misfolding disease. Trends Mol Med. 2015;21:439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matveyenko AV, Butler PC. β-Cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes. 2006;55:2106–2114. [DOI] [PubMed] [Google Scholar]
- 9. Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest. 2014;124:3489–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meier JJ, Butler AE, Saisho Y, et al. β-cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes. 2008;57:1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marselli L, Suleiman M, Masini M, et al. Are we overestimating the loss of β cells in type 2 diabetes? Diabetologia. 2014;57:362–365. [DOI] [PubMed] [Google Scholar]
- 14. Butler AE, Robertson RP, Hernandez R, Matveyenko AV, Gurlo T, Butler PC. β Cell nuclear musculoaponeurotic fibrosarcoma oncogene family A (MafA) is deficient in type 2 diabetes. Diabetologia. 2012;55:2985–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo S, Dai C, Guo M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest. 2013;123:3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spijker HS, Song H, Ellenbroek JH, et al. Loss of β-cell identity occurs in type 2 diabetes and is associated with islet amyloid deposits. Diabetes. 2015;64:2928–2938. [DOI] [PubMed] [Google Scholar]
- 17. Ritzel RA, Butler AE, Rizza RA, Veldhuis JD, Butler PC. Relationship between β-cell mass and fasting blood glucose concentration in humans. Diabetes Care. 2006;29:717–718. [DOI] [PubMed] [Google Scholar]
- 18. Meier JJ, Menge BA, Breuer TG, et al. Functional assessment of pancreatic β-cell area in humans. Diabetes. 2009;58:1595–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kjems LL, Kirby BM, Welsh EM, et al. Decrease in β-cell mass leads to impaired pulsatile insulin secretion, reduced postprandial hepatic insulin clearance, and relative hyperglucagonemia in the minipig. Diabetes. 2001;50:2001–2012. [DOI] [PubMed] [Google Scholar]
- 20. Saisho Y, Butler AE, Manesso E, et al. Relationship between fractional pancreatic β cell area and fasting plasma glucose concentration in monkeys. Diabetologia. 2010;53:111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matveyenko AV, Veldhuis JD, Butler PC. Mechanisms of impaired fasting glucose and glucose intolerance induced by an approximate 50% pancreatectomy. Diabetes. 2006;55:2347–2356. [DOI] [PubMed] [Google Scholar]
- 22. Laybutt DR, Preston AM, Akerfeldt MC, et al. Endoplasmic reticulum stress contributes to β cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. [DOI] [PubMed] [Google Scholar]
- 23. Huang CJ, Lin CY, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56:2016–2027. [DOI] [PubMed] [Google Scholar]
- 24. Marchetti P, Del Guerra S, Marselli L, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab. 2004;89:5535–5541. [DOI] [PubMed] [Google Scholar]
- 25. Gurlo T, Ryazantsev S, Huang CJ, et al. Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol. 2010;176:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Howard CF., Jr Longitudinal studies on the development of diabetes in individual Macaca nigra. Diabetologia. 1986;29:301–306. [DOI] [PubMed] [Google Scholar]
- 27. Butler AE, Dhawan S. β-Cell identity in type 2 diabetes: lost or found? Diabetes. 2015;64:2698–2700. [DOI] [PubMed] [Google Scholar]
- 28. Meier JJ, Kohler CU, Alkhatib B, et al. β-Cell development and turnover during prenatal life in humans. Eur J Endocrinol. 2010;162:559–568. [DOI] [PubMed] [Google Scholar]
- 29. Butler AE, Cao-Minh L, Galasso R, et al. Adaptive changes in pancreatic β cell fractional area and β cell turnover in human pregnancy. Diabetologia. 2010;53:2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gershengorn MC, Hardikar AA, Wei C, Geras-Raaka E, Marcus-Samuels B, Raaka BM. Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science. 2004;306:2261–2264. [DOI] [PubMed] [Google Scholar]
- 31. Talchai SC, Accili D. Legacy Effect of Foxo1 in pancreatic endocrine progenitors on adult β-cell mass and function. Diabetes. 2015;64:2868–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Costes S, Langen R, Gurlo T, Matveyenko AV, Butler PC. β-Cell failure in type 2 diabetes: a case of asking too much of too few? Diabetes. 2013;62:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]