

Abstract
ADAM10 and ADAM17 are two closely related members of the ADAM (a disintegrin and metalloprotease) family of membrane-bound sheddases, which proteolytically cleave surface membrane proteins. Both ADAM10 and ADAM17 have been implicated in the proteolytic cleavage of NOTCH receptors and as such regulators of NOTCH signaling. During retinal development, NOTCH signaling facilitates retinal neurogenesis by maintaining progenitor cells in a proliferative state and by mediating retinal cell fates. However, the roles of ADAM10 and ADAM17 in the retina are not well defined. In this study, we set out to clarify the roles of ADAM10 and ADAM17 during early retinal development. The retinal phenotype of conditionally abated Adam17 retinae (Adam17 CKO) did not differ from the controls whereas conditionally ablated Adam10 retinae (Adam10 CKO) exhibited abnormal morphogenesis characterized by the formation of rosettes and a loss of retinal laminae phenotypically similar to morphological abnormalities identified in mice with retinal NOTCH signaling deficiency. Additionally, Adam10 CKO retinae exhibited abnormal neurogenesis characterized by fewer proliferating progenitor cells and greater differentiation of early photoreceptors and retinal ganglion cells. Moreover, constitutive activation of the NOTCH1-intracellular domain (N1-ICD) rescued Adam10 CKO abnormal neurogenesis, as well as abnormal retinal morphology by maintaining retinal cells in the progenitor state. Collectively these findings provide in vivo genetic evidence that ADAM10, and not ADAM17, is indispensable for proper retinal development as a regulator of NOTCH signaling.
Introduction
During retinal development, all retinal cell types are derived from a single population of pluripotent retinal progenitor cells (RPCs). The birth order of retinal cells is unidirectional and highly conserved although at any given developmental time point there is an overlap in the generation of various retinal cell types [1–3]. In mice, retinal neurogenesis starts around E11 with the birth of ganglion cells followed by the birth of cone photoreceptors, horizontal and amacrine cells, with rod photoreceptors forming around birth and finally bipolar cells and Müller glia as the last retinal cell types born postnatally [1–3]. It has been proposed that RPCs undergo temporally regulated successive stages of competence to either generate a differentiated retinal cell type or to transit to the next stage of RPC competence that facilitates the birth of subsequent retinal cell types [1, 3].
NOTCH signaling is an evolutionarily conserved pathway involved in the development of most tissues. The role of NOTCH signaling is in the regulation of cell proliferation, cell death, cell fate determination, and differentiation [4, 5]. In mammals, there are four NOTCH receptors (NOTCH1-4) and five NOTCH ligands (JAG1, JAG2, DLL1, DLL3, DLL4) that exhibit both redundant and unique functions [4]. The canonical NOTCH pathway involves binding of a NOTCH ligand from the surface of adjacent cells to the NOTCH receptor thereby facilitating the subsequent NOTCH receptor cleavage at the S2 site followed by cleavage at the S3 and S4 sites resulting in the release of the NOTCH intracellular domain (NICD) from the cell membranes; once released the NICD translocates into the nucleus and forms a complex with RBPJ and MAML1 along with other cofactors to transcriptionally activate inhibitors of differentiation [6–8]. Therefore, one of the key roles of NOTCH signaling is maintaining progenitor cells in their undifferentiated state. Additionally, during retinal development NOTCH signaling facilitates neurogenesis by repressing retinal cell fates [9–15].
ADAM10 and ADAM17 are two closely related members of the ADAM family of proteins that proteolytically cleave or “shed” ectodomains of cell surface proteins [16, 17]. Both ADAM10 and ADAM17 have been implicated as sheddases of NOTCH receptors at the S2 cleavage site thereby facilitating subsequent cleavage at S3 and S4 sites by the γ-secretase complex [18–21]. In C. elegans, sup-17, the ortholog of ADAM10, and adm-4, the ortholog of ADAM17, are functionally redundant as NOTCH regulators [21]. In Drosophila, Kuzbanian (kuz), the fly ortholog of ADAM10, regulates NOTCH cleavage and signaling [18–20]. Phenotypically, kuz mutants exhibit neurogenic and ommatidial defects similar to those observed in the fly Notch mutants [18]. Mice deficient for Adam10 die at E9.5 and phenocopy Notch1 deficient mice [22] in contrast to Adam17-/- mice that die at birth without phenotypic similarities to Notch1 mouse mutants [23, 24]. Although findings from knock-out mice implicate ADAM10 in the proteolytic cleavage of NOTCH1, tissue culture studies have shown that ADAM17, and not ADAM10, cleaves NOTCH1 [25, 26]. Further studies determined that ADAM10 is indispensable for ligand-induced NOTCH1 signaling and ADAM17 mediates ligand-independent NOTCH1 cleavage [27, 28]. Therefore, it was proposed that different ADAMs might contribute to the NOTCH receptor cleavage in a tissue-specific manner with ADAM10 as the primary regulator of NOTCH1 cleavage in vivo [22]. Recent studies support this hypothesis showing that in vivo ADAM10 regulates NOTCH1 during brain [29], skin [30], intestinal [31], thymocyte [32], and vascular development [33].
During retinal development, the roles of ADAM10 and ADAM17 are unclear. Both ADAM10 and ADAM17 exhibit similar spatiotemporal expression in the developing chicken retina [34]. The early embryonic lethality of Adam10-/- mice prior to the optic cup formation has precluded any evaluation of the role of ADAM10 in the retina [22]. However, pharmacological inhibition of ADAM10 partially rescued upregulated NOTCH1 signaling in mouse retinae deficient for Srfp1 and Srfp2 implicating ADAM10 as a regulator of NOTCH1 signaling during early retinal development [35]. By contrast, at birth Adam17-/- mice do not exhibit any obvious retinal abnormalities suggesting that ADAM17 does not have an essential role during the early retinal development, although the perinatal death of Adam17-/- mice precluded evaluations of the postnatal Adam17 deficient mature retinae [23, 24].
The goal of this study was to determine the roles of ADAM10 and ADAM17 in the retina and establish whether ADAM10, ADAM17, or both regulate retinal NOTCH1 signaling. Our results demonstrate that ADAM10, and not ADAM17, is indispensable for early retinal development as a key regulator of NOTCH1 signaling.
Materials and Methods
Mice and clinical eye evaluation
All animal procedures were performed in accordance with NIH guidelines and approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee (IACUC). Mice carrying a floxed Adam10 allele (Adam10tm1.1Zhu) were generously provided by Tian Xu. Mice carrying a floxed Adam17 allele (Adam17tm1.2Bbl), were generously provided by Carl Blobel. Six3-Cre mice carrying a Cre allele driven by the promoter of the Six3 gene were generously provided by Yasuhide Furuta. Gt(ROSA)26Sor tm1(NOTCH1)Dam/J. Each mouse strain was genotyped as previously described [24, 32, 36, 37] using primers in S1 Table. Mice with Six3-Cre;Adam10tm1Zh/tm1Zhu or Six3-Cre; Adam17tm1Bbl/tm1Bbl genotypes were referred to here as Adam10 CKO mice and Adam17 CKO mice respectively. Littermates with genotypes Adam10tm1Zhu/+ and Adam10tm1Zhu/ tm1Zhu as well as Adam17tm1Bbl/+ and Adam17tm1Bbl/tm1Bbl are referred to as controls.
Mouse eyes from the eight week old control, Adam10 CKO, and Adam17 CKO mice were examined with a Topcon SL-D8Z slit lamp biomicroscope with a Nikon SLR-based Photo Slit Lamp imaging system following mydriasis with 1% atropine sulfate (Bausch & Lomb) as previously described [38]. Control (n = 3) and Adam17 CKO (n = 3) animals at 8 weeks of age were dark adapted for 2 hr and ERGs were recoded after a single flash of intensity sufficient to evoke the maximum retinal response as previously described [39].
RT-PCR
Eyes from newborn mice (n = 3) of each genotype were collected and the cornea, iris, and lens were removed. RNA was isolated and reverse transcribed as previously described [40]. Adam10 or Adam17 transcript levels were analyzed via semi-quantitative RT-PCR while in the exponential phase of the PCR amplification using Gapdh as an internal control as previously described [40] using primers in S1 Table. PCR band intensities were quantified using ImageJ software (http://rsbweb.nih.gov/ij/) and are expressed relative to Gapdh. The results represent at least three independent experiments performed in triplicates and the data is expressed as mean ± SEM. Comparison between the genotype groups was analyzed by Student’s t-test and P<0.05 was considered as statistically significant.
Histology and immunohistochemistry
Tissues were fixed in 4% PFA, or Davidson’s solution, embedded in paraffin, sectioned to 4 μm thickness and Hematoxylin and Eosin (H&E) stained as previously described [38]. For immunohistochemistry, following deparaffinization and rehydration, antigen retrieval was performed using 1x citrate buffer pH 6.0 (Life Technologies) as previously described [38] and then sections were blocked in 10% normal serum with 1% BSA in 1xPBS for 1–2 hr and incubated with primary antibody in 1xPBS with 1% BSA O/N at 4°C. 2x 5 min washes in 1xPBS, 0.025% Triton X-100 were followed by secondary antibody incubation in 1xPBS with 1% BSA for 1 hr at RT. Primary antibodies used in this study include: ADAM10 (R&D Systems), BRN3B, ADAM17 OTX2, and N-cadherin (Abcam), ISL1 (Hybridoma Bank), β-catenin, NOTCH1 and HES1 (Cell Signaling). Secondary antibodies used were Alexa Fluor 546 goat-anti rabbit and Alexa Fluor 488 goat-anti mouse (Life Technologies). All slides were counterstained with DAPI (Life Technologies), mounted using Prolong gold anti-fade reagent (Life Technologies) mounting media, and photographed with a Nikon DS-Fi1 camera on a Nikon eclipse 80i microscope. The percentage of OTX2 and ISL1positive cells were determined by counting the numbers of OTX2 and ISL1 positive cells and total DAPI positive cells from a minimum of 3 separate genotypes with at least 10 sections per genotype. Significance was calculated via Students t-test (GraphPad Prism), and P<0.05 was considered significant.
EdU, BrdU, and TUNEL assays
EdU (Life Technologies) or BrdU was injected intraperitoneally at a dose of 125 mg/kg of body weight into pregnant dams, 13.5 days after a vaginal plug was found and sacrificed 2 hr later. Embryos were fixed in 4% PFA for 2 hours and dehydrated through a graded series of ethanol washes, were paraffin embedded, and cut into 5 μm sections. EdU detection was performed with Click-iT EdU Alexa Fluor 488 Imaging Kit (Life Technologies) according to the manufacturer’s protocol and BrdU was detected using a mouse monoclonal anti-BrdU antibody conjugated to Alexa Flour 488 (Life Technologies). TUNEL staining was performed utilizing the ApopTag Plus In Situ Apoptosis Fluorescein Detection Kit (Millipore) according to the manufacturer’s recommendations. DAPI (Life Technologies) was used to stain the DNA, slides were mounted using Prolong gold anti-fade reagent (Life Technologies) and imaged using a Nikon DS-Fi1 camera on a Nikon Eclipse 80i microscope. The percentage of EdU, BrdU, and TUNEL positive cells after counting the numbers of EdU, BrdU or TUNEL positive cells and total DAPI positive cells from a minimum of 3 separate genotypes with at least 10 sections per genotype. Significance was calculated via a t-test (GraphPad Prism) and P<0.05 was considered significant.
Results
ADAM10 and ADAM17 are ubiquitously expressed retinal proteins
As the initial step, we evaluated the expression of ADAM10 and ADAM17 during retinal development in mice. In control embryonic E13.5, E16.5 and newborn P0.5 retinae both ADAM10 (Fig 1A, 1C and 1E) and ADAM17 (Fig 1B, 1D and 1F) exhibit ubiquitous expression in ganglion cell and neuroblastic layers. By P7, as the retina starts to mature, ADAM10 and ADAM17 are expressed in the ganglion cell layer, inner and outer plexiform layers, and in the photoreceptors (Fig 1G and 1H) and this pattern of expression is maintained in the adult retina for both ADAM10 (Fig 1I) and ADAM17 (Fig 1J).
Fig 1. Expression of ADAM10 and ADAM17 in the retina.
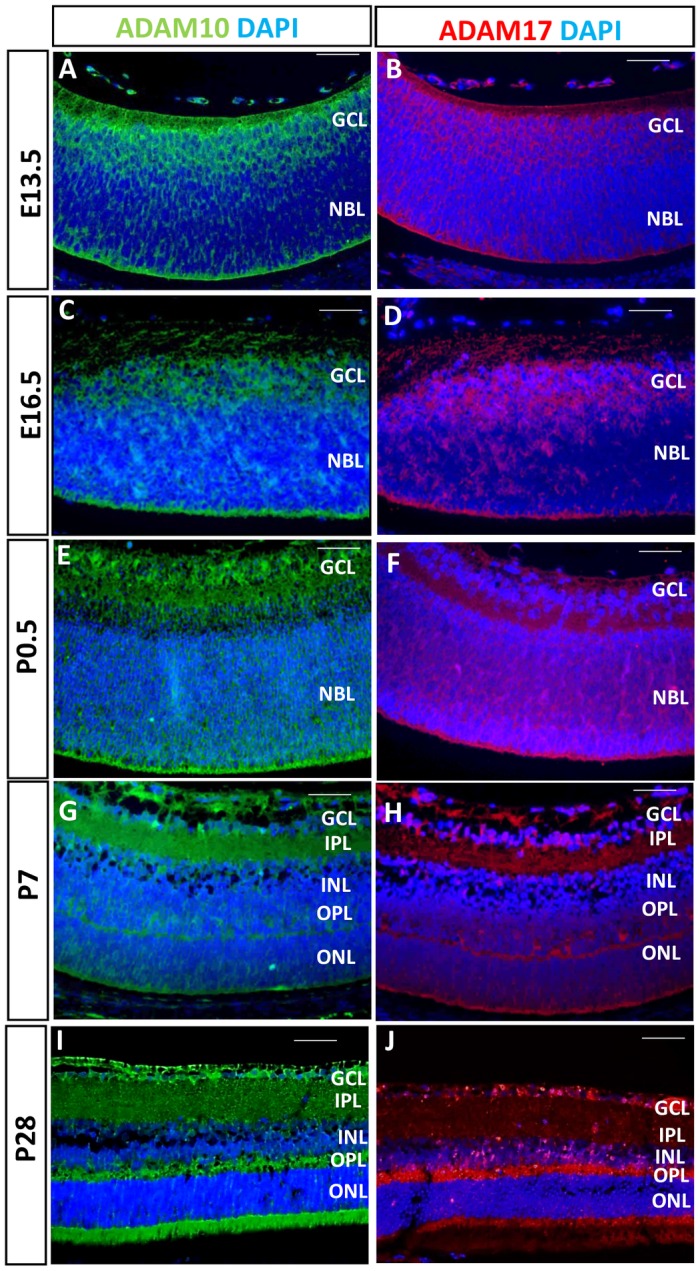
Immunostaining of control E13.5 retinae for ADAM10 (A) and ADAM17 (B), E16.5 retinae for ADAM10 (C) and ADAM17 (D) and P0.5 retinae for ADAM10 (E) and ADAM17 (F) revealed similar expression patterns in the ganglion cell layer (GCL) and neuroblastic layer (NBL). Immunostaining of P7 retinae for ADAM10 (G) and ADAM17 (H) and P28 retinae for ADAM10 (I) and ADAM17 (J) also revealed similar expression patterns in the inner plexiform layer (IPL), outer plexiform layer (OPL) and in the photoreceptors. INL = inner nuclear layer; ONL = outer nuclear layer. Nuclei were stained with DAPI. Scale bars = 50 μm.
ADAM10, but not ADAM17, is required for early retinal development
To determine the roles of ADAM10 and ADAM17 during early retinal development, we generated mice with retina-specific ablation of Adam10 (Adam10 CKO), as well as retina-specific ablation of Adam17 (Adam17 CKO) (Fig 2A and 2B). In newborn Adam10 CKO and Adam17 CKO retinae, only residual levels of Adam10 transcript containing exon 3 (Fig 2C) and Adam17 transcript containing exon 2 (Fig 2D) were identified when compared to the controls. Therefore, Adam10 and Adam17 were successfully disrupted in most retinal cells in Adam10 CKO and Adam17 CKO mice respectively.
Fig 2. Conditional ablation of Adam10 and Adam17 in the retina.

(A) Mice carrying homozygous floxed Adam10 allele (Adam10tm1.1Zhu) were crossed with Six3-Cre transgenic mice expressing Cre in the optic cup and optic stalk resulting in conditionally ablated Adam10 in the retina. (B) A similar approach was taken to generate Adam17 CKO mice after homozygote Adam17tm1.2Bbl mice were crossed to Six3-Cre mice. (C) Semi-quantitative analysis of the relative expression of Adam10 transcript containing exon 3 normalized to Gapdh expression from RNA isolated from newborn control and Adam10 CKO littermates revealed significantly reduced (P<0.0001; n = 3) levels in Adam10 CKO retinae when compared to the controls. (D) Semi-quantitative analysis of the relative expression of exon 2 containing Adam17 mRNA normalized to Gapdh expression from RNA isolated from newborn control and Adam17 CKO retinae also revealed significantly reduced (P<0.0002; n = 3) levels in Adam17 CKO retinae when compared to the controls. In both (C) and (D), the bars represent mean values ± SEM. Significance was established following Student’s t-test analysis and P<0.05 was considered significant. (F) Adam10 CKO eyes appeared smaller when compared to the eyes in the control mice (E) whereas Adam17 CKO eyes (H) that did not appear to differ from age-matched control littermates (G). H&E staining of Adam10 CKO eyes at 8 weeks of age (J) revealed severe retinal abnormalities characterized by the formation of rosettes (J’, arrow) in contrast to the retinal laminae in the age-matched controls (I-I’). Histological analysis of Adam17 CKO eyes (L-L’) did not identify any morphological differences when compared to the age-matched controls (K-K’). Figures shown in (I’) through (L’) are enlarged images of the areas depicted by the dashed boxes in Figures (I) through (L). GCL = ganglion cell layer, IPL = inner plexiform layer, INL = inner nuclear layer, OPL = outer plexiform layer, ONL = outer nuclear layer. Scale bars = 50 μm.
The evaluation of adult (8 weeks of age) Adam10 CKO eyes by observation revealed a smaller eye size (Fig 2F) when compared to age-matched control littermates (Fig 2E). Adam17 CKO mice did not exhibit any obvious eye size differences (Fig 2H) when compared to age-matched control littermates (Fig 2G). Histological analysis of adult Adam10 CKO eyes identified profoundly disrupted retinal laminae (Fig 2J and S1B Fig) characterized by the formation of rosettes (Fig 2J’). Histological analysis of Adam17 CKO eyes and retinae (Fig 2L and 2L’ and S1D Fig) did not identify any morphological differences when compared to age-matched control littermates (Fig 2K and 2K’ and S1C Fig). To exclude the possibility that Adam17 CKO retinae may exhibit functional, but not morphological abnormalities we performed electroretinograms (ERGs). Our analysis did not identify any differences in ERGs between the control and Adam17 CKO mice (not shown). Collectively these findings provided evidence that ADAM10, and not ADAM17, is required for early retinal development.
The onset and progression of retinal phenotypes in Adam10 CKO mice
The first morphological abnormalities in Adam10 CKO retinae were evident at E13.5 characterized by disrupted cellular architecture (Fig 3B). Along the apical-basal axis nuclei from Adam10 CKO retinae appeared rounded and disorganized resulting in misshapen apical and basal surfaces (Fig 3B’). By contrast, the E13.5 control retinae exhibited elongated and organized nuclei with distinct apical and basal surfaces (Fig 3A and 3A’). While morphological abnormalities were identified within the Adam10 CKO central retinae some regions of peripheral retinae retained undisrupted retinal cell organization (Fig 3B). It was reported previously that Six3-Cre is expressed across the entire retina although the highest expression is within the central retina [36]. To explore this further, we immunostained the control and the Adam10 CKO retinae for ADAM10. Within the central Adam10 CKO retinae, the majority of cells did not express ADAM10 (Fig 3D and S2B Fig). However, at the retinal periphery, we identified residual ADAM10 immunostaining (Fig 3D and S2B Fig). This finding suggested that milder retinal abnormalities identified at the retinal periphery of E13.5 Adam10 CKO may have been caused by the lower efficiency of CRE-mediated Adam10 ablation relative to the central retina. By E16.5 the Adam10 CKO retinae started to exhibit rosettes within the central retinae (Fig 3F); the identified rosettes were of different sizes (Fig 3F’). Immunostaining of E16.5 Adam10 CKO retinae for ADAM10 did not identify any residual staining (Fig 3H). We also morphologically evaluated the postnatal retinal development of Adam10 CKO mice. By P0.5 in Adam10 CKO retinae, the formation of rosettes expanded further towards the retinal periphery (S2D and S2D’ Fig) and by P7 the rosettes were present throughout the entire retina (S2F and S2F’ Fig).
Fig 3. Retinal morphology in control and Adam10 CKO mice.
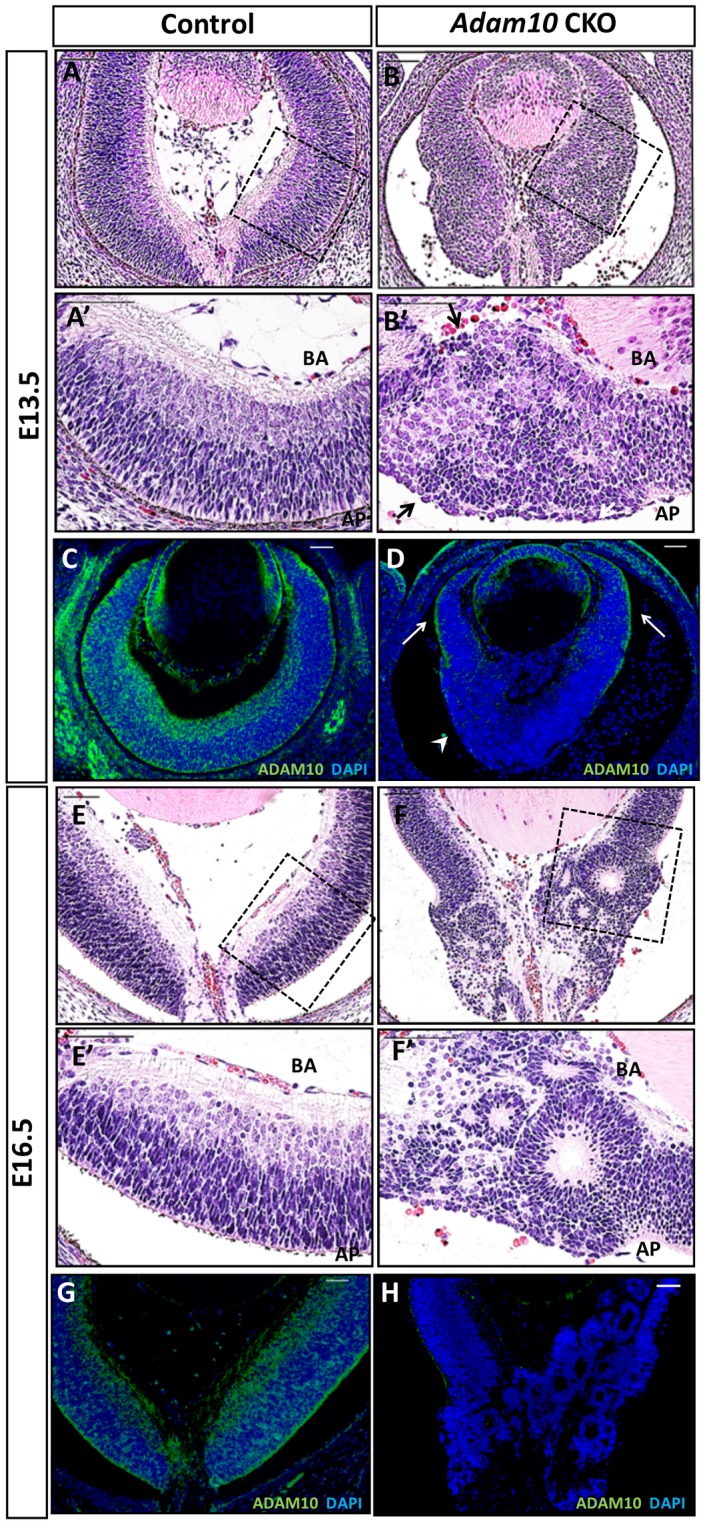
H&E staining revealed first abnormalities in E13.5 Adam10 CKO retinae (B) within the central region characterized by rounded and highly disorganized nuclei along the apical-basal axis resulting in disrupted basal and apical surfaces (B’, arrows) in contrast to the highly organized control retinae (A-A’). ADAM10 immunostaining of E13.5 Adam10 CKO retinae (D, arrowhead) revealed an absence of ADAM10 expressions in most cells within the central retina when compared to the age-matched controls (C). However, residual ADAM10 expression was detected in the peripheral retinae of Adam10 CKO mice (D, arrows). H&E staining of E16.5 Adam10 CKO retinae (F) identified within the central region rosettes of various sizes (F’) in contrast to the highly organized retinal laminae in the age-matched control retinae (E-E’). Immunostaining of E16.5 Adam10 CKO retinae (H) did not identify any ADAM10-positive cells in contrast to the age-matched controls (G). BA = basal surface, AP = apical surface. Scale bars = 50 μm.
ADAM10 inactivation disrupts retinal neurogenesis
Next, we wanted to determine if Adam10 CKO retinae exhibited disrupted neurogenesis. At E13.5 in control retinae, EdU-positive cells were highly organized and restricted to the neuroblastic layer (Fig 4A) whereas in E13.5 Adam10 CKO retinae EdU-positive cells were highly disorganized (Fig 4B). Quantification analysis revealed a significantly lower (P = 0.019; n = 3) percentage of EdU-positive cells in the Adam10 CKO retinae when compared to the controls (Fig 4C). Next, we hypothesized that the fewer proliferating cells identified in Adam10 CKO retinae (Fig 4A–4C) may be caused by precocious retinal cell differentiation. Immunostaining of the control retinae for orthodenticle homeobox 2 (OTX2), which is a marker for early photoreceptors [41], revealed sparse OTX2-positive staining in cells along the retinal apical surface consistent with the development of early photoreceptors (Fig 4D). By contrast, Adam10 CKO retinae exhibited highly disorganized OTX2-positive cells (Fig 4E). Quantification analysis revealed a significantly greater (P = 0.003; n = 3) percentage of OTX2-positive cells in the Adam10 CKO retinae when compared to the controls (Fig 4F). We also evaluated the ISL1 expression as a marker for early ganglion cells [42]. In control retinae, ISL1 immunostaining was restricted to the cells at the retinal basal layer consistent with the developing ganglion cells (Fig 4G). In the Adam10 CKO retinae, ISL1-positive cells were highly disorganized (Fig 4H). Quantification analysis revealed a significantly greater (P = 0.004; n = 3) percentage of ISL1-positive cells in E13.5 Adam10 CKO when compared to the control retinae (Fig 4I). Similar findings were observed following immunostaining with BRN3B (S3A–S3C Fig), a marker for early ganglion cells [42]. In both the control (Fig 4J) and Adam10 CKO retinae (Fig 4K), the OTX2-positive and ISL1-positive cells did not co-stain for EdU, indicating that OTX2-positive and ISL1-positive cells had exited the cell cycle.
Fig 4. Retinal neurogenesis at E13.5 in control and Adam10 CKO mice.

In the control retinae (A) the EdU-positive cells were highly organized along the apical surface with no EdU-positive cells present at the basal surface. By contrast, in the Adam10 CKO eyes (B) within the central retinae a few highly disorganized EdU-positive cells were identified. Quantification analysis (C) revealed in control retinae 33.50 ± 0.61% (gray bar) and in Adam10 CKO retinae 20.01± 2.5% (white bar) EdU-positive cells indicating a significantly lower (**P = 0.019; n = 3) percentage of EdU-positive cells in the Adam10 CKO retinae. In the control retinae (D), sparse OTX2-positive immunostaining was identified in cells along the apical surface. In Adam10 CKO retinae (E), OTX2-positive cells were present in all retinal layers. Quantification analysis (F) revealed in control retinae 7.95 ± 0.9% (gray bar) and in Adam10 CKO retinae 27.1 ± 2.8% (white bar) OTX2- positive cells indicating a significantly greater (**P = 0.003; n = 3) percentage of OTX2-positive cells in Adam10 CKO retinae. In the control retinae (G), ISL1-positive cells were identified within the basal layer whereas in Adam10 CKO retinae (H) ISL1-positive cells were highly disorganized and present in all retinal layers. Quantification analysis (I) revealed in the control retinae 23.4 ± 3.2% (gray bar) and in Adam10 CKO retinae 50.1 ± 1.3% (white bar) ISL1-positive cells indicating a significantly greater (**P = 0.004; n = 3) percentage of ISL1-positive cells in the Adam10 CKO retinae. In both the control (J) and Adam10 CKO retinae (K), the OTX2-positive and ISL1-positive cells did not co-stain for EdU. DAPI staining of the control and Adam10 CKO retinae are shown in (L) and (M) respectively. Bars in (C), (F), and (I) represent mean values ± SEM. AP = apical surface. BA = basal surface. Scale bar = 50 μm.
Characterization of rosettes in E16.5 Adam10 CKO retinae
Morphological evaluation of Adam10 CKO retinae revealed the formation of rosettes at E16.5 (Fig 3F and 3F’). Next we set to characterize which cells formed the rosettes in E16.5 Adam10 CKO retinae. In the control, the retinae BrdU-positive cells are positioned between the OTX2-positive photoreceptors on the apical side and ISL1-positive ganglion cells at the basal side (Fig 5A and 5C). Immunostaining of Adam10 CKO retinae revealed that BrdU-positive cells were forming the rosette outer cellular layers (Fig 5B) whereas OTX2 positive photoreceptors were forming the rosette inner cellular layers (Fig 5D). However, in the Adam10 CKO retinae, OTX2-positive photoreceptors were also identified as highly disorganized cells present at the apical and basal surface as well as cells randomly scattered throughout the retina (Fig 5D). The ISL1-positive cells did not associate with rosettes and appeared to be randomly distributed throughout the Adam10 CKO (Fig 5D). It was shown previously that both N-cadherin and β-catenin are indispensable for the formation of retinal laminae [43, 44], thus we wanted to determine if N-cadherin and β-catenin expression had been altered in the Adam10 CKO retinae. In the control retinae both N-cadherin and β-catenin (Fig 5E–5G) were expressed throughout the retina especially in the cells forming apical and basal surfaces consistent with previous reports [44]. In the Adam10 CKO retinae both N-cadherin (Fig 5F) and β-catenin (Fig 5H) were highly expressed in cells forming the rosette lumens whereas cells forming apical and basal surfaces intermittently expressed both N-cadherin and β-catenin. Next, we wanted to determine if cell death was contributing to the formation of rosettes. In E16.5 control retinae, we identified a very few TUNEL-positive cells (Fig 5I) whereas in Adam10 CKO the TUNEL-positive cells were scattered throughout all layers of the retinae (Fig 5J). Although a significantly greater (P = 0.003; n = 3) percentage of TUNEL-positive cells were identified in Adam10 CKO retinae (Fig 5H) when compared to controls, the overall percentage of TUNEL-positive cells was very small (<5%) in both the WT and Adam10 CKO retinae (Fig 5K). Consistent with this, the overall number of DAPI positive cells did not significantly differ (P = 0.19; n = 3) between the two genotypes (Fig 5L).
Fig 5. Characterization of rosettes in E16.5 Adam10 CKO retinae.
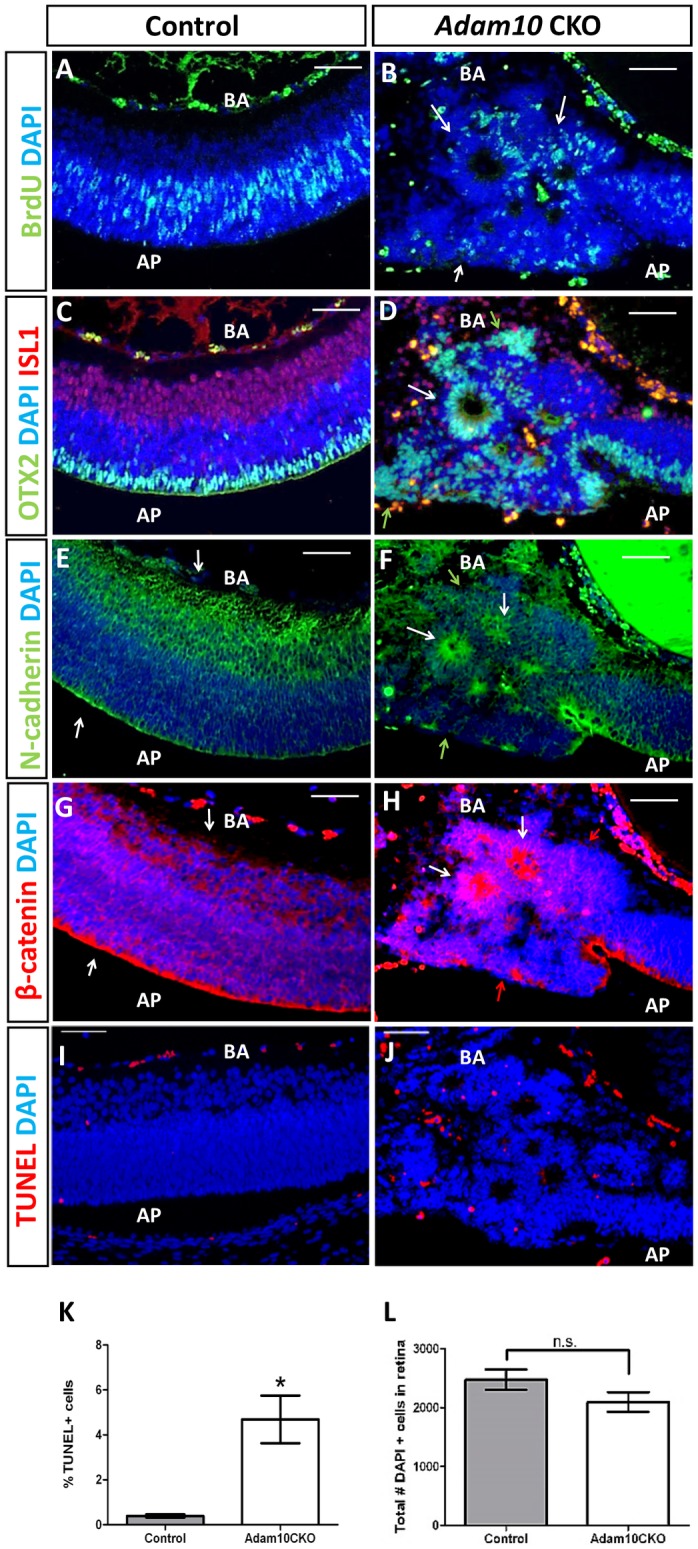
In controls (A) BrdU-positive cells were identified in the neuroblastic layer. In Adam10 CKO retinae, BrdU-positive cells were identified in the outer cell layer of rosettes (B, arrows). In controls (C) immunostaining for OTX2 (green) and ISL1 (red) marked early photoreceptor cells at the apical surface and ganglion cells at the basal surface. In Adam10 CKO retinae (D) the OTX2-positive cells (D, green) formed rosette lumens (D, white arrow), although OTX2-positive cells were also present at the apical and basal surfaces (green arrows in D). ISL1-positive cells (D, red) did not associate with rosettes and were scattered through the entire retina. In control retinae, N-cadherin (E) and β-catenin (G) were expressed in all retinal cells although the highest expression was in the cells forming the apical surface (white arrows in E and G). In Adam10 CKO retinae cells forming the rosette lumens were highly expressing N-cadherin and β-catenin (white arrows F and H); cells at the apical and basal surface were intermittently expressing N-cadherin (green arrows in F) and β-catenin (red arrows in H). Very few TUNEL-positive cells were identified in the control (I) and Adam10 CKO (J) retinae. Quantification analysis (K) identified in the controls 0.4 ± 0.01% (gray bar) and in Adam10 CKO 4.69 ± 0.11% (white bar) TUNEL-positive cells indicated a significantly greater (*P = 0.003; n = 3) percentage of TUNEL-positive cells in the Adam10 CKO retinae. Quantification analysis (L) identified 2476 ± 173 (gray bar) DAPI-positive cells in controls and 2096 ± 167 (white bar) DAPI-positive cells in Adam10 CKO retinae indicating that the overall number of retinal cells did not significantly differ (P = 0.19; n = 3) between the two genotypes. Bars in (K) and (L) represent mean values ± SEM. Significance was established following analysis with Student’s t-test and P<0.05 was considered significant. In all panels, nuclei were stained with DAPI. AP = apical surface. BA = basal surface. n.s. = not significant. Scale bar = 50 μm.
ADAM10 regulates NOTCH signaling
Next, we set out to determine if disrupted NOTCH1 signaling contributed to the retinal phenotypes identified in Adam10 CKO retinae. Immunostaining of E13.5 control retinae revealed that NOTCH1 positive immunostaining was restricted to the neuroblastic layer (Fig 6A). By contrast in E13.5 Adam10 CKO retinae very few cells within the central retina stained positive for NOTCH1, although towards the retinal periphery highly disorganized NOTCH1 positive cells were evident (Fig 6B). Quantification analysis confirmed a significantly (P = 0.001; n = 3) lower percentage of NOTCH1 positive cells in E13.5 Adam10 CKO retinae when compared to the age-matched controls (Fig 6C). Immunostaining for HES1, which is a NOTCH1 downstream target [15], revealed a very few HES1-immunopositive cells within the central E13.5 Adam10 CKO retinae that appeared highly disorganized (Fig 6D). However, at the retinal periphery HES1 positive cells were identified restricted to the neuroblastic layer (not shown). Quantification analysis confirmed a significantly (P = 0.0004; n = 3) lower percentage of HES1-positive in E13.5 Adam10 CKO retinae when compared to the age-matched controls.
Fig 6. Overexpression of N1-ICD in Adam10 CKO retinae rescues aberrant retinal morphogenesis and neurogenesis.

In controls (A) NOTCH1-positive cells were restricted to the neuroblastic layer. In Adam10 CKO retinae, within the central retinae only a very few NOTCH1-positive cells were identified associated with rosettes (B, arrow) although towards the retinal periphery NOTCH1-positive cells were identified restricted to the neuroblastic layer (B, arrowhead). Quantification analysis (C) identified in controls 42 ± 1.2% (gray bar) and in Adam10 CKO 22.79 ± 0.11% (white bar) NOTCH1-positive cells indicating a significantly fewer (**P = 0.001; n = 3) percentage of NOTCH1-positive cells in Adam10 CKO retinae when compared to the controls. In controls HES1, immunostaining was restricted to the neuroblastic layer (D) whereas in Adam10 CKO retinae a very few HES1-positive cells were identified within the central retinae and HES1-positve cells appeared disorganized (E). Quantification analysis (F) identified in controls 34 ± 0.01% (gray bar) and in Adam10 CKO retinae 4.7 ± 0.2% (white bar) HES1-positive cells indicating a significantly fewer (**P = 0.0004; n = 3) percentage of HES1-positive cells in Adam10 CKO retinae when compared to controls. (G) Adam10 CKO mice were crossed to RosaN1-IC (N1-ICD) mice harboring a Neo/STOP cassette flanked by loxP sites upstream of a sequence encoding the intracellular domain of the NOTCH1 gene thereby generating Adam10 CKO; N1-ICD mice that exhibit constitutive expression of N1-ICD in the same retinal cells in which Six3-CRE led to ablation of Adam10. H&E staining revealed in N1-ICD mice (H) and Adam10 CKO;N1-IC mice (I) highly organized retinae. In N1-ICD retinae (J) and Adam10 CKO;N1-IC retinae (K) EdU-positive cells were present in all retinal layers. Quantification analysis (L) revealed in N1-ICD retinae 41.99 ± 2.78% (gray bar) and in Adam10 CKO retinae 44.15 ± 3.76% (white bar) EdU-positive cells indicating that the percentage of EdU positive retinal cells did not significantly differ (P = 0.24; n = 3). Bars in (C), (F), and (L) represent mean values ± SEM. Significance was established with the Student’s t-test and P<0.05 was considered significant. AP = apical surface. BA = basal surface. Scale bars = 50 μm.
In order to determine if disrupted NOTCH1 signaling is directly caused by a functional loss of ADAM10, we utilized a genetic approach. We focused on establishing if overexpression of the NOTCH1-intracellular domain (N1-ICD) can rescue aberrant morphogenesis and/or neurogenesis identified in Adam10 CKO retinae. We crossed Adam10 CKO mice to Gt(ROSA)26Sor tm1(NOTCH1)Dam/J mice and generated mice with genotypes Six3-Cre;Adam10 CKO;RosaN1-IC/+ (Adam10 CKO;N1-ICD) wherein the same retinal cells Six3-driven CRE recombinase activity deleted Adam10 exon 3 and overexpressed N1-ICD; mice with Six3-Cre;RosaN1-IC/+ (N1-ICD) genotypes, where Six3-CRE-mediated constitutive retinal activation of N1ICD, were used as controls (Fig 6G). At E13.5 cells, both N1-ICD retinae (Fig 6H) and Adam10 CKO;N1-ICD retinae (Fig 6I), were highly organized along the apical-basal axis with well-defined apical and basal surfaces. EdU incorporation revealed that in both N1-ICD retinae (Fig 6J) and Adam10 CKO; N1-ICD retinae (Fig 6K) proliferative cells were present in all retinal layers suggesting that all retinal cells in these mice were maintained in the progenitor state. The percentage of EdU positive cells did not significantly differ between the genotypes (Fig 6L). To explore this further, we immunostained E13.5 N1-ICD retinae and Adam10 CKO; N1-ICD retinae for OTX2 and ISL1 as early photoreceptor and ganglion cell markers respectively. Within the central region of both N1-ICD retinae (S4A Fig) and Adam10 CKO; N1-ICD (S4B Fig) we did not identify any OTX2-positive or ISL1-positive cells. However, at the retinal periphery very few OTX2-positive (<1%) and ISL1-positive cells (<3%) were identified in both N1-ICD (S4C Fig) and Adam10 CKO; N1-ICD retinae (S4D Fig). Quantification analysis did not identify a significant difference between N1-ICD and Adam10 CKO;N1-ICD retinae in the percentage of OTX2-positive and ISL1-positive cells (S4E and S4F Fig).
Discussion
In this study, we show that ADAM10 is indispensable for retinal neurogenesis as well as the formation of retinal laminae. The rescue of aberrant retinal phenotypes in ADAM10-deficient retinae by overexpression of the N1-ICD provides in vivo genetic evidence that ADAM10 is a key regulator of NOTCH1 signaling. In mice during early retinal development NOTCH1 maintains retinal cells in the undifferentiated progenitor state, but it also has a role in repressing photoreceptor cell fate [10, 11]. As such the role of NOTCH1 signaling has been proposed to have a key role in assuring the sufficient number of progenitor cells necessary for the development of all retinal cell types [10, 11]. Results from this study showed that ADAM10 deficiency decreased the pool of retinal progenitor cells and increased the production of early photoreceptors further supporting the idea that ADAM10 is a regulator of NOTCH1 signaling. However, our results also show that ADAM10 retinal deficiency increased the production of ganglion cells, a phenotype not identified in mice with NOTCH1-deficient retinae [10, 11]. ADAM10 ablation in the developing retina phenocopies aberrant retinal neurogenesis in mice with retinae deficient for both NOTCH1 and NOTCH3 [45] as well as in mice with retinae deficient for RPBJ [12]. While it is well established that NOTCH1 is a regulator of the progenitor cell pool and represses photoreceptor fate, recent studies have shown both NOTCH3 and NOTCH1 contribute to the repression of ganglion cell fate [45]. Furthermore both NOTCH1 and NOTCH3 signal through RBPJ as a common downstream target [12]. Therefore, it was hypothesized that NOTCH1+NOTCH3>RBPJ repress retinal ganglion cell fate. We propose that ADAM10 is a common NOTCH signaling regulator by facilitating the cleavage of both NOTCH1 and NOTCH3 which in turn facilitates maintenance of the progenitor cell pool and repression of both photoreceptors and ganglion cells. In support of this hypothesis are recent reports showing that in cell culture ADAM10 cleaves NOTCH3 via a similar mechanism as NOTCH1 [46]. Additionally, during retinal development expression of NOTCH1 and NOTCH3 spatially and temporally coincide [47]. However, further studies are needed to unequivocally establish if ADAM10 mediates the cleavage of both NOTCH1 and NOTCH3 during early retinal neurogenesis.
In addition to aberrant neurogenesis, ADAM10 retinal ablation also results in severely disrupted formation of retinal laminae. Prior to differentiation, RPCs are positioned along the apical-basal axis and are attached to both the apical and basal surfaces [48, 49]. As RPCs start to differentiate, they undergo detachment allowing the cells to migrate to the appropriate retinal layer thereby establishing the retinal laminae and neuronal circuitry required for vision [48, 49]. Our results show that ADAM10 retinal deficiency results in the formation of rosettes similar to the rosettes reported for mice with disrupted retinal NOTCH1 signaling [9–15]. It was shown that RBPJ retinal deficiency causes discontinuous expression of β-catenin resulting in the formation of rosettes; ectopic overexpression of β-catenin in RBPJ-deficient retinae rescued the rosette formation without rescuing neurogenesis defects [13]. How disrupted retinal NOTCH1 signaling alters β-catenin expression still remains unclear. Active β-catenin is a downstream effector of Wnt signaling [50], but active β-catenin is also bound to N-cadherin at the cell membranes [51, 52]. A functional deficiency of either N-cadherin or β-catenin disrupts the formation of retinal laminae without disrupted neurogenesis [43, 44, 53, 54]. However, it was shown previously that ADAM10 mediates the cleavage of N-cadherin and as such regulates the release of active β-catenin into the cytoplasmic pool [51]. At this point, it is unclear if the rosetting phenotypes observed in ADAM10-deficient retinae are solely caused by the disrupted NOTCH1 signaling or if disrupted ADAM10-mediated cleavage of N-cadherin also contributes to the aberrant retinal morphogenesis. The rescue of aberrant retinal morphology in ADAM10-deficeint retinae by overexpression of N1-ICD supports the idea that the failure in the formation of laminae in the ADAM10-deficeint retinae is solely caused by disrupted NOTCH1 signaling. However, we cannot exclude the possibility that ADAM10 may be regulating the cleavage of N-cadherin as a downstream event following cleavage of the NOTCH1 receptor.
While this study establishes ADAM10 as indispensable for the early retinal development, the role of ADAM17 still remains unclear. Our results show that ADAM10 and ADAM17 exhibit an overlapping spatiotemporal expression pattern in the developing mouse retinae consistent with the expression of ADAM10 and ADAM17 previously reported for the developing chick retinae [34]. However, it should be pointed out that another study using a different antibody identified that in mice, the retinal expression of ADAM17 starts at P7 [55]. Furthermore, the same study revealed that in the adult (P175) mouse retinae ADAM17 expression is restricted to the ganglion cell layer, the inner and outer edge of the inner nuclear layer [55]. This discrepancy may have been caused by the variable specificity of the antibodies used as well as different time points selected for the analysis. Nonetheless, our findings unequivocally indicate that ADAM17 is dispensable for retinal development. In the developing chick retina, in addition to ADAM10, ADAM17 exhibits similar spatiotemporal expression pattern with ADAM9, ADAM12, ADAM13, ADAM22, and ADAM23 [34]. Except for ADAM9, that has been shown to have a role in the development of retinal vasculature and homeostasis [56, 57], the roles of other ADAM proteins during retinal development and homeostasis are unknown providing no information if ADAM17 may have functional overlaps with other ADAM proteins. Studies focusing on the role of ADAM17 as a regulator of retinal homeostasis are currently in progress.
Supporting Information
(TIF)
(TIF)
(TIF)
(TIF)
(DOCX)
Acknowledgments
We would like to thank Mr. Peter Volberding and Dr. Joseph Besharse from the Department of Cell Biology, Neurobiology and Anatomy, Medical College of Wisconsin for their assistance with performing, recording, and interpretation of ERGs in mice.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by EY018872, EY001931, EY014537, and EY018872, National Eye Institute, National Institutes of Health, http://www.nih.gov/. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Cepko CL, Austin CP, Yang X, Alexiades M, Ezzeddine D. Cell fate determination in the vertebrate retina. Proc Natl Acad Sci U S A. 1996;93(2):589–95. Epub 1996/01/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212(2):199–205. Epub 1985/06/01. 10.1002/ar.1092120215 . [DOI] [PubMed] [Google Scholar]
- 3.Cepko C. Intrinsically different retinal progenitor cells produce specific types of progeny. Nature reviews Neuroscience. 2014;15(9):615–27. Epub 2014/08/07. 10.1038/nrn3767 . [DOI] [PubMed] [Google Scholar]
- 4.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–33. Epub 2009/04/22. 10.1016/j.cell.2009.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nature reviews Genetics. 2012;13(9):654–66. Epub 2012/08/08. 10.1038/nrg3272 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R. Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. The EMBO journal. 1999;18(8):2196–207. Epub 1999/04/16. 10.1093/emboj/18.8.2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohsawa R, Kageyama R. Regulation of retinal cell fate specification by multiple transcription factors. Brain research. 2008;1192:90–8. Epub 2007/05/10. S0006-8993(07)00814-1 [pii] 10.1016/j.brainres.2007.04.014 . [DOI] [PubMed] [Google Scholar]
- 8.Gazave E, Lapebie P, Richards GS, Brunet F, Ereskovsky AV, Degnan BM, et al. Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC evolutionary biology. 2009;9:249 Epub 2009/10/15. 10.1186/1471-2148-9-249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rocha SF, Lopes SS, Gossler A, Henrique D. Dll1 and Dll4 function sequentially in the retina and pV2 domain of the spinal cord to regulate neurogenesis and create cell diversity. Developmental biology. 2009;328(1):54–65. Epub 2009/04/25. 10.1016/j.ydbio.2009.01.011 . [DOI] [PubMed] [Google Scholar]
- 10.Yaron O, Farhy C, Marquardt T, Applebury M, Ashery-Padan R. Notch1 functions to suppress cone-photoreceptor fate specification in the developing mouse retina. Development. 2006;133(7):1367–78. Epub 2006/03/03. 10.1242/dev.02311 . [DOI] [PubMed] [Google Scholar]
- 11.Jadhav AP, Mason HA, Cepko CL. Notch 1 inhibits photoreceptor production in the developing mammalian retina. Development. 2006;133(5):913–23. Epub 2006/02/03. dev.02245 [pii] 10.1242/dev.02245 . [DOI] [PubMed] [Google Scholar]
- 12.Riesenberg AN, Liu Z, Kopan R, Brown NL. Rbpj cell autonomous regulation of retinal ganglion cell and cone photoreceptor fates in the mouse retina. J Neurosci. 2009;29(41):12865–77. Epub 2009/10/16. 29/41/12865 [pii] 10.1523/JNEUROSCI.3382-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng MH, Shi M, Pei Z, Gao F, Han H, Ding YQ. The transcription factor RBP-J is essential for retinal cell differentiation and lamination. Molecular brain. 2009;2:38 Epub 2009/12/19. 10.1186/1756-6606-2-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomita K, Ishibashi M, Nakahara K, Ang SL, Nakanishi S, Guillemot F, et al. Mammalian hairy and Enhancer of split homolog 1 regulates differentiation of retinal neurons and is essential for eye morphogenesis. Neuron. 1996;16(4):723–34. Epub 1996/04/01. . [DOI] [PubMed] [Google Scholar]
- 15.Takatsuka K, Hatakeyama J, Bessho Y, Kageyama R. Roles of the bHLH gene Hes1 in retinal morphogenesis. Brain research. 2004;1004(1–2):148–55. Epub 2004/03/23. 10.1016/j.brainres.2004.01.045 . [DOI] [PubMed] [Google Scholar]
- 16.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Molecular aspects of medicine. 2008;29(5):258–89. Epub 2008/09/03. 10.1016/j.mam.2008.08.001 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reiss K, Saftig P. The "a disintegrin and metalloprotease" (ADAM) family of sheddases: physiological and cellular functions. Seminars in cell & developmental biology. 2009;20(2):126–37. Epub 2008/12/04. 10.1016/j.semcdb.2008.11.002 . [DOI] [PubMed] [Google Scholar]
- 18.Rooke J, Pan D, Xu T, Rubin GM. KUZ, a conserved metalloprotease-disintegrin protein with two roles in Drosophila neurogenesis. Science. 1996;273(5279):1227–31. Epub 1996/08/30. . [DOI] [PubMed] [Google Scholar]
- 19.Sotillos S, Roch F, Campuzano S. The metalloprotease-disintegrin Kuzbanian participates in Notch activation during growth and patterning of Drosophila imaginal discs. Development. 1997;124(23):4769–79. Epub 1998/01/15. . [DOI] [PubMed] [Google Scholar]
- 20.Lieber T, Kidd S, Young MW. kuzbanian-mediated cleavage of Drosophila Notch. Genes & development. 2002;16(2):209–21. Epub 2002/01/19. 10.1101/gad.942302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarriault S, Greenwald I. Evidence for functional redundancy between C. elegans ADAM proteins SUP-17/Kuzbanian and ADM-4/TACE. Developmental biology. 2005;287(1):1–10. Epub 2005/10/04. 10.1016/j.ydbio.2005.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Human molecular genetics. 2002;11(21):2615–24. Epub 2002/10/02. . [DOI] [PubMed] [Google Scholar]
- 23.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282(5392):1281–4. Epub 1998/11/13. . [DOI] [PubMed] [Google Scholar]
- 24.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, et al. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. Journal of immunology. 2007;179(5):2686–9. Epub 2007/08/22. . [DOI] [PubMed] [Google Scholar]
- 25.Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, et al. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5(2):207–16. Epub 2000/07/06. . [DOI] [PubMed] [Google Scholar]
- 26.Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, et al. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5(2):197–206. Epub 2000/07/06. S1097-2765(00)80416-5 [pii]. . [DOI] [PubMed] [Google Scholar]
- 27.Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Molecular and cellular biology. 2009;29(21):5679–95. Epub 2009/08/26. MCB.00406-09 [pii] 10.1128/MCB.00406-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R, Vooijs M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. The Journal of biological chemistry. 2009;284(45):31018–27. Epub 2009/09/04. M109.006775 [pii] 10.1074/jbc.M109.006775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30(14):4833–44. Epub 2010/04/08. 10.1523/JNEUROSCI.5221-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber S, Niessen MT, Prox J, Lullmann-Rauch R, Schmitz A, Schwanbeck R, et al. The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development. 2011;138(3):495–505. Epub 2011/01/06. 10.1242/dev.055210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai YH, VanDussen KL, Sawey ET, Wade AW, Kasper C, Rakshit S, et al. ADAM10 regulates Notch function in intestinal stem cells of mice. Gastroenterology. 2014;147(4):822–34 e13 Epub 2014/07/20. 10.1053/j.gastro.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian L, Wu X, Chi C, Han M, Xu T, Zhuang Y. ADAM10 is essential for proteolytic activation of Notch during thymocyte development. International immunology. 2008;20(9):1181–7. Epub 2008/07/19. dxn076 [pii] 10.1093/intimm/dxn076 . [DOI] [PubMed] [Google Scholar]
- 33.Glomski K, Monette S, Manova K, De Strooper B, Saftig P, Blobel CP. Deletion of Adam10 in endothelial cells leads to defects in organ-specific vascular structures. Blood. 2011;118(4):1163–74. Epub 2011/06/10. blood-2011-04-348557 [pii] 10.1182/blood-2011-04-348557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan X, Lin J, Rolfs A, Luo J. Differential expression of the ADAMs in developing chicken retina. Dev Growth Differ. 2011;53(5):726–39. Epub 2011/06/16. 10.1111/j.1440-169X.2011.01282.x . [DOI] [PubMed] [Google Scholar]
- 35.Esteve P, Sandonis A, Cardozo M, Malapeira J, Ibanez C, Crespo I, et al. SFRPs act as negative modulators of ADAM10 to regulate retinal neurogenesis. Nat Neurosci. 2011;14(5):562–9. Epub 2011/04/12. nn.2794 [pii] 10.1038/nn.2794 . [DOI] [PubMed] [Google Scholar]
- 36.Furuta Y, Lagutin O, Hogan BL, Oliver GC. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis. 2000;26(2):130–2. Epub 2000/03/21. 10.1002/(SICI)1526-968X(200002)26:2<130::AID-GENE9>3.0.CO;2-I [pii]. . [DOI] [PubMed] [Google Scholar]
- 37.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100(25):14920–5. Epub 2003/12/06. 10.1073/pnas.2436557100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toonen JA, Liang L, Sidjanin DJ. Waved with open eyelids 2 (woe2) is a novel spontaneous mouse mutation in the Protein Phosphatase 1, Regulatory (inhibitor) Subunit 13 like (Ppp1r13l) gene. BMC genetics. 2012;13(1):76 Epub 2012/08/30. 10.1186/1471-2156-13-76 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fogerty J, Besharse JC. 174delG mutation in mouse MFRP causes photoreceptor degeneration and RPE atrophy. Investigative ophthalmology & visual science. 2011;52(10):7256–66. Epub 2011/08/04. 10.1167/iovs.11-8112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hassemer EL, Le Gall SM, Liegel R, McNally M, Chang B, Zeiss CJ, et al. The waved with open eyelids (woe) locus is a hypomorphic mouse mutation in Adam17. Genetics. 2010;185(1):245–55. Epub 2010/03/03. 10.1534/genetics.109.113167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishida A, Furukawa A, Koike C, Tano Y, Aizawa S, Matsuo I, et al. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat Neurosci. 2003;6(12):1255–63. Epub 2003/11/20. . [DOI] [PubMed] [Google Scholar]
- 42.Pan L, Deng M, Xie X, Gan L. ISL1 and BRN3B co-regulate the differentiation of murine retinal ganglion cells. Development. 2008;135(11):1981–90. Epub 2008/04/25. dev.010751 [pii] 10.1242/dev.010751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malicki J, Jo H, Pujic Z. Zebrafish N-cadherin, encoded by the glass onion locus, plays an essential role in retinal patterning. Developmental biology. 2003;259(1):95–108. Epub 2003/06/19. . [DOI] [PubMed] [Google Scholar]
- 44.Fu X, Sun H, Klein WH, Mu X. Beta-catenin is essential for lamination but not neurogenesis in mouse retinal development. Developmental biology. 2006;299(2):424–37. Epub 2006/09/09. 10.1016/j.ydbio.2006.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maurer KA, Riesenberg AN, Brown NL. Notch signaling differentially regulates Atoh7 and Neurog2 in the distal mouse retina. Development. 2014;141(16):3243–54. Epub 2014/08/08. 10.1242/dev.106245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Groot AJ, Habets R, Yahyanejad S, Hodin CM, Reiss K, Saftig P, et al. Regulated proteolysis of NOTCH2 and NOTCH3 receptors by ADAM10 and presenilins. Molecular and cellular biology. 2014;34(15):2822–32. Epub 2014/05/21. 10.1128/MCB.00206-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindsell CE, Boulter J, diSibio G, Gossler A, Weinmaster G. Expression patterns of Jagged, Delta1, Notch1, Notch2, and Notch3 genes identify ligand-receptor pairs that may function in neural development. Mol Cell Neurosci. 1996;8(1):14–27. Epub 1996/01/01. . [DOI] [PubMed] [Google Scholar]
- 48.Galli-Resta L, Leone P, Bottari D, Ensini M, Rigosi E, Novelli E. The genesis of retinal architecture: an emerging role for mechanical interactions? Prog Retin Eye Res. 2008;27(3):260–83. Epub 2008/04/01. 10.1016/j.preteyeres.2008.02.001 . [DOI] [PubMed] [Google Scholar]
- 49.Reese BE. Development of the retina and optic pathway. Vision research. 2011;51(7):613–32. Epub 2010/07/22. 10.1016/j.visres.2010.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. Epub 2004/10/12. 10.1146/annurev.cellbio.20.010403.113126 . [DOI] [PubMed] [Google Scholar]
- 51.Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, et al. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. The EMBO journal. 2005;24(4):742–52. Epub 2005/02/05. 10.1038/sj.emboj.7600548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, et al. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci U S A. 2005;102(26):9182–7. Epub 2005/06/17. 10.1073/pnas.0500918102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masai I, Lele Z, Yamaguchi M, Komori A, Nakata A, Nishiwaki Y, et al. N-cadherin mediates retinal lamination, maintenance of forebrain compartments and patterning of retinal neurites. Development. 2003;130(11):2479–94. Epub 2003/04/19. . [DOI] [PubMed] [Google Scholar]
- 54.Erdmann B, Kirsch FP, Rathjen FG, More MI. N-cadherin is essential for retinal lamination in the zebrafish. Developmental dynamics: an official publication of the American Association of Anatomists. 2003;226(3):570–7. Epub 2003/03/06. 10.1002/dvdy.10266 . [DOI] [PubMed] [Google Scholar]
- 55.Sel S, Kalinski T, Enssen I, Kaiser M, Nass N, Trau S, et al. Expression analysis of ADAM17 during mouse eye development. Annals of anatomy = Anatomischer Anzeiger: official organ of the Anatomische Gesellschaft. 2012;194(4):334–8. Epub 2011/11/08. 10.1016/j.aanat.2011.10.008 . [DOI] [PubMed] [Google Scholar]
- 56.Guaiquil V, Swendeman S, Yoshida T, Chavala S, Campochiaro PA, Blobel CP. ADAM9 is involved in pathological retinal neovascularization. Molecular and cellular biology. 2009;29(10):2694–703. Epub 2009/03/11. 10.1128/MCB.01460-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parry DA, Toomes C, Bida L, Danciger M, Towns KV, McKibbin M, et al. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am J Hum Genet. 2009;84(5):683–91. Epub 2009/05/05. 10.1016/j.ajhg.2009.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(TIF)
(TIF)
(TIF)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.