

Abstract
Isomorphous crystals of MnV(O) and CrV(O) corrolazines were characterized by single crystal X-ray diffraction. Reactivity studies with H-atom donors and separated PCET reagents show a dramatic difference in H-atom abstracting abilities for these two complexes. The implied large difference in driving force is opposite to the trend in redox potentials, indicating that basicity is a key factor in determining the striking difference in reactivity for two metal-oxo species in identical ligand environments.
TOC image
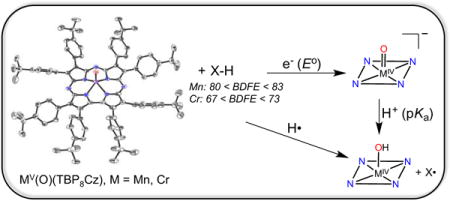
The ability of high-valent metal-oxo complexes to abstract hydrogen atoms from organic compounds is of critical importance to the functioning of metal-based oxidation catalysts. Included among these catalysts are enzymatic systems that utilize both heme and nonheme metal active sites.1,2 How the metal ion, the coordinating ligands, and surrounding protein matrix in the case of biological catalysts, control the reactivity of metal-oxo intermediates in H-atom abstraction is a question of fundamental importance. In heme enzymes, the Cytochrome P450s are among the most powerful H-atom abstractors, utilizing Compound I ((FeIV(O)(porph•+)(cys)) for strong C-H cleavage.1 The large driving force presented for H-atom abstraction by P450 can be related to the bond dissociation free energy (BDFE) of the O-H bond of Compound II (FeIV(OH)(porph)(cys)), formed after H-atom transfer (HAT). The O-H BDFE can be further dissected into electron (E°) and proton (pKa) affinities (or basicity), and evidence indicates that it is the elevated basicity of the FeIV=O unit in Cpd-II (pKa ~ 12) that provides an advantage in driving force for HAT.1c,e
Attempts to synthesize biomimetic high-valent metal-oxo species and examine their propensity for HAT has led to parallel insights regarding the thermodynamic control of these reactions.3,4 These studies have helped support the analysis of the biological systems, and supplied information for the design of synthetic oxidation catalysts. However, much remains to be learned regarding how the metal ion and ligand(s) of M(O)(Ln) complexes tune M(O-H) BDFEs, redox potentials, pKas, and ultimately HAT reactivity.
In this report we compare the H-atom abstraction abilities of MnV(O) and CrV(O) porphyrinoid complexes. Both of these complexes are characterized by single crystal X-ray diffraction (XRD). To our knowledge, the manganese complex is the first example of a structurally characterized MnV(O) complex in a heme-type environment. The Cr and Mn complexes are isomorphous, providing a unique opportunity to determine the inherent HAT reactivity of MnV(O) versus CrV(O) moieties. Although CrV(O) complexes, including porphyrins, are known, little information is available regarding their H-atom abstraction abilities.5 In this report we show that CrV(O) is a better 1-e− oxidant than MnV(O), but is a much weaker H-atom abstractor.
The synthesis of MnV(O)(TBP8Cz) (TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato3−) was carried out in a manner similar to that previously reported.4a Addition of freshly prepared PhIO (10 equiv) to MnIII(TBP8Cz) (1) in CH2Cl2 gives the oxidized MnV(O)(TBP8Cz) (2), which can be purified by silica gel chromatography. Dissolution of 2 in toluene to give a dark green solution followed by slow vapor diffusion of CH3CN leads to the growth of dark green needles after a few days. These crystals were suitable for X-ray structure determination, and the structure of 2 is shown in Figure 1. Previous attempts at growing crystals of 2 in other solvent combinations were plagued by decomposition of the complex. However, we observed that 2 was stable in toluene/CH3CN for at least two weeks, and favored slow crystal growth. This crystallization method was also highly reproducible. As depicted in Figure 1, the MnV ion is 5-coordinate, with Mn–Npyrrole distances between 1.873(2)–1.8974(19) Å, and a short Mn–O distance of 1.5455(18) Å consistent with an Mn≡O triple bond. These distances are in agreement with those determined previously by EXAFS (d(Mn–O) = 1.56 Å, d(Mn–Npyrrole) = 1.88 Å).4a Structurally characterized non-heme complexes have Mn–O bond distances of 1.548(4) Å – 1.558(4) Å, which are comparable with that of 2.6 The manganese ion in 2 is significantly displaced by ca. 0.59 Å from the plane of the four pyrrole N atoms toward the terminal oxo ligand. For the isoelectronic MnV(NMes)(TBP8Cz), the terminal mesitylimido Mn–N distance is slightly longer at 1.595(4) – 1.611(4) Å, while the Mn ion is less displaced out of the Npyrrole plane (Mn – N4(plane) = 0.55 Å).4c The structure unequivocally shows that the MnV(O) complex is 5-coordinate, as opposed to the proposed structures for related MnV(O) porphyrins.7
Figure 1.
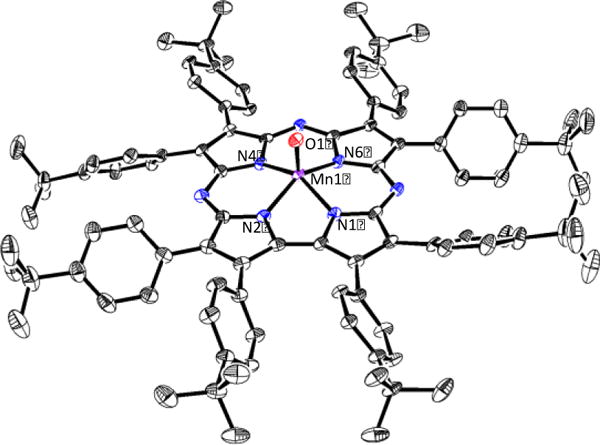
Displacement ellipsoid plot (50% probability level) of MnV(O)(TBP8Cz) (2) at 110(2) K. H-atoms and disorder are omitted for clarity.
The analogous CrV(O)(TBP8Cz) (3) was synthesized by aerobic, oxidative metallation of the metal-free corrolazine, TBP8CzH3, with Cr(CO)6 in refluxing toluene.5d No significant color change from the deep green of the metal-free starting material (λmax = 456, 679 nm) was noted, but monitoring the reaction by UV-vis revealed distinct shifts in both the Soret and Q-band regions (3: λmax = 448, 653 nm). Complex 3 was purified by flash chromatography (eluent: 60:40, CH2Cl2:hexanes), and recrystallized from vapor diffusion of acetonitrile into a toluene solution of 3 over 1 week. X-ray structure determination was carried out and its structure, which is isomorphous with the MnV(O) complex, is shown in Figure S1. The CrV ion is 5-coordinate as seen for Mn, and Cr–O = 1.553(2) Å. This distance is similar to that seen for CrV(O) corroles (~1.57 Å).5d,8 The out of plane displacement of the chromium in 3 (Cr–N4(plane) = 0.61 Å) is slightly larger than that seen for the corrole analogs (0.56–0.58), probably arising from the smaller cavity size of corrolazine (trans Npyrrole–Npyrrole: 3.61 Å) versus corrole (trans Npyrrole–Npyrrole: 3.67–3.69 Å). The metal-oxo distance for 3 is identical to that of 2, but the M–Npyrrole distances are slightly longer for 3 versus 2 (see Table S1). The out-of-plane distance for 3 is also slightly larger than 2. These observations are consistent with the larger ionic radius of the CrV versus MnV ion.9
The EPR spectrum (9.44 GHz, 294 K) of 3 is shown in Figure S3. A 9 line signal centered at g = 1.987 is observed, consistent with a CrV (d1, S = ½) ion with hyperfine coupling to four equivalent pyrrole nitrogen atoms (14N, I = 1). The satellite signals at high and low fields are due to hyperfine splitting from 53Cr (9.5% abundant, I = 3/2). Evans method NMR measurement gave a magnetic moment of μeff = 1.36 μB, which is close to the predicted spin-only value of 1.73 μB for an S = ½ ion. These data confirm the +5 oxidation state of the Cr ion, and rule out the involvement of other potential ground state electronic configurations, such as CrIV(O)(TBP8Cz•+).4g,h
Prior to examining the reactivity of the CrV(O) complex 3, we synthesized the reduced chromium(III) analog to obtain a spectroscopic benchmark for this species. Addition of triphenylphosphine, an oxygen atom acceptor, to 3 in toluene, followed by slow vapor diffusion of CH3CN over the course of 2 weeks led to X-ray quality crystals of CrIII(TBP8Cz)(CH3CN)2 (4). The crystal structure of 4 is shown in Figure S5. Complex 4 is 6-coordinate, with the CrIII ion bound by 2 axial CH3CN molecules. Unlike in 3, the Cr ion is displaced from the N4(plane) by only 0.013 Å to accommodate the sixth ligand. The UV-vis spectrum of 4 in CH2Cl2 gives Soret and Q-bands at 465 and 709 nm, respectively, which are easily distinguished from those observed for the CrV(O) complex. The low-temperature EPR spectrum of 4 (9.44 GHz, 12 K) is consistent with the S =3/2 ground state expected for the CrIII (d3) ion (Figure S7).8a
The MnV(O) and CrV(O) complexes are ideal candidates for examining the relative reactivity of high-valent, biomimetic metal-oxo complexes bound in identical ligand environments. Previously, we showed that 2 reacts with a range of H-atom donors, including both substituted phenols (O–H bonds) and hydrocarbon (C–H) substrates, which exhibited bond dissociation energies (BDEs) from 66 – 80 kcal/mol.4b,d We examined complex 3 for its potential reactivity toward substrates with similar bond strengths. A rapid reaction between 3 and excess TEMPOH in CH2Cl2 was observed by UV-vis spectroscopy, resulting in the isosbestic conversion of 3 (448, 653 nm) to a CrIII product with a spectrum similar to 4 in the presence of excess TEMPOH (Figure 2a, Figure S10). The corresponding yield of TEMPO• was 82% (EPR quantitation) or 1.62 equiv relative to 3. These data indicate the stoichiometry for this reaction follows that shown in Scheme 1, in which two equiv of TEMPOH react with 3 to give one equiv of reduced CrIII product and two equiv of TEMPO• product. No CrIV intermediates were observed. This reaction exhibited pseudo-first-order behavior over 5 half-lives, and a plot of kobs (s−1) values correlated linearly with [TEMPOH] to give a second-order rate constant of k2 = 16 ± 1 M−1 s−1 (Figure 2b). A kinetic isotope effect of kH/kD = 5.2 ± 0.6 was measured for TEMPOH/D (Figure S9). These observations are consistent with a concerted H-atom transfer (HAT) mechanism for the reaction of 2 with TEMPOH.
Figure 2.
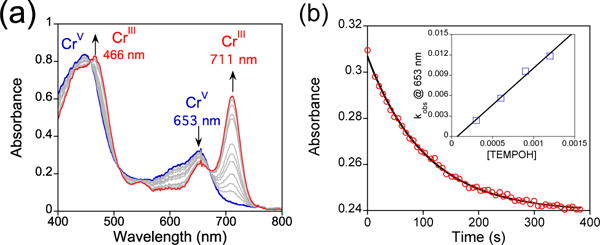
(a) UV-vis spectral changes (0 – 3 min) for the reaction of 3 (12 μM) with TEMPOH (150 equiv) at 25 °C. (b) Change in absorbance at 653 nm versus time corresponding to the decay of 3 (red circles) and best fit (black line). Inset: second-order rate plot.
Scheme 1.
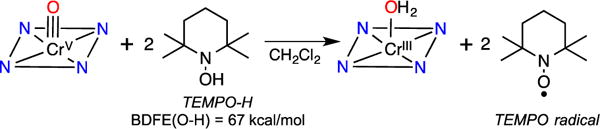
As seen in Table 1, complex 3 was only capable of oxidizing TEMPOH, with a weak O–H bond of 67 kcal/mol. It was unreactive toward other H-atom donors, even under higher temperatures and prolonged reaction times. For example, a mixture of 3 and excess xanthene (BDFE = 73.3 kcal/mol) in toluene at 70 °C for 40 h gives back only starting material. In contrast, the MnV(O) complex 2 reacts with substrates that have BDFEs up to 80 kcal/mol.
Table 1.
Reaction of 2 (MnV(O)) or 3 (CrV(O)) with H-atom donors (C-H and O-H) with a range of BDFEs
| Substrate | MnV(O) | CrV(O) | BDFE3a |
|---|---|---|---|
| HMBa | no | No | 83 |
| 2,4,6-TTBPb | yes | no | 80 |
| DHAc | yes | no | 77 |
| xanthene | yes | no | 73 |
| TEMPOH | yes | yes | 67 |
HMB = hexamethylbenzene.
2,4,6-TTBP = 2,4,6-tri-tert-butylphenol.
DHA = 9,10-dihydroanthracene.
A thermodynamic analysis of H-atom abstraction for 2 and 3 can provide insight into the differences in reactivity seen for these two complexes. H-atom abstraction by the metal-oxo complexes can be described as shown in Scheme 2, where HAT follows either the concerted (diagonal) or step-wise electron-transfer (ET, horizontal) and proton-transfer (PT, vertical) steps shown in the square scheme. The thermodynamic parameters (E°, pKa) associated with the ET and PT steps combined with the free energy of formation of the hydrogen atom (CG), can be used to calculate the bond dissociation free energy (BDFE) for M(O-H) (Eq 1).10a The difference in BDFE (ΔBDFE) for MnIV(OH) versus CrIV(OH) is expressed in Eq 2 and relies only on the differences in E° and pKa, eliminating the requirement for an accurate measure of CG. Assuming the reaction is under thermodynamic control, the BDFE of the M(O-H) bond must be similar to or greater than the X–H bond being cleaved in the substrate. The correlation of HAT reactivity with BDFE has been observed for metal-oxo complexes.3 The results in Table 1 indicate that the BDFE for MnIV(OH) should be between 80 – 83 kcal/mol, whereas for CrIV(OH) the BDFE is 67 – 73 kcal/mol. These data imply a ΔBDFE of at least 8 kcal/mol for these two complexes.11
| (1) |
| (2) |
Scheme 2.
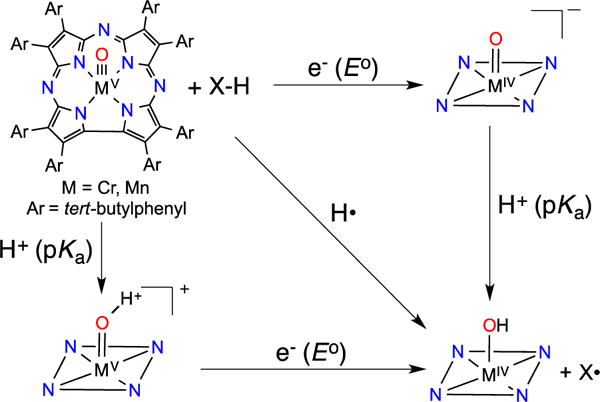
The MnV(O) and CrV(O) complexes were also capable of reacting with separated electron-transfer/proton-transfer reagents through a proton-coupled electron-transfer (PCET) mechanism. It has been shown that an effective ‘BDFE’ for separate reductant/acid pairs can be calculated from their individual E° and pKa values.10b,c Complex 2 reacts with the reductant dimethylferrocene (Me2Fc) (E1/2 = −0.24 V versus Fc+/Fc in CH3CN) in the presence of the H+ donor acetic acid (pKa = 23.5), to give MnIII(TBP8Cz)(OH2) in CH2Cl2 as shown by UV-vis (Figure S13). However, no reaction occurs with either Me2Fc or CH3CO2H alone, supporting a PCET process.4e The BDFE for the Me2Fc/CH3CO2H pair is 81.6 kcal/mol in CH3CN (CG = 54.9 kcal/mol). However, replacement of the Me2Fc reductant with unsubstituted Fc (E1/2 = 0.00 V) leads to no reaction. An effective BDFE = 87.1 kcal/mol is calculated for Fc/CH3CO2H. Similar experiments with the CrV(O) complex showed efficient PCET from Me2Fc and trifluoroacetic acid (TFA) (pKa = 12.6) to give CrIII(TBP8Cz), but no reaction was observed for Fc/TFA. These reductant/acid pairs have effective BDFEs of 66.5 and 75.5 kcal/mol, respectively. The results obtained for the separated PCET reagents provide good support for the BDFE range predicted for both 2 and 3 from the reactivity pattern with the H-atom donors in Table 1.
Insight into the origin of the large difference in BDFE and related reactivity for 2 and 3, comes from cyclic voltammetry (Figure 3). The quasi-reversible wave at E1/2 = −0.43 V is assigned to the CrV/CrIV redox potential based on previous assignments for metal-locorrolazines, including 2.4a Interestingly, the CrV/CrIV potential is ~100 mV more positive than that seen for MnV/MnIV (E1/2(2) = −0.55 V). According to eq 1, the larger redox potential for 3 should provide a 2.3 kcal/mol (23.06 × (0.1 V)) increase in BDFE compared to 2. However, the BDFE for CrIV(OH) appears to be weaker than the BDFE for MnIV(OH) by at least 8 kcal/mol based on the observed HAT and PCET reactivity. Assuming a ΔBDFE of 8 kcal/mol, and including the measured ΔE° = −0.1 V, we find that ΔpKa must be ~8 according to eq 2. This result indicates that a reduced [MnIV(O)−] species is at least 8 orders of magnitude more basic than the corresponding [CrIV(O)−], and this basicity dominates the difference in driving force for H-atom abstraction. We previously have suggested that the basicity of [MnIV(O)−] was a potential key factor in HAT,4b and more recent studies have supported this conclusion.12 However, this study provides a rare direct comparison of two high-valent metal-oxo species in identical ligand environments, and demonstrates that a dramatic difference in reactivity can be assigned to the different basicities of the metal-oxo units.
Figure 3.
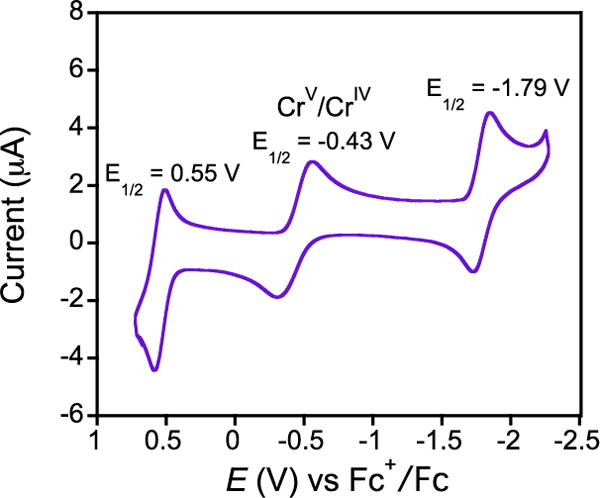
CV of 3 in CH2Cl2 with 0.1 M TBAPF6 as supporting electrolyte, scan rate 25 mV/s.
In summary, we report a new CrV(O) porphyrinoid complex, and the first X-ray structure of an MnV(O) porphyrinoid complex. A comparison of HAT/PCET reactivity for these two adjacent first-row metal-oxo complexes (Mn, Cr) in identical ligand environments implies that MnV(O) must have a much larger driving force (~8 kcal/mol) for H• abstraction than the corresponding CrV(O), despite the latter complex having a 100 mV larger E1/2 value. The larger driving force can be attributed to the basicity of the one-electron reduced [MnIV(O)−], which we estimate to be ~8 orders of magnitude more basic than [CrIV(O)−]. This work supports the hypothesis that the basicity of high-valent metal-oxo species in heme enzymes is a critical factor in tuning reactivity.
Supplementary Material
Acknowledgments
We thank the NIH (GM101153 to D.P.G.) for financial support. R.A.B. is grateful for the E2SHI, George E. Owen, and Harry and Cleio Greer Fellowships.
Footnotes
Supporting Information. Experimental procedures, kinetic studies, EPR, CV, Figures S1–S14, Table S1, and CIF files. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.(a) Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]; (b) Rittle J, Green MT. Science. 2010;330:933. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]; (c) Yosca TH, Rittle J, Krest CM, Onderko EL, Silakov A, Calixto JC, Behan RK, Green MT. Science. 2013;342:825. doi: 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Poulos TL. Chem Rev. 2014;114:3919. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yosca TH, Behan RK, Krest CM, Onderko EL, Langston MC, Green MT. J Am Chem Soc. 2014;136:9124. doi: 10.1021/ja503588n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM. Acc Chem Res. 2007;40:484. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bruijnincx PC, van Koten G, Gebbink RJK. Chem Soc Rev. 2008;37:2716. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]
- 3.(a) Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Borovik AS. Chem Soc Rev. 2011;40:1870. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bigi JP, Harman WH, Lassalle-Kaiser B, Robles DM, Stich TA, Yano J, Britt RD, Chang CJ. J Am Chem Soc. 2012;134:1536. doi: 10.1021/ja207048h. [DOI] [PubMed] [Google Scholar]; (d) Usharani D, Lacy DC, Borovik AS, Shaik S. J Am Chem Soc. 2013;135:17090. doi: 10.1021/ja408073m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nam W, Lee Y-M, Fukuzumi S. Acc Chem Res. 2014;47:1146. doi: 10.1021/ar400258p. [DOI] [PubMed] [Google Scholar]; (f) Chen Z, Yin G. Chem Soc Rev. 2015;44:1083. doi: 10.1039/c4cs00244j. [DOI] [PubMed] [Google Scholar]; (g) Puri M, Que L. Acc Chem Res. 2015 doi: 10.1021/acs.accounts.5b00244. Article ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Lansky DE, Mandimutsira B, Ramdhanie B, Clausén M, Penner-Hahn J, Zvyagin SA, Telser J, Krzystek J, Zhan R, Ou Z, Kadish KM, Zakharov L, Rheingold AL, Goldberg DP. Inorg Chem. 2005;44:4485. doi: 10.1021/ic0503636. [DOI] [PubMed] [Google Scholar]; (b) Lansky DE, Goldberg DP. Inorg Chem. 2006;45:5119. doi: 10.1021/ic060491+. [DOI] [PubMed] [Google Scholar]; (c) Lansky DE, Kosack JR, Narducci Sarjeant AA, Goldberg DP. Inorg Chem. 2006;45:8477. doi: 10.1021/ic0609251. [DOI] [PubMed] [Google Scholar]; (d) Prokop KA, de Visser SP, Goldberg DP. Angew Chem, Int Ed. 2010;49:5091. doi: 10.1002/anie.201001172. [DOI] [PubMed] [Google Scholar]; (e) Fukuzumi S, Kotani H, Prokop KA, Goldberg DP. J Am Chem Soc. 2011;133:1859. doi: 10.1021/ja108395g. [DOI] [PubMed] [Google Scholar]; (f) Cho K, Leeladee P, McGown AJ, DeBeer S, Goldberg DP. J Am Chem Soc. 2012;134:7392. doi: 10.1021/ja3018658. [DOI] [PubMed] [Google Scholar]; (g) Leeladee P, Baglia RA, Prokop KA, Latifi R, de Visser SP, Goldberg DP. J Am Chem Soc. 2012;134:10397. doi: 10.1021/ja304609n. [DOI] [PubMed] [Google Scholar]; (h) Baglia RA, Dürr M, Ivanović-Burmazović I, Goldberg DP. Inorg Chem. 2014;53:5893. doi: 10.1021/ic500901y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Groves JT, Haushalter RC. J Chem Soc, Chem Commun. 1981:1165. [Google Scholar]; (b) Garrison JM, Bruice TC. J Am Chem Soc. 1989;111:191. [Google Scholar]; (c) Collins TJ, Slebodnick C, Uffelman ES. Inorg Chem. 1990;29:3433. [Google Scholar]; (d) Meier-Callahan AE, Gray HB, Gross Z. Inorg Chem. 2000;39:3605. doi: 10.1021/ic000180d. [DOI] [PubMed] [Google Scholar]; (e) Bakac A, Guzei IA. Inorg Chem. 2000;39:736. doi: 10.1021/ic991098p. [DOI] [PubMed] [Google Scholar]; (f) Hess JS, Leelasubcharoen S, Rheingold AL, Doren DJ, Theopold KH. J Am Chem Soc. 2002;124:2454. doi: 10.1021/ja0167553. [DOI] [PubMed] [Google Scholar]; (g) O’Reilly M, Falkowski JM, Ramachandran V, Pati M, Abboud KA, Dalal NS, Gray TG, Veige AS. Inorg Chem. 2009;48:10901. doi: 10.1021/ic9019469. [DOI] [PubMed] [Google Scholar]; (h) Cho J, Woo J, Eun Han J, Kubo M, Ogura T, Nam W. Chem Sci. 2011;2:2057. [Google Scholar]; (i) Kotani H, Kaida S, Ishizuka T, Sakaguchi M, Ogura T, Shiota Y, Yoshizawa K, Kojima T. Chem Sci. 2015;6:945. doi: 10.1039/c4sc02285h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Collins TJ, Gordon-Wylie SW. J Am Chem Soc. 1989;111:4511. [Google Scholar]; (b) MacDonnell FM, Fackler NLP, Stern C, O’Halloran TV. J Am Chem Soc. 1994;116:7431. [Google Scholar]
- 7.(a) Groves JT, Lee J, Marla SS. J Am Chem Soc. 1997;119:6269. [Google Scholar]; (b) Arunkumar C, Lee Y-M, Lee JY, Fukuzumi S, Nam W. Chem - Eur J. 2009;15:11482. doi: 10.1002/chem.200901362. [DOI] [PubMed] [Google Scholar]
- 8.(a) Meier-Callahan AE, Di Bilio AJ, Simkhovich L, Mahammed A, Goldberg I, Gray HB, Gross Z. Inorg Chem. 2001;40:6788. doi: 10.1021/ic010723z. [DOI] [PubMed] [Google Scholar]; (b) Egorova OA, Tsay OG, Khatua S, Huh JO, Churchill DG. Inorg Chem. 2009;48:4634. doi: 10.1021/ic900393v. [DOI] [PubMed] [Google Scholar]; (c) Egorova OA, Tsay OG, Khatua S, Meka B, Maiti N, Kim M-K, Kwon SJ, Huh JO, Bucella D, Kang S-O, Kwak J, Churchill DG. Inorg Chem. 2010;49:502. doi: 10.1021/ic9021432. [DOI] [PubMed] [Google Scholar]
- 9.Shannon RD. Acta Crystallogr, Sect A. 1976;32:751. [Google Scholar]
- 10.(a) Bordwell FG, Cheng JP, Harrelson JA. J Am Chem Soc. 1988;110:1229. [Google Scholar]; (b) Waidmann CR, Miller AJM, Ng C-WA, Scheuermann ML, Porter TR, Tronic TA, Mayer JM. Energy Environ Sci. 2012;5:7771. [Google Scholar]; (c) Tarantino KT, Liu P, Knowles RR. J Am Chem Soc. 2013;135:10022. doi: 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]
- 11.This ΔBDFE assumes that the BDFE of CrIV(OH) is below that of the xanthene, ie 72 kcal/mol or less, whereas the BDFE of MnIV(OH) is assumed to be ≥ 80 kcal/mol.
- 12.Green MT. Curr Opin Chem Biol. 2009;13:84. doi: 10.1016/j.cbpa.2009.02.028. and references therein. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.