

Abstract
Iron is an essential transition metal ion for virtually all aerobic organisms, yet its dysregulation (iron overload or anemia) is a harbinger of many pathologic conditions. Hence, iron homeostasis is tightly regulated to prevent the generation of catalytic iron (CI) which can damage cellular biomolecules. In this study, we investigated the role of iron-binding/trafficking innate immune protein, lipocalin 2 (Lcn2, aka siderocalin) on iron and CI homeostasis using Lcn2 knockout (KO) mice and their WT littermates. Administration of iron either systemically or via dietary intake strikingly upregulated Lcn2 in the serum, urine, feces, and liver of WT mice. However, similarly-treated Lcn2KO mice displayed elevated CI, augmented lipid peroxidation and other indices of organ damage markers, implicating that Lcn2 responses may be protective against iron-induced toxicity. Herein, we also show a negative association between serum Lcn2 and CI in the murine model of dextran sodium sulfate (DSS)-induced colitis. The inability of DSS-treated Lcn2KO mice to elicit hypoferremic response to acute colitis, implicate the involvement of Lcn2 in iron homeostasis during inflammation. Using bone marrow chimeras, we further show that Lcn2 derived from both immune and non-immune cells participate in CI regulation. Remarkably, exogenous rec-Lcn2 supplementation suppressed CI levels in Lcn2KO serum and urine. Collectively, our results suggest that Lcn2 may facilitate hypoferremia, suppress CI generation and prevent iron-mediated adverse effects.
Keywords: Lipocalin 2, Iron, Catalytic iron, Inflammation, Anemia of inflammation, Oxidative stress
Introduction
Iron is an essential micronutrient required by almost all aerobic organisms except Lactobacillus and Borrelia species (Weinberg 1997; Posey et al. 2000). As a component of hemoglobin it plays critical roles in oxygen transportation, energy metabolism, and cellular proliferation. The biological properties of iron result from its flexibility to transition from reduced ferrous (Fe+2) and oxidized (Fe+3) forms, thus this chemical property results in iron’s catalytic role in a multitude of redox reactions (Wang et al. 2011). It has been estimated that of the 3–5g of total iron in the human body, 70–90mg is in the form of catalytic iron [(CI) (aka: reactive or labile)](Kakhlon et al. 2002). Further, reactions catalyzed by Cytochrome P450 and myeloperoxidase are potential endogenous sources of CI (Huang et al. 2002; Maitra et al. 2013). CI is an essential mediator of damage to almost all cellular macromolecules via generation of reactive oxygen species (ROS) which occurs via Fenton’s reaction and leads to multiple organ dysfunctions (Hentze et al. 2004; Koskenkorva-Frank et al. 2013; Cabantchik 2014). CI levels are substantially elevated in a number of metabolic diseases including obesity (Sulieman et al. 2004; Boddaert et al. 2007; Mitchell et al. 2007; Lele et al. 2009; Sullivan 2009; Thethi et al. 2011), which is believed to be a consequence of low-grade chronic inflammation. Further, CI plays a critical role in inducing pro-inflammatory cytokines (Darshan et al. 2010), and one of the pathways is through activation of NLRP3 inflammasome (Nakamura et al. 2015). In addition, CI is also known to induce cell death via ‘ferroptosis’ (Dixon et al. 2012). Therefore, regulation of iron homeostasis occurs at the levels in absorption, transportation and storage (Ratledge 2007; Shi et al. 2008; Weiss 2009; Martines et al. 2013). The hepatic hormone hepcidin plays a major role in iron homeostasis by regulating iron absorption by ferroportin expressed basolaterally (serosal) in intestinal epithelia. Hepcidin levels are regulated by both iron overload and inflammation, which controls the iron levels in the circulation and tissues by inducing the degradation of ferroportin (Ganz 2003).
Lipocalin 2 (Lcn2; human ortholog neutrophil gelatinase-associated lipocalin [NGAL]) is a multifaceted innate immune protein expressed by both immune and non-immune cells. Recently, the roles for Lcn2 in iron homeostasis and cellular iron transport has emerged (Yang et al. 2002; Bao et al. 2010; Devireddy et al. 2010). Intriguingly, Lcn2 alone cannot bind Fe+3 but it can chelate siderophore-bound Fe+3 (e.g. bacterial siderophore enterobactin), which exerts a direct antibacterial activity (Goetz et al. 2002; Flo et al. 2004). Several in vitro studies have demonstrated that constitutive Lcn2 protects against cellular stress and exposure to H2O2 and over-expression allows cells to tolerate superphysiological iron concentrations (Roudkenar et al. 2008; Hu et al. 2009; Roudkenar et al. 2011). In comparison, Lcn2 knockout (Lcn2KO) macrophages exhibit elevated intracellular iron levels (Nairz et al. 2009). Our previous study demonstrated that Lcn2KO mice display a delayed LPS-induced hypoferremic response and Lcn2-deficient immune cells are sensitive to LPS-induced apoptosis (Srinivasan et al. 2012). We hypothesize that Lcn2 is not only involved in iron transport, but also plays a critical role in inducing hypoferremia of inflammation.
In this study, we tested the hypothesis that Lcn2 protects against iron-induced toxicity. We observed that both intraperitoneal iron administration and dietary iron intake dramatically induced Lcn2 in WT mice. WT mice were able to maintain iron homeostasis under these conditions, whereas Lcn2-deficient (Lcn2KO) mice exhibited elevated iron and CI levels, serum aspartate transaminase and liver oxidative stress. By using a well-established murine model of dextran sulfate sodium (DSS)-induced acute colitis, we further demonstrated that Lcn2 play important role in modulating iron and CI homeostasis during acute inflammation, specifically by facilitating the hypoferremic response. More importantly, we showed that exogenous Lcn2 treatment could attenuate the high CI levels in serum and urine Lcn2KO mice, implicating that Lcn2 also participate in reducing CI generation. Collectively, our results suggest that the elevated Lcn2 expression could be a host protective response to dysregulated iron homeostasis, perhaps by reducing CI generation and protect against oxidative stress caused by ROS. Accordingly, Lcn2 can be a promising therapeutic target to treat iron-induced toxicity and other CI-associated complications.
Materials and methods
Materials
Duoset mouse Lcn2 and CXCL1 (KC) ELISA kit, and anti-mouse Lcn2 antibody were obtained from R&D Systems. Aspartate transaminase (AST), alanine transaminase (ALT) triglycerides (TG), cholesterol (CHOL), lactate dehydrogenase (LDH) and creatinine kits were procured from Randox Laboratories. Glucometer and glucose test strips were obtained from Nova Max. Iron atomic absorption (AA) standard was purchased from RICCA Chemical Company. Ferrous sulfate was obtained from Alfa Aesar. SYBR® Green mix and qScript cDNA synthesis kit was procured from Quanta Biosciences. Bleomycin sulfate was procured from Biotang. Reagent grade Dextran Sulfate Sodium salt (DSS, reagent grade, M.W. 36–50 kDa, Ref = 160110) was purchased from MP Biomedicals. Mouse recombinant (rec)-Lcn2 was obtained from Cell Signaling and human rec-NGAL was acquired from R&D Systems; both of which are free from endotoxin, siderophore, and iron. All other fine chemicals used in present study were reagent grade and procured from Sigma.
Mice
Lcn2KO mice on C57BL/6 background generated by Dr. Shizuo Akira (Japan) were obtained via Dr. Alan Aderem (University of Washington) and bred with C57BL/6 wild type (WT) mice. The resulting offsprings were crossed to generate homozygous Lcn2KO mice and their WT littermates. These mice bred in-house in the animal facility at The Pennsylvania State University. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at The Pennsylvania State University.
Bone marrow chimera generation
Eight weeks old Lcn2KO and their WT littermates were exposed to whole body radiation (850 rads) using Shephered Cesium Irradiator and then stabilize them for 2h. Femurs and tibias from four weeks old female WT and Lcn2KO donor mice were flushed with complete RPMI 1640 media. Subsequently, the bone marrow (BM) cells were suspended in the RBC lysis buffer and incubate for 5 min to lyse erythrocytes. After incubation, cells were immediately pelleted down and re-suspended in complete RPMI 1640 media. Before injection the cells were washed and suspended in sterile HBSS. After stabilization period BM cells (107cells/mouse) in HBSS were administered to irradiated-mice via retro-orbital injection to generate WT→Lcn2KO and Lcn2KO→WT bone marrow chimera (BMC). We also generated WT→WT and Lcn2KO→Lcn2KO BMC as controls. Subsequently, mice were immediately given neomycin (2 mg/ml) contained drinking water for initial 2 weeks and then allowed 6 more weeks for reconstitution. Our bone marrow chimera model generates 95–99% chimerism as determined by the differential expression of the CD45.1 and CD45.2 alleles in PBMC of recipient mice by flow cytometry (Sanders et al. 2008; Carvalho et al. 2011).
Bone marrow-derived macrophages (BMDMs) generation and treatment
Bone marrow cells obtained as above were used to generate BMDM as described previously (Zhang et al. 2008). In brief, cells were cultured in 90 mm culture dish in presence of macrophage colony–stimulating factor (M-CSF; 100U/ml) (Stanley 1985) in 37 °C, 5% CO2. 60% of media was replaced on every third day with fresh complete medium containing M-CSF. After 7 days in culture, contaminating non-adherent cells were eliminated and adherent cells were harvested for assays when F4/80-positivity was found to be more than 90%. BMDMs (1 × 106 cells) were seeded in 30 mm culture wells in incomplete RPMI media. After 3h, BMDMs were incubated with or without FeSO4 (100 μM) for 6h. Culture supernatant was collected to measure Lcn2 levels by ELISA.
Systemic iron administration
Six weeks old male Lcn2KO mice and WT littermates were administered ferrous sulfate (i.p.; 25 mg/kg BW) and control mice were given sterile phosphate buffer saline (PBS). After 4 and 24h mice were bled to analyze for serum organ damage and inflammation markers.
Carbonyl iron enriched diet
Four weeks old male Lcn2KO mice and their WT littermates were either fed on a lab chow (TestDiet 5001) containing 280 ppm iron, or lab chow fortified with 2% carbonyl iron (TestDiet 1816708-201; 20,000 ppm iron) for 12 weeks. Diets were formulated by TestDiet (St. Louis, MO). Mice were monitored for body weights and food intake weekly.
Dextran-sodium sulfate (DSS)-induced colitis
Eight weeks old male Lcn2KO mice and their WT littermates were administered 1.8% DSS in drinking water over a period of 7 days. The induction of colonic inflammation was confirmed via fecal occult blood, diarrhea and loss in body weight as described previously (Chassaing et al. 2014). Serum and urine were collected on day 7 for analysis.
Euthanasia and blood collection
At termination of the experiment, mice were euthanized via CO2 asphyxiation and analyzed for standard colitis parameters as described previously (Singh et al. 2015). Blood was collected at the time of mice euthanasia in BD microtainer® (Becton, Dickinson), via cardiac puncture. Hemolysis-free serum was obtained after centrifugation and stored at −80°C until further analysis. 5 hours fasting serum was collected one week before termination via retro-orbital plexus under isoflurane anesthesia for lipid analysis. Serum collected from hemochromatosis protein (HFE)-deficient mice and their WT littermates were a kind gift from Dr. Nagendra Singh (Augusta University, Augusta, GA).
Enzyme-linked immunosorbent assay (ELISA)
Reconstitution of fecal samples was performed as described previously (Chassaing et al. 2012). Briefly, frozen or freshly collected feces were reconstituted in PBS containing 0.1% Tween 20 to make 100 mg/ml fecal suspension and vortexed for 30 min at room temperature. Fecal suspensions were centrifuged at 4°C for 10,000g for 10 min and the clear supernatants were collected and diluted (1:10) for controls and titrated for optimal dilution for treated samples for Lcn2 ELISA. Liver samples were homogenized in RIPA buffer with protease inhibitor to make 100 mg/ml suspension and centrifuged (4°C, 10,000g for 10 min). Clear supernatants were collected and diluted (1:500) for Lcn2 ELISA. Serum and urine samples were diluted, 1:250 or 1:2500; depending on severity of inflammation, for Lcn2 ELISA whereas for KC ELISA serum was diluted 1:5. All samples were diluted in kit recommended reagent diluent and analyzed using Duoset ELISA kits according to manufacturer instructions.
Blood glucose estimation
Mice were fasted for overnight (15h) and blood glucose was measured by Nova Max blood glucose meter and test strips via the tail vein.
Assay of serum transaminases, lipids and urinary creatinine
Serum aspartate transaminase (AST), alanine transaminase (ALT), triglycerides (TG) and cholesterol (CHOL), lactate dehydrogenase (LDH) and urinary creatinine levels were measured using kits from Randox (Crumlin, UK) according to the manufacturer’s instructions.
Measurement of serum total iron
Total iron was measured in serum samples as described previously (Walmsley et al. 1992). Briefly, serum samples were mixed with an equal volume of protein precipitation solution containing hydrochloric acid (0.1mg/ml), trichloroacetic acid (1.0 mol/L) and thioglycolic acid (30 ml/L). This step precipitates the serum proteins and liberates any protein bound iron. After centrifugation at room temperature (6,200g for 15 min), the supernatants were collected and mixed with an equal volume of chromogen solution containing 1.5M ferrozine and 1.5M sodium acetate. The optical density of the chromogen was measured at 562 nm. Total iron levels were estimated using a standard curve generated with the iron AA standard.
Measurement of non-heme iron in liver
Hepatic iron concentration was measured by the non-heme iron assay protocol as described by Torrance and Bothwell (Torrance et al. 1968). Briefly, 50 mg/ml of liver samples were digested in acid solution (3M HCl containing 10% trichloroacetic acid) and incubated at 65 °C for 20h. Then, samples were centrifuged and 25μL of the supernatant was applied to a 96-well microplate (Corning). Working chromogen reagent was prepared fresh on the day of assay from chromogen reagent stock (0.1% bathophenanthroline sulphonate and 1% thioglycolic acid). 250μL of working chromogen reagent (1 volume of chromogen reagent stock, 5 volumes of saturated sodium acetate and 5 volumes of double deionized water) was added to the samples. After 10 min incubation at RT, sample absorbance was measured at 535 nm. Iron concentration was determined using a standard curve generated using the iron AA standard.
Bleomycin-detectable iron (Fe+2) assay
Serum (1:2) and urine (1:2) samples were diluted with Chelex 100 (Sigma) pre-treated water. In some experiments, serum and urine samples were pre-incubated with endotoxin-, siderophore-, iron and carrier free rec-Lcn2 (500nM) for 15 min. The bleomycin assay for CI was performed as outlined by Burkitt et al. (Burkitt, Milne, and Raafat 2001) in 96-well plate (Corning) with the following modifications. Briefly, samples were added to a mixture containing 50 μg/mL calf thymus DNA (Sigma), 50 mU/mL bleomycin sulfate, 0.1 M Tris/HCl (Sigma), 5 mM MgCl2·6H2O (Sigma) in water treated with Chelex 100 (Sigma). The reaction was initiated by adding 1.0 mM ascorbic acid. For control, the mixture was prepared as above but without bleomycin sulfate. After incubation at 37 °C for 2 h, the reaction was stopped by adding 0.1 mM EDTA (Sigma) and 0.1 mM ethidium bromide (Sigma). Fluorescence readings were taken (excitation 510 nm; emission 590 nm). In principle, the conversion of Fe3+ to Fe2+ by ascorbic acid is detected as bleomycin-induced DNA damage that is proportional to the amount of available Fe2+. However, proteins- and chelators- bound Fe3+ is not detectable, because ascorbic acid cannot reduce tightly bound Fe3+ to Fe2+. The percent of DNA damage was calculated by comparing the readings from the sample group to the control group. All indicated concentrations above represent final system concentrations. Iron AA standard was used to prepare the standard curve.
Quantitative RT-PCR
Mouse liver and duodenum [first segment of small intestine, approximately one six of whole small intestine length (Duan et al. 2004) ] were collected in RNALater (Sigma) and stored in −80 °C. Total mRNA was extracted by using Trizol reagent (Sigma) as described in the manufacturer’s protocol. mRNA (0.8 μg) was used to synthesize cDNA for qRT-PCR using SYBR green (Quanta) according to manufacturer’s protocol. The following primers were used to assess gene expression: Fpn (ferroportin) 5′-TTGTTGTTGTGGCAGGAGAA-3′ and 5′-AGCTGGTCAATCCTTCTAATGG-3′ (Masaratana et al. 2013); Hamp (hepcidin) 5′-AGAAAGCAGGGCAGACATTG-3′ and 5′-CACTGGGAATTGTTACAGCATT-3′ (Masaratana et al. 2013); Dmt-1 (divalent metal transporter-1) -5′-GGCTTTCTTATGAGCATTGCCTA-3′ and 5′-GGAGCACCCAGAGCAGCTTA-3′ (Dupic et al. 2002); Dcytb (duodenum cytochrome b) 5′-GCAGCGGGCTCGAGTTTA-3′ and 5′-TTCCAGGTCCATGGCAGTCT-3′ (Dupic et al. 2002); 36B4 5′–TCCAGGCTTTGGGCATCA–3′ and 5′–CTTTATTCAGCTGCACATCACTCAGA–3′ (Chassaing et al. 2012). 36B4 was used to normalize relative mRNA expression using Ct (2ΔΔCt) method. Fold change was determined by comparison to the untreated control group.
Lipid peroxidation
Lipid peroxidation in liver was assayed by estimating the formation of thiobarbituric acid (TBA) reactive substances according to the method described by Buege and Aust (Buege et al. 1978). Briefly, weighted liver tissues were homogenized in ice cold 1.15% KCl to make 10% homogenate. Liver homogenates were incubated with 1mM FeSO4 and 1.5 mM ascorbic acid in 150 mM Tris-HCl buffer for 15min at 37°C. Next, TBA reagent (0.375% TBA and 10%TCA) was added to the samples and then incubated for 15 min in 80°C water bath. Sample absorbance at 535 nm was measured with a spectrophotometer. The amount of malondialdehyde (MDA) formed was quantified using the MDA molar extinction coefficient of 1.56×105 M−1.
Immunoblotting
Serum prepared in loading buffer were fractionated by 4–20% SDS-PAGE (Bio-Rad) and transferred to PVDF membrane (Bio-Rad). Recombinant Lcn2 was used as a positive control. Next, PVDF membrane was stained with Ponceau S solution (Sigma) to confirm equal loading of samples and then washed with H2O. The blots were blocked for 1 hour in 5% non-fat milk (Bio-Rad) at room temperature and then incubated overnight at 4°C with primary antibody biotinylated anti-mouse Lcn2. After 3 washes, blots were incubated with streptavidin HRP (Invitrogen) and developed by chemiluminiscent reagent.
Statistical analysis
Student unpaired and paired t test, and one-way ANOVA with Tukey’s post hoc test were used for statistical analysis. All data are shown as mean ± SEM. The correlations between Lcn2 and CI or total iron levels were assessed by Pearson correlation test and linear regression was used to plot the best-fit line (with 95% confidence interval). GraphPad Prism 6 software was used to calculate statistical significance with p value <0.05 considered as significant.
Results
Iron upregulates Lcn2 expression in vitro and in vivo
Lcn2 is highly upregulated during inflammation as a mechanism to sequester iron-laden bacterial siderophores, yet it is still unclear whether iron status can directly modulate Lcn2 levels. To investigate this possibility, we administered exogenous iron to bone marrow-derived macrophages from C57BL/6 WT mice and measure their Lcn2 secretion. Elevated Lcn2 secretion was observed in ferrous iron (Fe2+)-treated macrophages compared to vehicle-treated macrophages (Fig. 1a). We did not observe significant changes in Lcn2 levels when macrophages were treated with ferric iron (Fe3+) (data not shown), suggesting that ferrous and ferric iron differentially induce Lcn2 response from macrophages.
Fig. 1. Iron induces Lcn2 in vitro and in vivo.
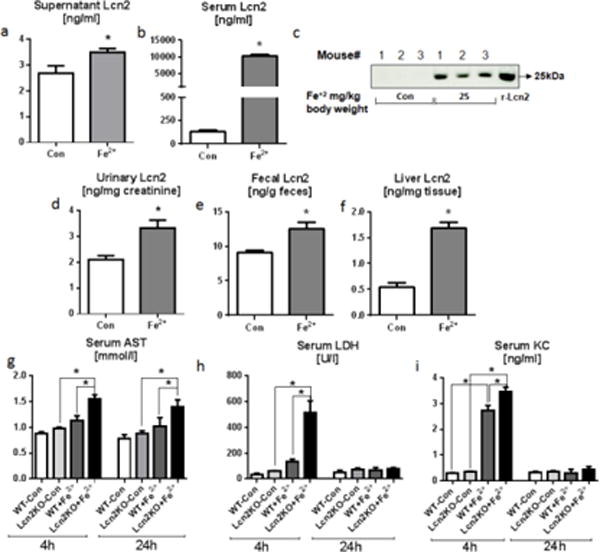
For the in vitro studies, bone marrow-derived macrophages (1.0×106 cells/well) were either incubated with or without Fe2+ (100 μM) in triplicates for 6h. a Bar graph represents Lcn2 (ng/ml) level in the supernatants obtained from Fe2+ treated macrophages. For the in vivo studies, six weeks old WT mice (n=4–5) were given either PBS or FeSO4 (25 mg/kg body weight) intraperitoneally. After 24h serum, urine feces and liver were collected for estimating Lcn2 using ELISA and immunoblotting. b–c Bar graph and immunoblot display the Lcn2 level in the serum. d–f Lcn2 level in urine, feces and liver after 24h. Organ damage and inflammation markers were analyzed at both 4h and 24h after FeSO4 (25 mg/kg body weight) administration. g AST, h LDH, and i KC were measured in serum obtained from control and FeSO4 treated Lcn2KO and their WT littermates using commercial kits. Results presented as mean ± SEM. unpaired t-test (a–b, d–f) and Tukey’s post hoc test (g–i) *p< 0.05
Iron supplementation is routinely used to treat iron-deficient anemia observed in inflammatory bowel diseases (IBD) (Stein et al. 2010). Next, we asked whether administration of ferrous iron could also upregulate systemic Lcn2 levels in vivo as well. WT mice treated with ferrous iron (25 mg/kg bodyweight via i.p.) displayed 100-fold upregulation of serum Lcn2 at 24h post-treatment (Fig. 1b). The elevation of Lcn2 levels in serum was further confirmed by immunoblotting (Fig. 1c). Congruently, the urinary, fecal and liver Lcn2 levels were increased by 58%, 37% and 208%, respectively (Fig. 1d–f). These results suggest that the upregulation of Lcn2 may be beneficial, perhaps as a mechanism to restore iron homeostasis. The elevated Lcn2 level was observed in hemochromatosis protein (HFE)-deficient mice with iron overload (Supplementary Fig. 1a–b). Specially, a significant negative correlation was observed between serum Lcn2 and iron in HFE-KO mice (Supplementary Fig. 1c) further implicates a potential role of Lcn2 in mediating tolerance to excess iron. To study whether iron-induced Lcn2 response is protective, we administered Fe2+ (25 mg/kg body weight via i.p.) to age- and sex-matched Lcn2KO mice and their WT littermates. Intriguingly, the serum levels of aspartate transaminase (AST, Fig 1g), lactate dehydrogenase (LDH, Fig 1h), and KC (aka CXCL1, Fig 1i) were significantly elevated at 4h time point. However, the levels of LDH and KC subsided to basal levels by 24h except AST (Fig. 1g–i), which was still elevated at 24h post iron challenge. Remarkably, both organ damage and inflammation markers were relatively elevated in Lcn2KO mice indicating that the loss of Lcn2 could potentiate iron-induced organ damage. However, we did not find any notable difference in the serum levels of alanine aminotransferase (ALT) between WT and Lcn2KO mice with iron treatment at both 4h and 24h time points (data not shown).
Lcn2 deficiency results in adverse effects in response to dietary iron
To explore the role of Lcn2 in regulating dietary iron, we placed age- and sex-matched WT and Lcn2KO mice (Fig. 2a) on either control diet (lab chow; contains 280 ppm iron) or high-iron diet (fortified with 2% carbonyl iron, 20,000 ppm) which has been used to induce iron overload (Handa et al. 2016). Lcn2KO mice on control diet gained significantly more body weight compared to all other groups, but this phenotype is lost in Lcn2KO mice fed on high-iron diet. We did not observe any significant difference in the food intake between all groups of mice (Mean, WT-Con: 3.4g/d, Lcn2KO-Con: 3.5g/d, WT+Iron: 3.2g/d and Lcn2KO+Iron: 3.4g/d). Nonetheless, the disparity between control and high-iron diet-fed Lcn2KO mice indicate a possible altered metabolic/inflammatory state in the latter that is also reflected by their reduced adipose tissue weight, augmented splenomegaly, elevated fasting blood glucose and serum triglycerides when compared to all other mice groups (Fig. 2b–e). Moreover, Lcn2KO mice on high iron diet displayed elevated serum levels of AST and an increasing trend in serum ALT levels (Fig. 2f, g) compared to other groups. There was no difference between groups for serum cholesterol (data not shown).
Fig. 2. Lcn2 deficiency potentiates dietary iron-induced toxicity.
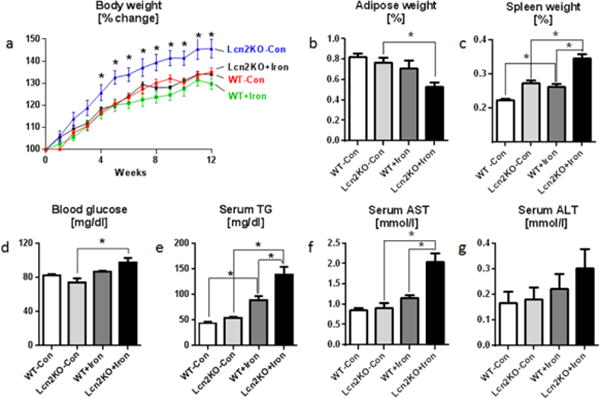
Four weeks old male Lcn2KO mice and their WT littermates (n=5) were maintained on 2% carbonyl iron diet for 12 weeks, and monitored for body weight and food intake regularly. Serum, adipose tissue and spleen were collected for analysis. Line graph represents a body weight. Bar graphs represent b adipose tissue weight, c spleen weight, d overnight fasting blood glucose, e serum triglycerides (TG), f serum AST, and g ALT. Results presented as mean ± SEM. unpaired t-test (a, Lcn2KO+Iron to Lcn2KO-Con) and Tukey’s post hoc test (b–g) *p< 0.05
The increased dietary iron intake also elevated the Lcn2 levels in the serum, urine, feces and liver by 121%, 386%, 71% and 85% respectively in WT mice on high iron diet (Fig. 3a–d). These Lcn2 responses appear to exert important roles in maintaining iron homeostasis in WT mice, but such protective mechanism is abrogated in Lcn2KO mice. Accordingly, high-iron diet-fed Lcn2KO exhibited increased total iron in the serum by 70% (Fig. 3e), and increased CI in serum and urine by 220% and 60% respectively (Fig. 3f–g) when compared to similarly-fed WT mice. Since the liver is the primary organ for iron homeostasis (Ganz 2011), we next measured the levels of non-heme iron in the liver of WT and Lcn2KO mice. Interestingly, the levels of hepatic iron was already higher in control diet-fed Lcn2KO (than WT mice on control diet), which become even more prominently elevated in high-iron diet-fed Lcn2KO mice (than all other mice groups) (Fig. 3h). Such excessive iron accumulation in the liver of these mice might enhance their susceptibility to oxidative stress. To confirm that, we analyzed liver MDA level (a marker of lipid peroxidation) and observed that high-iron diet caused lipid peroxidation in both WT and Lcn2KO group (Fig. 3i), albeit to a greater extent in high-iron diet-fed Lcn2KO mice.
Fig. 3. Dietary iron induces hyperferremia and elevated catalytic iron in Lcn2KO mice.
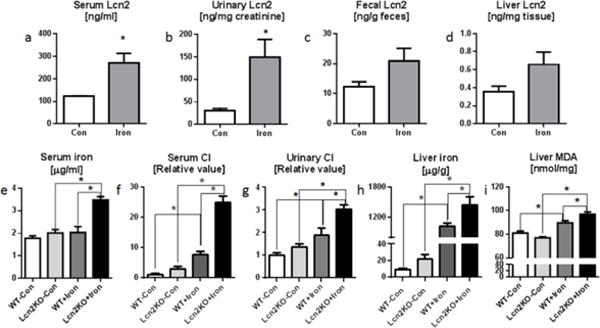
Four weeks old male Lcn2KO mice and their WT littermates (n=5) were maintained on 2% carbonyl iron diet for 12 weeks and monitored regularly for body weights and food intake. Serum, urine, feces and liver tissue were collected on the day of euthanasia. Lcn2 was analyzed by ELISA in a serum, b urine, c feces, and d liver. e Bar graph represents serum total iron. Catalytic iron (CI) levels were quantified by bleomycin-detectable iron assay. Bar graphs represent CI levels in f serum and g urine. h Liver tissue iron was measured by non-heme tissue iron method. i Liver lipid peroxidation marker liver malondialdehyde (MDA) was estimated by MDA-TBA assay. Results presented as mean ± SEM. unpaired t-test (a–d) and Tukey’s post hoc test (e–i). *p< 0.05
Dietary iron is tightly regulated at the levels of absorption, transportation and storage (Ratledge 2007, Shi et al. 2008, Weiss 2009, Martines et al. 2013). Our results have suggested a potential role for Lcn2 in modulating circulating and storage forms of iron and therefore, we next evaluated whether Lcn2 deficiency could also dysregulate iron absorption. High iron intake downregulates the expression of genes associated with iron absorption, including ferroportin (Fpn), duodenal cytochrome-b (Dyctb) and divalent metal transporter 1 (Dmt1), in the duodenum of both WT and Lcn2KO mice (Supplementary Fig. 2a–c). Despite so, the high-iron diet-fed Lcn2KO mice were not able to downregulate Fpn and Dyctb to the extent that was observed in similarly-fed WT mice. The upregulation of hepcidin (master iron regulator) expression in the liver was comparable between high-iron diet-fed WT and Lcn2KO mice, (Supplementary Fig. 2d) indicating that hepcidin response to excess iron was not impaired in Lcn2KO mice.
Lcn2KO mice fail to reduce systemic iron levels and display higher levels of catalytic iron (CI) in murine model of acute colitis
Inflammation is accompanied with systemic hypoferremia as an innate immune mechanism to dampen iron-mediated oxidative stress and also to deplete iron sources for opportunistic pathogens (Nemeth et al. 2004). Hence, we next investigated whether there is any correlation between Lcn2 and the levels of total and CI (CI aka bleomycin-detectable iron) by employing the murine model of DSS-induced acute colitis. Briefly, intestinal inflammation was induced in 6 weeks old male WT and Lcn2KO mice by administration of 1.8% DSS in drinking water for 7 days. The induction of acute colitis was confirmed by the presence of bloody diarrhea and body weight loss. Disease was more severe in Lcn2KO mice as analyzed by greater loss in body weight, and colon shortening (data not shown). Consistent with our previous results (Chassaing et al. 2012), serum Lcn2 levels were increased significantly in DSS-treated WT mice by day 7 (Fig. 4a). Although the serum total iron levels were decreased in DSS-treated WT mice compared to healthy WT mice, no significant change in serum total iron levels were observed between healthy and colitic Lcn2KO mice (Fig. 4b). Lcn2 and total iron in DSS-treated WT mice were negatively correlated (Fig. 4c; r=−0.5826, p=0.0997). In a similar fashion, the serum CI levels were markedly reduced in DSS-treated WT mice, whereas the CI levels remain unchanged in DSS-treated Lcn2KO mice (Fig. 4d). Nonetheless, a negative correlation was observed between serum Lcn2 and CI in the WT group (Fig. 4e; r=−0.9186, p=0.0002). Furthermore, a significantly increased urinary Lcn2 and a considerably decreased level of urinary CI were observed in DSS-treated WT mice (Fig. 4f–g). However, we did not observe any correlation, between urinary Lcn2 and CI in DSS-treated WT mice (data not shown).
Fig. 4. Lcn2KO mice fail to display hypoferremia during DSS-induced acute colitis.
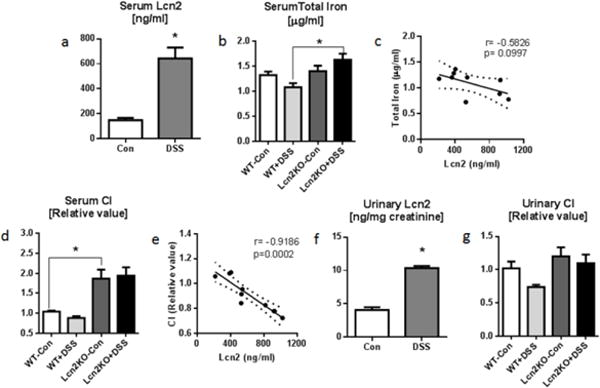
Eight weeks old male Lcn2KO mice and their WT littermates (n=6–9) were administered 1.8% DSS drinking water for 7 days. Serum and urine were collected on the day of euthanasia for analysis. Bar graphs represent a serum Lcn2 and b serum total iron. c Line graph represents Pearson correlation analysis of serum Lcn2 and serum total iron. d Bar graph represents serum CI. e Line graph shows Pearson correlation analysis of serum Lcn2 and serum CI. f–g Urinary Lcn2 and CI are shown by bar graphs. Pearson correlations between serum Lcn2, total iron and CI were plotted by linear regression (Best-fit line with 95% confidence interval). Results presented as mean ± SEM. unpaired t-test (a, f) and Tukey’s post hoc test (b, d and g). *p< 0.05
Exogenous Lcn2 reduces the CI levels in serum and urine from Lcn2KO mice
Next we hypothesize that Lcn2 chelated siderophore-bound iron is more stable and unable to participate in Fenton reaction, thus limiting free radical generation. To assess our hypothesis that Lcn2 can effectively chelate CI, we exogenously added siderophore-, iron-, endotoxin- and carrier-free mouse rec-Lcn2 (500nM) to serum and urine from WT mice and Lcn2KO mice fed on lab chow diet. The addition of mouse rec-Lcn2 reduced the CI levels (Fig. 5a, b) in serum and urine from both WT and Lcn2KO mice. More importantly, the decrease in CI is much higher in the serum and urine from Lcn2KO mice. Moreover, serum CI from Lcn2KO mice fed on high-iron diet was reduced by 42% after treating with rec-Lcn2 (data not shown). Similar results were observed when the experiments were repeated using the human Lcn2 (NGAL). Collectively, our findings suggest that Lcn2 is a host protective factor that acts against generation of CI.
Fig. 5. Exogenous rec-Lcn2 reduces the CI levels.
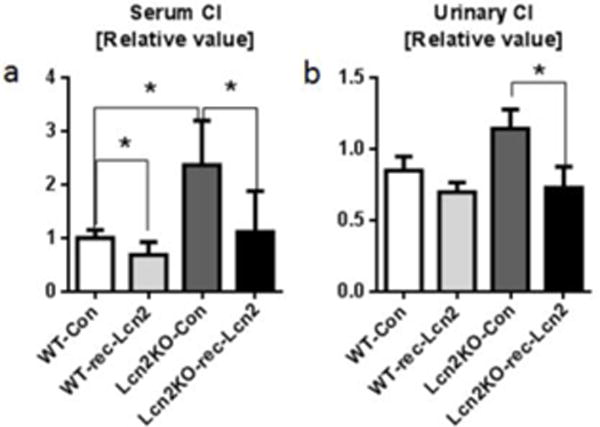
Serum and urine were collected from age matched male Lcn2KO and their WT littermates fed on lab chow. Samples were incubated with endotoxin-, siderophore-, iron- and carrier-free rec-Lcn2 (500nM) for 15 min and subjected to bleomycin-detectable iron assay. Bar graphs represent a serum CI and b urinary CI. Results presented as mean ± SEM., paired t-test (with or without rec-Lcn2 treatment on same sample and same group) and unpaired t-test. *p< 0.05
Both immune and non-immune cell-derived Lcn2 play a role in iron regulation
To further dissect the relative contribution of immune and non-immune cells to the pool of systemic Lcn2, we generated bone marrow chimeras and measure their basal serum Lcn2 and iron levels. Lcn2KO→WT group of mice showed higher level of serum Lcn2 than WT→Lcn2KO group, indicating non-immune cells are the major contributor of circulating Lcn2 (Fig. 6a) when compared to immune cells. We did not observe significant differences in total serum iron levels among all groups (data not shown). However, WT→Lcn2KO group displayed higher levels of serum CI levels when compared to Lcn2KO→WT (Fig. 6b). WT→WT and Lcn2KO→Lcn2KO mice displayed normal and elevated levels of CI, respectively, which are consistent with our previous observations. Collectively, our results suggest that Lcn2 from non-immune cells may play a major role in regulation of CI generation.
Fig. 6. Both immune and non-immune cell-derived Lcn2 play role in CI regulation.
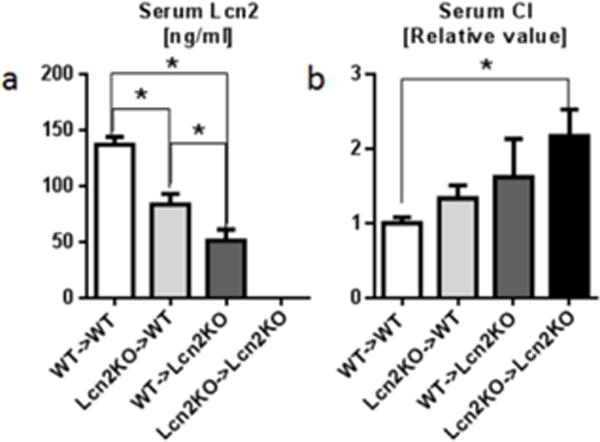
To investigate the relative contribution of immune and non-immune cell-derived Lcn2 in maintaining serum CI levels, serum from bone marrow chimeras was analyzed for a serum Lcn2 by ELISA and b serum CI by bleomycin-detectable iron assay. Results presented as mean ± SEM., Tukey’s post hoc test. *p< 0.05
Discussion
Iron is an essential trace metal ion required by virtually all aerobic cells for their biologic functions. The versatility of iron to transition between its Fe2+ and Fe3+ redox states enables it to catalyze various chemical reactions that are essential to life. Yet, due to its highly reactive nature, free iron (aka catalytic iron, CI) can readily participate in Fenton reaction and induces adverse oxidative stress. Accordingly, iron-dependent organisms have evolved to express a plethora of iron-binding proteins to regulate iron homeostasis throughout absorption, transportation and storage. Lcn2 is one such iron-binding protein known to be highly elevated in various pathophysiological conditions. (Jiang et al. 2008; Roudkenar et al. 2008; Chakraborty et al. 2012). Herein, we demonstrated that administration of iron either systemically or through dietary intake could also upregulate Lcn2 levels. Consistent with a previous report (Nairz et al. 2009), we also observed elevated serum Lcn2 in HFE-KO mice that are routinely used to study hereditary iron overload. Such systemic elevation of Lcn2 may be a beneficial response to restore iron homeostasis. Indeed, the overexpression of Lcn2 in colonic epithelia was previously shown to protect against iron overload, whereas the loss of Lcn2 expression aggravates iron-induced toxicity and cell death (Hu et al. 2009). Likewise, Lcn2KO mice displayed impaired tolerance to excess dietary iron and were more susceptible to iron-induced toxicity and oxidative stress. The mechanisms by which Lcn2 confer protection against iron-induced toxicity are likely to be multifaceted, which could possibly involve the sequestration of CI and/or mediated via the anti-oxidative property of Lcn2 (Yamada et al. 2016). However, a decreasing trend in body weights and adiposity were observed in WT mice fed with high-iron diet. This might be due to excess dietary iron causing low-grade inflammation. Notably, the loss in body weight and adipose tissue was more pronounced in Lcn2KO mice. Similarly, iron-rich diet fed mice displayed hypertriglyceridemia in both WT and Lcn2KO mice but the TG levels were significantly more in Lcn2KO mice. Such elevated serum TG [lipemia of inflammation (Barcia et al. 2005)] could be due to low-grade inflammation caused by high-iron diet resulting in increased lipolysis and/or hepatic TG output.
Labile iron, also known as catalytic or free iron, is considered harmful due to its propensity to catalyze the formation of free radicals that could damage cellular macromolecular components (Halliwell et al. 1990). Indeed, iron-catalyzed tissue damage had been reported in a variety of disease conditions, most notably in acute and chronic kidney disease (Shah et al. 2011). In a seminal study by Mishra et al., ischemia-reperfusion injury in mice was shown to be ameliorated upon intravenous administration of Lcn2, yet the role of Lcn2 on CI modulation was only speculative at the time of the study (Mishra et al. 2004). Further studies were subsequently carried out to elucidate the interplay between Lcn2 and CI, although these studies were restricted to in vitro conditions (Bao et al. 2010; Bao et al. 2013). By using Lcn2KO mice, we herein sought to further elucidate the potential role of Lcn2 in regulating CI in vivo. Indeed, Lcn2KO mice on high-iron diet displayed substantially elevated levels of CI in both serum and urine, when compared to similarly-fed WT mice. Their inability to suppress CI may be detrimental and might, in part, explain the enhanced susceptibility of Lcn2KO mice to LPS-induced sepsis and mortality (Srinivasan et al. 2012).
Despite its role in transporting iron, Lcn2 by itself could not directly bind to iron without the aid of small molecules known as siderophores. The bacterial siderophore, enterobactin and its monomeric form 2,3-dihydroxybenzoic acid are among those extensively studied in the context of inflammation, whereby Lcn2 sequesters these siderophores to suppress bacterial growth (Flo et al. 2004). Lcn2 could also form complexes with 2, 5-dihydroxybenzoic acid, which had been proposed to be a putative mammalian-derived siderophore (Devireddy et al. 2010). Other ferric ligands including ionic compounds such as citrate, polyphosphate and phospholipids have also been proposed to interact with Lcn2 (Kruszewski 2003). It is perhaps important to note that iron bound to some siderophores still retain its capacity to participate in redox reactions (Iwahashi et al. 1989). Bao et al. demonstrated that catechol-bound ferric iron remains catalytic, but Lcn2 blocked the reactivity of iron upon forming a ternary complex with catechol-iron in vitro (Bao et al. 2010). The tight binding by Lcn2 to siderophores at subnanomolar affinities may contribute to stabilize the Fe3+ (Goetz et al. 2002; Hoette et al. 2008; Coudevylle et al. 2010). Current technology, however, does not easily permit in-depth study of the iron binding states of Lcn2 in in vivo conditions. Future exploration on which siderophores are involved in Lcn2-mediated protection against iron-induced toxicity could reveal key insights regarding Lcn2 signaling, iron homeostasis and regulation of CI.
Iron status and inflammation are tightly linked and regulated. For instance, the iron overload state of HFE-KO mice markedly attenuated their inflammatory responses to LPS and Salmonella infection (Wang et al. 2008; Wang et al. 2009). Further, inflammation is robustly associated with the induction of hypoferremia (Kaitha et al. 2015) and elevated systemic levels of Lcn2 (Srinivasan et al. 2012). Hence, we sought to elucidate whether Lcn2 could play a role in the acute systemic hypoferremic response by using the murine model of DSS-induced colitis. Our analysis revealed a negative correlation between (i) Lcn2 and total iron, and (ii) Lcn2 and CI in the serum of colitic WT mice. These results suggest that Lcn2 contributes to hypoferremia at early time points induced by acute inflammation, which corroborated our previous observations that Lcn2KO mice showed delayed LPS-induced hypoferremia (Srinivasan et al. 2012). Even though our study did not rule out the possible contribution of hepcidin and other iron-binding proteins/factors in facilitating hypoferremia in colitic WT mice, it is interesting to note that deficiency of Lcn2 may be sufficient to disrupt early hypoferremic response in Lcn2KO mice. Further studies, however, are required to investigate whether other mechanisms may supersede Lcn2 to induce/maintain hypoferremia during the later phase of chronic colitis. Although our study did not detect significant correlation between urinary CI and Lcn2, one study reported a positive correlation in patients with high risk in developing acute kidney injury after undergoing open heart surgery (Akrawinthawong et al. 2013). Such discrepancy could be due to the differences in (1) the model or pathological condition (murine vs human), (2) the time points in which samples were collected, (3) type of tissue analyzed, and (4) use of any medication.
That dramatic induction of Lcn2 protects mice against iron administration and inflammation suggests the possibility that Lcn2, and its human ortholog NGAL, may have therapeutic potential in the treatment of iron induced toxicity. Our results suggest that Lcn2 not only display anti-inflammatory agent by regulating hypoferremia of inflammation, but also acts as CI chelator, which help harmful CI clearance. Collectively, our results show that Lcn2 can be targeted to develop novel therapeutics (e.g. anticalins)(Schiefner et al. 2015) to treat iron-related disorders.
Supplementary Material
Acknowledgments
We thank Dr. Gregory Shearer for his critical input.
Funding Sources
This work was supported by grants from the National Institutes of Health (NIH) R01 (DK097865) and PSU Dean’s Schultz endowment, College of Health and Human Development Seed grant to M.V.-K. B.S.Y. is supported by NIH T32 (T32AI074551).
Abbreviations
- Lcn2
lipocalin2
- Lcn2KO
lipocalin 2 knockout
- WT
wild-type
- NGAL
neutrophil gelatinase-associated lipocalin
- TG
triglycerides
- LDH
lactate dehydrogenase
- ALT
alanine transaminase
- AST
aspartate transaminase
- CI
catalytic iron
Footnotes
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
- Akrawinthawong K, Shaw MK, Kachner J, et al. Urine catalytic iron and neutrophil gelatinase-associated lipocalin as companion early markers of acute kidney injury after cardiac surgery: a prospective pilot study. Cardiorenal medicine. 2013;3:7–16. doi: 10.1159/000346815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao G, Clifton M, Hoette TM, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nature chemical biology. 2010;6:602–609. doi: 10.1038/nchembio.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao GH, Xu J, Hu FL, Wan XC, Deng SX, Barasch J. EGCG inhibit chemical reactivity of iron through forming an Ngal-EGCG-iron complex. Biometals : an international journal on the role of metal ions in biology, biochemistry, and medicine. 2013;26:1041–1050. doi: 10.1007/s10534-013-9681-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia AM, Harris HW. Triglyceride-rich lipoproteins as agents of innate immunity. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2005;41(Suppl 7):S498–503. doi: 10.1086/432005. [DOI] [PubMed] [Google Scholar]
- Boddaert N, Le Quan Sang KH, Rotig A, et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–408. doi: 10.1182/blood-2006-12-065433. [DOI] [PubMed] [Google Scholar]
- Buege JA, Aust SD. Microsomal lipid peroxidation. Methods in enzymology. 1978;52:302–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- Cabantchik ZI. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Frontiers in pharmacology. 2014;5:45. doi: 10.3389/fphar.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Aitken JD, Gewirtz AT, Vijay-Kumar M. TLR5 activation induces secretory interleukin-1 receptor antagonist (sIL-1Ra) and reduces inflammasome-associated tissue damage. Mucosal immunology. 2011;4:102–111. doi: 10.1038/mi.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochimica et biophysica acta. 2012;1826:129–169. doi: 10.1016/j.bbcan.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. In: Coligan John E, et al., editors. Current protocols in immunology. 2014. p. 104. Unit 15 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PloS one. 2012;7:e44328. doi: 10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudevylle N, Geist L, Hotzinger M, et al. The v-myc-induced Q83 lipocalin is a siderocalin. The Journal of biological chemistry. 2010;285:41646–41652. doi: 10.1074/jbc.M110.123331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darshan D, Frazer DM, Wilkins SJ, Anderson GJ. Severe iron deficiency blunts the response of the iron regulatory gene Hamp and pro-inflammatory cytokines to lipopolysaccharide. Haematologica. 2010;95:1660–1667. doi: 10.3324/haematol.2010.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devireddy LR, Hart DO, Goetz DH, Green MR. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell. 2010;141:1006–1017. doi: 10.1016/j.cell.2010.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan LP, Wang HH, Wang DQ. Cholesterol absorption is mainly regulated by the jejunal and ileal ATP-binding cassette sterol efflux transporters Abcg5 and Abcg8 in mice. Journal of lipid research. 2004;45:1312–1323. doi: 10.1194/jlr.M400030-JLR200. [DOI] [PubMed] [Google Scholar]
- Dupic F, Fruchon S, Bensaid M, et al. Duodenal mRNA expression of iron related genes in response to iron loading and iron deficiency in four strains of mice. Gut. 2002;51:648–653. doi: 10.1136/gut.51.5.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102:783–788. doi: 10.1182/blood-2003-03-0672. [DOI] [PubMed] [Google Scholar]
- Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117:4425–4433. doi: 10.1182/blood-2011-01-258467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Molecular cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions in human disease: an overview. Methods in enzymology. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- Handa P, Morgan-Stevenson V, Maliken BD, et al. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. American journal of physiology Gastrointestinal and liver physiology. 2016;310:G117–127. doi: 10.1152/ajpgi.00246.2015. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Hoette TM, Abergel RJ, Xu J, Strong RK, Raymond KN. The role of electrostatics in siderophore recognition by the immunoprotein Siderocalin. Journal of the American Chemical Society. 2008;130:17584–17592. doi: 10.1021/ja8074665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Hittelman W, Lu T, et al. NGAL decreases E-cadherin-mediated cell-cell adhesion and increases cell motility and invasion through Rac1 in colon carcinoma cells. Laboratory investigation; a journal of technical methods and pathology. 2009;89:531–548. doi: 10.1038/labinvest.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Salahudeen AK. Cold induces catalytic iron release of cytochrome P-450 origin: a critical step in cold storage-induced renal injury. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2002;2:631–639. doi: 10.1034/j.1600-6143.2002.20708.x. [DOI] [PubMed] [Google Scholar]
- Iwahashi H, Morishita H, Ishii T, Sugata R, Kido R. Enhancement by catechols of hydroxyl-radical formation in the presence of ferric ions and hydrogen peroxide. Journal of biochemistry. 1989;105:429–434. doi: 10.1093/oxfordjournals.jbchem.a122681. [DOI] [PubMed] [Google Scholar]
- Jiang W, Constante M, Santos MM. Anemia upregulates lipocalin 2 in the liver and serum. Blood cells, molecules & diseases. 2008;41:169–174. doi: 10.1016/j.bcmd.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World journal of gastrointestinal pathophysiology. 2015;6:62–72. doi: 10.4291/wjgp.v6.i3.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes(1) Free radical biology & medicine. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- Koskenkorva-Frank TS, Weiss G, Koppenol WH, Burckhardt S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free radical biology & medicine. 2013;65:1174–1194. doi: 10.1016/j.freeradbiomed.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutation research. 2003;531:81–92. doi: 10.1016/j.mrfmmm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Lele S, Shah S, McCullough PA, Rajapurkar M. Serum catalytic iron as a novel biomarker of vascular injury in acute coronary syndromes. EuroIntervention : journal of EuroPCR in collaboration with the Working Group on Interventional Cardiology of the European Society of Cardiology. 2009;5:336–342. doi: 10.4244/v5i3a53. [DOI] [PubMed] [Google Scholar]
- Maitra D, Shaeib F, Abdulhamid I, et al. Myeloperoxidase acts as a source of free iron during steady-state catalysis by a feedback inhibitory pathway. Free radical biology & medicine. 2013;63:90–98. doi: 10.1016/j.freeradbiomed.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martines AM, Masereeuw R, Tjalsma H, Hoenderop JG, Wetzels JF, Swinkels DW. Iron metabolism in the pathogenesis of iron-induced kidney injury. Nature reviews Nephrology. 2013;9:385–398. doi: 10.1038/nrneph.2013.98. [DOI] [PubMed] [Google Scholar]
- Masaratana P, Patel N, Latunde-Dada GO, Vaulont S, Simpson RJ, McKie AT. Regulation of iron metabolism in Hamp (−/−) mice in response to iron-deficient diet. European journal of nutrition. 2013;52:135–143. doi: 10.1007/s00394-011-0295-z. [DOI] [PubMed] [Google Scholar]
- Mishra J, Mori K, Ma Q, et al. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. Journal of the American Society of Nephrology : JASN. 2004;15:3073–3082. doi: 10.1097/01.ASN.0000145013.44578.45. [DOI] [PubMed] [Google Scholar]
- Mitchell KM, Dotson AL, Cool KM, Chakrabarty A, Benedict SH, LeVine SM. Deferiprone, an orally deliverable iron chelator, ameliorates experimental autoimmune encephalomyelitis. Multiple sclerosis. 2007;13:1118–1126. doi: 10.1177/1352458507078916. [DOI] [PubMed] [Google Scholar]
- Nairz M, Theurl I, Schroll A, et al. Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood. 2009;114:3642–3651. doi: 10.1182/blood-2009-05-223354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kawakami T, Yamamoto N, et al. Activation of the NLRP3 inflammasome by cellular labile iron. Experimental hematology. 2015 doi: 10.1016/j.exphem.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. The Journal of clinical investigation. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE, Gherardini FC. Lack of a role for iron in the Lyme disease pathogen. Science. 2000;288:1651–1653. doi: 10.1126/science.288.5471.1651. [DOI] [PubMed] [Google Scholar]
- Ratledge C. Iron metabolism and infection. Food and nutrition bulletin. 2007;28:S515–523. doi: 10.1177/15648265070284S405. [DOI] [PubMed] [Google Scholar]
- Roudkenar MH, Halabian R, Bahmani P, Roushandeh AM, Kuwahara Y, Fukumoto M. Neutrophil gelatinase-associated lipocalin: a new antioxidant that exerts its cytoprotective effect independent on Heme Oxygenase-1. Free radical research. 2011;45:810–819. doi: 10.3109/10715762.2011.581279. [DOI] [PubMed] [Google Scholar]
- Roudkenar MH, Halabian R, Ghasemipour Z, et al. Neutrophil gelatinase-associated lipocalin acts as a protective factor against H(2)O(2) toxicity. Archives of medical research. 2008;39:560–566. doi: 10.1016/j.arcmed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Sanders CJ, Moore DA, 3rd, Williams IR, Gewirtz AT. Both radioresistant and hemopoietic cells promote innate and adaptive immune responses to flagellin. Journal of immunology. 2008;180:7184–7192. doi: 10.4049/jimmunol.180.11.7184. [DOI] [PubMed] [Google Scholar]
- Schiefner A, Skerra A. The menagerie of human lipocalins: a natural protein scaffold for molecular recognition of physiological compounds. Accounts of chemical research. 2015;48:976–985. doi: 10.1021/ar5003973. [DOI] [PubMed] [Google Scholar]
- Shah SV, Rajapurkar MM, Baliga R. The role of catalytic iron in acute kidney injury. Clinical journal of the American Society of Nephrology : CJASN. 2011;6:2329–2331. doi: 10.2215/CJN.08340811. [DOI] [PubMed] [Google Scholar]
- Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320:1207–1210. doi: 10.1126/science.1157643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Yeoh BS, Carvalho F, Gewirtz AT, Vijay-Kumar M. Proneness of TLR5 deficient mice to develop colitis is microbiota dependent. Gut Microbes. 2015;6:279–283. doi: 10.1080/19490976.2015.1060390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan G, Aitken JD, Zhang B, et al. Lipocalin 2 deficiency dysregulates iron homeostasis and exacerbates endotoxin-induced sepsis. Journal of immunology. 2012;189:1911–1919. doi: 10.4049/jimmunol.1200892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley ER. The macrophage colony-stimulating factor, CSF-1. Methods in enzymology. 1985;116:564–587. doi: 10.1016/s0076-6879(85)16044-1. [DOI] [PubMed] [Google Scholar]
- Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nature reviews Gastroenterology & hepatology. 2010;7:599–610. doi: 10.1038/nrgastro.2010.151. [DOI] [PubMed] [Google Scholar]
- Sulieman M, Asleh R, Cabantchik ZI, et al. Serum chelatable redox-active iron is an independent predictor of mortality after myocardial infarction in individuals with diabetes. Diabetes care. 2004;27:2730–2732. doi: 10.2337/diacare.27.11.2730. [DOI] [PubMed] [Google Scholar]
- Sullivan JL. Iron in arterial plaque: modifiable risk factor for atherosclerosis. Biochimica et biophysica acta. 2009;1790:718–723. doi: 10.1016/j.bbagen.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Thethi TK, Parsha K, Rajapurkar M, et al. Urinary catalytic iron in obesity. Clinical chemistry. 2011;57:272–278. doi: 10.1373/clinchem.2010.154757. [DOI] [PubMed] [Google Scholar]
- Torrance JD, Bothwell TH. A simple technique for measuring storage iron concentrations in formalinised liver samples. The South African journal of medical sciences. 1968;33:9–11. [PubMed] [Google Scholar]
- Walmsley TA, George PM, Fowler RT. Colorimetric measurement of iron in plasma samples anticoagulated with EDTA. Journal of clinical pathology. 1992;45:151–154. doi: 10.1136/jcp.45.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Pantopoulos K. Regulation of cellular iron metabolism. The Biochemical journal. 2011;434:365–381. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Harrington L, Trebicka E, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. The Journal of clinical investigation. 2009;119:3322–3328. doi: 10.1172/JCI39939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Johnson EE, Shi HN, Walker WA, Wessling-Resnick M, Cherayil BJ. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. Journal of immunology. 2008;181:2723–2731. doi: 10.4049/jimmunol.181.4.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg ED. The Lactobacillus anomaly: total iron abstinence. Perspectives in biology and medicine. 1997;40:578–583. doi: 10.1353/pbm.1997.0072. [DOI] [PubMed] [Google Scholar]
- Weiss G. Iron metabolism in the anemia of chronic disease. Biochimica et biophysica acta. 2009;1790:682–693. doi: 10.1016/j.bbagen.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Miyamoto T, Kashima H, et al. Lipocalin 2 attenuates iron-related oxidative stress and prolongs the survival of ovarian clear cell carcinoma cells by up-regulating the CD44 variant. Free radical research. 2016:1–36. doi: 10.3109/10715762.2015.1134795. [DOI] [PubMed] [Google Scholar]
- Yang J, Goetz D, Li JY, et al. An iron delivery pathway mediated by a lipocalin. Molecular cell. 2002;10:1045–1056. doi: 10.1016/s1097-2765(02)00710-4. [DOI] [PubMed] [Google Scholar]
- Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. In: Coligan John E, et al., editors. Current protocols in immunology. 2008. Chapter 14 Unit 14 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.