

Abstract
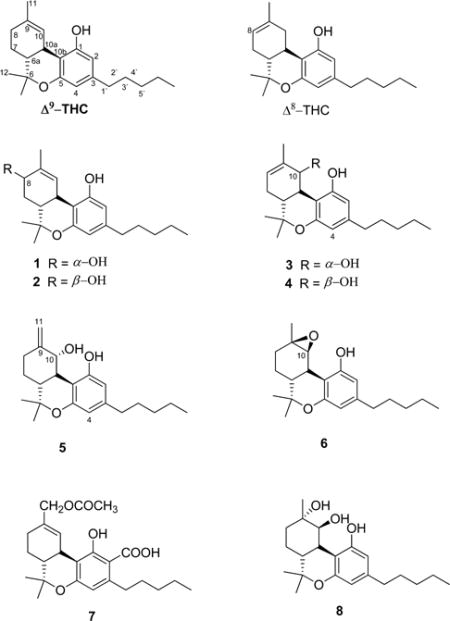
Seven new naturally occurring hydroxylated cannabinoids (1–7), along with the known cannabiripsol (8), have been isolated from the aerial parts of high-potency Cannabis sativa. The structures of the new compounds were determined by 1D and 2D NMR spectroscopic analysis, GC-MS, and HRESIMS as 8α-hydroxy-Δ9-tetrahydrocannabinol (1), 8β-hydroxy-Δ9-tetrahydrocannabinol (2), 10α-hydroxy-Δ8-tetrahydrocannabinol (3), 10β-hydroxy-Δ8-tetrahydrocannabinol (4), 10α-hydroxy-Δ9,11-hexahydrocannabinol (5), 9β,10β-epoxyhexahydrocannabinol (6), and 11-acetoxy-Δ9-tetrahydrocannabinolic acid A (7). The binding affinity of isolated compounds 1–8, Δ9-tetrahydrocannabinol, and Δ8-tetrahydrocannabinol toward CB1 and CB2 receptors as well as their behavioral effects in a mouse tetrad assay were studied. The results indicated that compound 3, with the highest affinity to the CB1 receptors, exerted the most potent cannabimimetic-like actions in the tetrad assay, while compound 4 showed partial cannabimimetic actions. Compound 2, on the other hand, displayed a dose-dependent hypolocomotive effect only.
Graphical abstract
Many natural product classes besides the cannabinoids have been identified from Cannabis sativa L. (Cannabaceae), including monoterpenes, sesquiterpenes, flavonoids, steroids, and nitrogenous compounds. However, the C21 terpenophenolics (cannabinoids) are characteristic structures found in C. sativa. To date, more than 750 constituents have been identified from cannabis, out of which 104 are classified as cannabinoids.1–14
The medicinal properties of cannabis have been much debated from scientific and political points of view, and the subject has gained much interest over the years. After the discovery of the primary active constituent of marijuana, (−)-trans-Δ9-tetrahydrocannabinol (Δ9-THC), in 1964,15 various clinical trials were undertaken to determine its efficacy as an analgesic,16 antiemetic,17 antidepressant,18 and appetite suppressant/stimulant,19 as a treatment of glaucoma,20 and for the management of chemotherapy-induced nausea and vomiting.21 Research in the area of the use of C. sativa for medicinal purposes has gained worldwide interest following the discovery of the endogenous endocannabinoid system that interacts with the constituents of this plant.
Features of this endogenous system include cannabinoid receptors, of which there are two types reported (CB1 and CB2), and endogenous ligands that act as receptor agonists and antagonists.22,23 The CB1 receptors, recognized by the cannabinoids, are found in the brain and peripheral tissue of the central nervous system (CNS),24 while CB2 receptors are found primarily outside the CNS in tissues associated with the immune system of the body.25
The availability of high-potency marijuana with unprecedented Δ9-THC concentrations (>20% by dry weight)26 has afforded an opportunity to discover novel constituents from C. sativa. As part of a program to study the constituents of high-potency cannabis and its pharmacological effects,2–5,7,13 reported herein are the isolation and structure elucidation of eight minor cannabinoid constituents, including seven new compounds (1–7) and one known compound (8). Binding affinities of these compounds to both CB1 and CB2 receptors as well as their pharmacological evaluation in a mouse tetrad assay are described.
RESULTS AND DISCUSSION
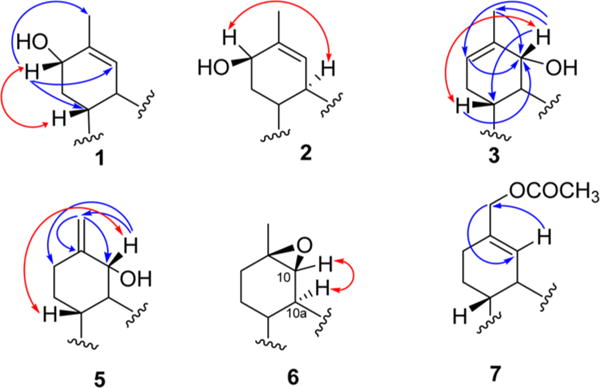
Compound 1 was isolated as a yellow oil. Its molecular formula was determined as C21H30O3 from the positive-mode HRESIMS ion at m/z 331.2262 [M + H]+ and from the GC-MS and 13C NMR spectra. The 1H NMR spectrum (Table 1) of 1 displayed four methyl signals (δH 0.87, 1.09, 1.42, and 1.80), two aromatic proton signals (δH 6.11 and 6.27), five methylenes, and three methines. The 13C NMR spectrum (Table 2) displayed 21 carbon resonances, in good agreement with Δ9-THC.3,27 The presence of an oxymethine carbon at δC 68.8 corresponding to a proton resonance at δH 4.10 in the HMQC spectrum indicated that 1 is a hydroxylated Δ9-THC derivative. The location of the hydroxy group was determined at C-8 based on the HMBC correlations of H-8 (δH 4.10) with C-6a (δC 40.8), C-11 (δC 20.8), and C-10 (δC 128.6) (Figure 1). The α-orientation of the hydroxy group at C-8 was assigned from the ROESY correlation of H-8 with H-6a (δH 1.81) (Figure 1). On the basis of these experiments, the chemical structure of 1 was elucidated as 8α-hydroxy-Δ9-tetrahydrocannabinol.
Table 1.
1H NMR Spectroscopic Data for Compounds 1–7 in CDCl3.
| position | 1
|
2
|
3
|
4
|
5
|
6
|
7
|
|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | |
| 2 | 6.11, s | 6.15, s | 6.12 s | 6.12 s | 6.23, s | 6.25, s | |
| 4 | 6.27, s | 6.24, s | 6.30 s | 6.27, s | 6.15, s | 6.25, s | 6.25, s |
| 6a | 1.81, m | 1.80, m | 1.85, m | 1.85, m | 1.75, dt (10.6, 3.6) | 1.85 m | 1.62 m |
| 7 | 2.41, m | 2.41, m | 2.10, m | 2.38, m | 1.86, m | 1.56, m | 1.94, m 1.35, m |
| 8 | 4.10, d (4.8) | 4.31, bt | 5.68, d (5.6) | 5.62, bs | 2.21, m | 1.82, m | 2.26, m |
| 10 | 6.66, bs | 6.54, bs | 4.27, d (8.4) | 5.08, d (2.4) | 4.22, d (8.8) | 3.88, bs | 6.79, s |
| 10a | 3.11, d (11.2) | 3.30, d (10) | 2.77, t (8.1) | 2.70, m | 2.69, t (10.2) | 2.79, d (12.0) | 3.27, m |
| 11 | 1.80 s | 1.76 s | 1.76 s | 1.84 s | 5.10 bs | 1.36 s | 4.48 s |
| 4.94 bs | |||||||
| 12 | 1.42, s | 1.39, s | 1.37, s | 1.40, s | 1.35, s | 1.39, s | 1.45, s |
| 13 | 1.09, s | 1.09, s | 1.04, s | 1.11, s | 1.01, s | 1.03, s | 1.11, s |
| 1′ | 2.41, dd (6.4, 8.4) | 2.41, dd (6, 8.4) | 2.42, m | 2.40, m | 2.43, dd (8.4, 6.0) | 2.44, m | 2.93, m |
| 2′ | 1.55, m | 1.54, m | 1.55, m | 1.55, m | 1.56, m | 1.54, m | 1.58, m |
| 3′ | 1.29, m | 1.29, m | 1.29, m | 1.29, m | 1.30, m | 1.31, m | 1.23, m |
| 4′ | 1.29, m | 1.29, m | 1.29, m | 1.29, m | 1.30, m | 1.31, m | 1.23, m |
| 5′ | 0.87, t (6.8) | 0.87, t (7.1) | 0.89, t (6.8) | 0.89, t (6.8) | 0.87, t (6.4) | 0.88, t (6.8) | 0.89, t (6.9) |
| OCOCH3 | 2.07, s |
Table 2.
13C NMR Spectroscopic Data for Compounds 1‒8 in CDCl3
| position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| 1 | 155.1 | 154.9 | 156.2 | 154.8 | 156.4 | 155.0 | 164.7 | 156.9 |
| 2 | 107.9 | 107.9 | 109.9 | 107.8 | 110.3 | 107.9 | 102.7 | 108.5 |
| 3 | 143.3 | 143.2 | 144.0 | 143.6 | 144.1 | 143.7 | 147.5 | 143.4 |
| 4 | 110.4 | 110.1 | 110.1 | 110.5 | 110.1 | 109.3 | 112.8 | 110.3 |
| 5 | 154.4 | 154.5 | 155.1 | 156.7 | 155.1 | 155.0 | 159.8 | 154.9 |
| 6 | 77.0 | 76.7 | 75.3 | 76.6 | 76.2 | 77.3 | 78.9 | 77.1 |
| 6a | 40.8 | 46.1 | 45.0 | 37.4 | 46.0 | 44.5 | 45.3 | 39.7 |
| 7 | 35.7 | 36.2 | 28.1 | 28.2 | 27.3 | 22.9 | 24.6 | 23.3 |
| 8 | 68.8 | 72.4 | 125.6 | 124.0 | 30.7 | 31.1 | 29.9 | 33.9 |
| 9 | 134.6 | 135.7 | 135.1 | 135.3 | 148.8 | 59.9 | 132.5 | 77.1 |
| 10 | 128.6 | 128.4 | 77.5 | 68.3 | 79.1 | 65.4 | 129.5 | 73.1 |
| 10a | 34.7 | 34.4 | 41.0 | 38.5 | 41.7 | 36.4 | 33.7 | 36.4 |
| 10b | 107.8 | 108.4 | 108.9 | 106.5 | 109.1 | 107.2 | 109.0 | 107.0 |
| 11 | 20.8 | 19.7 | 19.5 | 21.8 | 110.4 | 24.2 | 68.9 | 28.0 |
| 12 | 27.7 | 27.6 | 27.9 | 27.8 | 28.0 | 27.9 | 27.6 | 28.6 |
| 13 | 19.9 | 19.6 | 18.7 | 19.2 | 18.8 | 19.2 | 19.7 | 19.1 |
| 1′ | 35.7 | 35.7 | 35.6 | 35.6 | 35.6 | 35.9 | 35.6 | 35.7 |
| 2′ | 30.9 | 30.9 | 30.7 | 30.7 | 30.4 | 30.9 | 30.4 | 30.8 |
| 3′ | 31.7 | 31.7 | 31.8 | 31.9 | 31.8 | 31.8 | 31.8 | 31.9 |
| 4′ | 22.7 | 22.7 | 22.8 | 22.8 | 22.8 | 22.7 | 22.8 | 22.8 |
| 5′ | 14.3 | 14.3 | 14.3 | 14.3 | 14.3 | 14.3 | 14.3 | 14.2 |
| OCOCH3 | 21.3 | |||||||
| OCOCH3 | 171.5 | |||||||
| COOH | 176.5 |
Figure 1.
Key HMBC (→) and ROESY (↔) correlations compounds 1–3 and 5‒7.
Compound 2 was found to possess the same molecular formula as 1, C21H30O3, and was obtained as a yellow oil. The 1H and 13C NMR data of 2 (Tables 1 and 2) were quite similar to those of 1 except for the higher frequency shifts of H-8 (δH 4.31) and C-8 (δC 72.4), indicating the β-orientation of the hydroxy group at C-8, which was confirmed by the ROESY correlation of H-8 and H-10a (Figure 1). Thus, the structure of 2 was established as 8β-hydroxy-Δ9-tetrahydrocannabinol.
Compound 3 was isolated as a yellow oil. The molecular formula was established as C21H30O3 based on the [M + H]+ ion peak at m/z 331.2266. The 1H and 13C NMR spectroscopic data of 3 (Tables 1 and 2) were similar to those reported for Δ8-THC28 but with an extra hydroxy group (δH 4.27, δC 77.5). The location of this hydroxy group was determined at C-10 based on the COSY correlation between H-10 (δH 4.27) and H-10a (δH 2.77), and this was confirmed by HMBC (H-10/C-6a, C-11, C-8; H-8/C-10; H3-11/C-10; H-6a/C-10) correlations (Figure 1). The configuration at C-10 was determined by a ROESY correlation between H-10 (δH 4.27) and H-6a (δH 1.85), which supported the structural assignment of 3 as 10α-hydroxy-Δ8-tetrahydrocannabinol.
Compound 4 was obtained as a yellow oil. Its NMR spectroscopic data (Tables 1 and 2) were quite similar to those of 3 except for the downfield shift of H-10 (δH 5.08) and the upfield shift of C-10 (δC 68.3). The upfield shift of C-10 was due to the syn relationship with the aryl moiety at C-10a, which indicated the β-orientation of the hydroxy group at C-10. This was confirmed by a small coupling constant (2.4 Hz) between H-10 and H-10a and ROESY correlations between H-10 and H-10a. The structure of 4 was thus established as 10β-hydroxy-Δ8-tetrahydrocannabinol.
Compound 5 was obtained as a yellow resin with a molecular formula of C21H30O3, as indicated by the HRESIMS (m/z 331.2266, [M + H]+). The 1D NMR data were found to be closely similar to those of 3, with the only difference being the presence of signals for an exocyclic methylene (δH 4.94, 5.10, 1H each, s; δC 110.4, 148.8) instead of a double bond between C-8 and C-9 (Tables 1 and 2). This was supported by the HMBC correlations between H2-11 (δH 4.94, 5.10) and C-9 (δC 148.8), C-10 (δH 79.1), and C-8 (δH 30.7). The NOESY correlations between H-10 (δH 4.22) and H-6a (δH 1.74) (Figure 1) indicated the α-orientation of the hydroxy group at C-10. Hence, the structure of compound 5 was determined as 10α-hydroxy-Δ9,11-hexahydrocannabinol.
Compound 6 was isolated as a yellow oil. The molecular formula was determined as C21H30O3 from its HRESIMS [M + H]+ ion at m/z 331.2256, indicating seven degrees of unsaturation. The 1H, 13C, DEPT, and HMQC NMR spectroscopic data displayed four methyls, six methylenes, and five methines (Tables 1 and 2). The spectroscopic data were similar to those of Δ9-THC,3,27 except for the epoxidation of the olefinic group at C-9 and C-10 [δH 3.88 (H-10, s), δC 65.4 (C-10) and δC 59.9 (C-9)]. The ROESY correlation between H-10 (δH 3.88) and H-10a (δH 2.78) was used to determine the orientation of the epoxy moiety as β (Figure 1). Therefore, compound 6 was identified as 9β,10β-epoxyhexahydrocannabinol.
Compound 7 was isolated as a brown oil. Its HRESIMS exhibited a sodiated molecular ion peak at m/z 439.2092 [M + Na]+, corresponding to the molecular formula C24H32O6Na (calcd m/z 439.2097). Thus, 7 was assigned with eight degrees of unsaturation. Its IR spectrum showed strong absorptions at νmax 1734 and 1644 cm−1, indicative of ester and hydrogen-bonded carbonyl groups, respectively. The spectroscopic data of 7 were similar to those of Δ9-tetrahydrocannabinolic acid A,3,27 with the absence of the CH3-11 signals and the presence of an acetoxy methyl moiety instead [δH 2.07 (H3, s), 4.48 (H2, s); δC 21.3, 68.9, and 176.5]. The J3-HMBC correlation between H-10 (δH 6.79) and the oxymethylene carbon at δC 68.9 as well as that between oxymethylene protons (δH 4.48, H2-11) and C-10 (δC 129.5) (Figure 1) was used to place the acetoxy group at C-11. The structure of 7 was assigned as 11-acetoxy-Δ9-tetrahydrocannabinoic acid A.
Compound 8 was identified as cannabiripsol by comparing its specific rotation, GC-MS, and 1H NMR data with literature values,28 but this is the first time its 13C NMR spectroscopic data (Table 2) have been reported.
Compounds 1, 2, and 6 were previously reported as metabolites of Δ9-THC, and they have been synthesized, but their identification was established by GC and 1H NMR spectroscopy only.29–31 Compounds 3 and 5 were synthesized by Pitt et al. in 1975, but their chemical structures were determined only by 1H NMR spectroscopy.32 This is the first time that compounds 1–3 and 5 and 6 have been isolated from a natural source and have had their full spectroscopic data reported.
Cell lines were established expressing either the full-length human cannabinoid-1 receptor or the full-length human cannabinoid-2 receptor. Radioligand binding assays were performed to assess the affinity of these hydroxylated cannabinoid compounds.33 Most of the tested compounds exhibited a submicromolar affinity toward the receptors and, in some cases, demonstrated a several-fold selectivity toward the CB-1 receptor. Of these, 10-α-hydroxy-Δ8-tetrahydrocannabinol (3) was found to bind to both the cannabinoid receptors nearly as tightly as THC and is a partial agonist at both receptors (Table 3).
Table 3.
Binding Affinities of Compounds 1–8, Δ9-THC, and Δ8-THC
| compound | binding affinity (nM)
|
|
|---|---|---|
| CB1 | CB2 | |
| 1 | 1906 ± 578 | 3219 ± 876 |
| 2 | 65 ± 16 | 88 ± 19 |
| 3 | 31 ± 6 | 30 ± 4 |
| 4 | 830 ± 94 | 3274 ± 515 |
| 5 | 117 ± 16 | 129 ± 13 |
| 6 | 224 ± 20 | 335 ± 36 |
| 7 | 47 ± 18 | 912 ± 77 |
| 8 | 5668 ± 1324 | 2143 ± 353 |
| Δ8-THC | 78 ± 5 | 12 ± 2 |
| Δ9-THC | 18 ± 4 | 42 ± 9 |
The mouse tetrad assay is a four-point behavioral assay that characterizes the effect on locomotor activity, catalepsy, body temperature, and nociception. The neurobehavioral effects of Δ9-THC in the mouse tetrad assay are well established and are typically referred to as classical cannabimimetic action. These activities manifest as a reduction in locomotor activity, catalepsy, hypothermia, and antinociceptive effects.34–36 The results shown in Table 4 indicate that compounds 3 and 6 showed the most potent cannabimimetic-like actions in this mouse tetrad assay. Both compounds exerted significant hypolocomotive, cataleptic, and hypothermic as well as antinociceptive effects in both the tail-flick and hot-plate assays. On the other hand, compound 2 lacked typical cannabimimetic-like action at doses up to 20 mg/kg, but exhibited a significant dose-dependent hypolocomotive activity. Interestingly, compound 4 administration resulted in hypothermic and antinociceptive activity at 40 mg/kg, while a significant locomotor stimulant effect was produced at a 10 mg/kg dose.
Table 4.
Behavioral Effects of the Intraperitoneal Administration of Selected Cannabinoids in a Mouse Tetrad Assay
| treatment group (mg/kg) | locomotor activity | catalepsy latency (s) | tail-flick latency (% MPE) | hot-plate latency (% MPE) | decrease in rectal temperature (°C) |
|---|---|---|---|---|---|
| vehicle | 2091 ± 209.3 | 2.1 ± 0.60 | 0.84 ± 3.58 | 8.32 ± 2.20 | 0.58 ± 0.26 |
| Δ9-THC | |||||
| 10 | 190.9 ± 33.8c | 14.4 ± 4.16 | 6.52 ± 2.00 | 29.04 ± 5.37c | 2.05 ± 0.25 |
| 20 | 276.2 ± 52.6c | 47.9 ± 20.04b | 42.21 ± 17.04b | 46.85 ± 9.08c | 4.25 ± 0.78c |
| 40 | 194.0 ± 49.2c | 37.7 ± 14.64a | 51.96 ± 13.22c | 38.18 ± 4.05c | 4.80 ± 0.28c |
| Δ8-THC | |||||
| 5 | 1480 ± 184.3 | 1.6 ± 0.60 | 8.01 ± 5.88 | 12.82 ± 1.84 | 0.12 ± 0.09 |
| 20 | 377.4 ± 169.7c | 5.3 ± 2.68 | 5.97 ± 1.63 | 16.98 ± 3.53 | 1.96 ± 0.37a |
| compound 1 | |||||
| 20 | 908.90 ± 245.10 | 1.50 ± 0.33 | 5.01 ± 3.59 | 4.95 ± 1.89 | 0.58 ± 0.18 |
| compound 2 | |||||
| 10 | 1617 ± 364.50 | 1.143 ± 0.14 | 0.13 ± 3.50 | 11.83 ± 3.16 | 0.49 ± 0.49 |
| 20 | 1148 ± 242.60a | 1.43 ± 0.29 | 1.62 ± 3.92 | 2.04 ± 2.48 | 0.50 ± 0.31 |
| 40 | 777.40 ± 185.10b | 11.43 ± 3.92 | 12.69 ± 6.62 | 11.79 ± 2.48 | 0.91 ± 0.18 |
| compound 3 | |||||
| 5 | 318.1 ± 96.29c | 22.0 ± 12 | 37.40 ± 12.62a | 21.04 ± 6.27 | 2.46 ± 0.52b |
| 10 | 288.3 ± 98.7c | 23.9 ± 6.71 | 31.46 ± 12.64 | 56.33 ± 8.61c | 3.21 ± 0.3c |
| 20 | 301.9 ± 62.9c | 51.1 ± 18.59a | 90.16 ± 6.16c | 47.63 ± 7.80c | 5.38 ± 0.38c |
| compound 4 | |||||
| 10 | 3045 ± 266.3a | 1.4 ± 0.31 | 2.20 ± 1.16 | 6.819 ± 3.18 | 0.47 ± 0.18 |
| 40 | 2537 ± 316.1 | 13.70 ± 4.55 | 69.95 ± 14.02c | 55.65 ± 7.74c | 5.32 ± 0.29c |
| compound 6 | |||||
| 5 | 213.40 ± 49.77c | 23.80 ± 10.55a | 16.25 ± 7.08 | 36.98 ± 8.87a | 3.75 ± 0.42c |
| 10 | 291.20 ± 69.89c | 8.80 ± 3.39 | 82.71 ± 11.31c | 35.89 ± 7.56a | 3.11 ± 0.25c |
| 20 | 380.30 ± 109.70c | 27.50 ± 7.83a | 50.37 ± 15.99b | 49.39 ± 13.81b | 4.58 ± 0.65c |
| compound 8 | |||||
| 20 | 1892 ± 121.20 | 2.25 ± 1.25 | 2.98 ± 2.36 | 2.40 ± 6.71 | 0.45 ± 0.20 |
< 0.05.
< 0.01.
< 0.001 versus vehicle (Dunnett’s posthoc test).
The data collected from the in vivo tetrad activity assay (Table 4) correlated with the order of in vitro binding affinities of the isolated compounds with the CB1 receptors (Table 3). Thus, compound 3 showed typical cannabimimetic actions in vivo and possessed the highest binding affinities to the CB1 and CB2 receptors, with Ki values of 31.0 ± 6.0 and 30.0 ± 4.0 nM, respectively.33 Additionally, compound 3 proved to act as a partial CB1 agonist in functional assays, similar to the classical cannabinoid, Δ9-THC. In contrast, compound 2 lacked any cannabimimietic activity at the doses tested and showed poor binding affinity to the CB1 receptors. On the other hand, compound 4 showed a partial cannabimimetic action and at the highest tested dose (40 mg/kg) exerted only antinociceptive and hypothermic actions. While compound 1 showed a binding affinity to the CB1 (Ki = 1.91 ± 0.58 μM) receptor, it did not exhibit typical cannabimimetic activity in the mouse tetrad assay at a dose of 20 mg/kg. Additional quantities of this compound need to be secured by isolation or synthetic techniques in order to further evaluate its functional activity at the CB1 receptor as well as to characterize its full in vivo dose–response activity. It is crucial to determine if compound 1 acts as an agonist or antagonist of CB1. An antagonist effect would result in the lack of cannabimimetic activity in the tetrad assay, while an antagonist effect would block an agonist effect (e.g., Δ9-THC) in this four-point behavioral assay. In addition, a full receptor binding panel evaluation should be conducted to determine if compound 1 interacts with other CNS receptors that might mask the cannabimimetic activity inferred by CB1 binding. Future binding studies of the isolated compounds to targets other than the CB1 and CB2 receptors, like the transient receptor potential (TRP) channels of both the vanilloid type-3 (TRPV3) and the ankyrin type-1 (TRPA1), are needed to better establish the mechanism and potential value of the pharmacological effects observed in this study.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were measured on an Autoplot IV automatic polarimeter. IR spectra were recorded on a Bruker Tensor 27 IR spectrometer. 1H NMR (400 MHz), 13C NMR (100 MHz), DEPT-135, APT, and 2D NMR spectra were recorded using the residual solvent signal as an internal standard on a Varian AS 400 NMR spectrometer. High-resolution mass spectra were measured using a Bruker BioApex instrument. HPLC was performed on a Waters Delta Prep 4000 preparative chromatography system connected to a Waters 486 tunable UV absorbance detector using Phenomenex Luna C18 and Si gel columns (250 × 21.2 mm, 5 μm, 100 Å). GC-MS analysis was carried out on an HP 6890 series GC, equipped with a split/splitless capillary injector, an HP 6890 series injector autosampler, and an Agliant DB-5 ms column (30 m × 0.25 mm × 0.25 μm), interfaced to an HP 5973 mass selective detector. The injector temperature was 250 °C, and 1 μL injections were performed in the splitless mode, with the splitless time set at 60 s, the split flow set at 50 mL/min, and the septum purge valve set to close 60 s after the injection occurred. The oven temperature was raised from 70 to 270 °C (held for 20 min) at a rate of 5 °C/min, for a total run time of 60 min; the transfer line temperature was 280 °C. Chemicals and reagents for pharmacological studies were obtained from Sigma-Aldrich (St. Louis, MO, USA) except fetal bovine serum (Midwest Scientific, Valley Park, MO, USA) and Biorad Bradford dye (Fisher Scientific, Pittsburgh, PA, USA).
Plant Material
Cannabis sativa plants were grown from high-potency Mexican seeds at The University of Mississippi growing field (University, MS, USA) and were harvested in December 2007. The plants were authenticated by Dr. Suman Chandra at The University of Mississippi, and a voucher specimen (S1310 V1) has been deposited at the Coy Waller Complex, National Center for Natural Products Research, School of Pharmacy, The University of Mississippi.
Extraction and Isolation
The dried plant material (9.0 kg) was extracted sequentially with hexanes (48 L), CH2Cl2 (40 L), EtOAc (40 L), EtOH (40 L), EtOH−H2O (36 L, 1:1), and H2O (40 L) at room temperature. The extracts were evaporated under reduced pressure at 40 °C to afford hexanes (1.48 kg), CH2Cl2 (0.15 kg), EtOAc (0.13 kg), EtOH (0.09 kg), EtOH−H2O (0.77 kg), and H2O (0.54 kg) extracts, for a total extract weight of 3.16 kg (35.1%, w/w). Portions of the CH2Cl2, EtOAc, and EtOH extracts were combined (191.0 g) since they showed similar TLC profiles (EtOAc-n-hexane, 4:6) and were subjected to silica gel VLC, eluting with EtOAc-n-hexane [0:100, 10:90, 20:80, 30:70, 40:60, 50:50, 75:25, 100:0 (2 L of each mixture)] followed by EtOH (4 L), yielding nine pooled fractions (A–I). Fraction D (14.3 g) was fractionated on a Si gel column using EtOAc−petroleum ether (5:95 to 50:50) for elution to afford four fractions (D1−D4). Fraction D2 (2 g) was subjected to passage over a C18 flash column (MeOH−H2O, 6:4), followed by a C18-SPE column, and was finally purified by Si gel HPLC (EtOAc-n-hexane, 10:90) to give 3 (14.0 mg, tR 15.0 min), 4 (25.0 mg, tR 12.3 min), and 5 (11.9 mg, tR 11.3 min). Compound 7 (9.8 mg) was isolated from fraction D3 (626 mg) by repeated C18 flash column chromatography (MeCN−H2O, 70–100%), passage over Sephadex LH-20 (MeOH), and Si gel solid-phase extraction (MeOH−CH2Cl2, 0–10%). Fraction G (6.95 g) was subjected to Si gel column chromatography using a stepwise gradient system using mixtures of EtOAc-n-hexane (0:100 to 100:0), giving 13 fractions (G1−G13). Fraction G11 (2.78 g) was further purified by passage over Sephadex LH-20 eluted with MeOH, followed by reversed-phase HPLC 12.9 min), 6 (21.2 mg, tR 13.2 min), and 8 (35.5 mg, tR 9.1 min).
8α-Hydroxy-Δ9-tetrahydrocannabinol (1): yellow oil; [α]25D −56.6 (c 0.21, CHCl3); 1H NMR and 13C NMR, see Tables 1 and 2; HRESIMS m/z 331.2262 [M + H]+ (calcd for C21H31O3, 331.2273); GC-MS m/z 330 (M+, 35%), 315 (16%), 297 (16%), 271 (100%).
8β-Hydroxy-Δ9-tetrahydrocannabinol (2): yellow oil; [α]25D −122.8 (c 0.17, CHCl3); 1H NMR and 13C NMR, see Tables 1 and 2; HRESIMS m/z 331.2274 [M + H]+ (calcd for C21H31O3, 331.2273); GC-MS m/z 330 (M+, 35%), 315 (51%), 312 (98%), 297 (89%), 271 (100%), 214 (42%).
10α-Hydroxy-Δ8-tetrahydrocannabinol (3): yellow oil; [α]25D −29.6 (c 0.34, CHCl3); 1H NMR and 13C NMR, see Tables 1 and 2; HRESIMS m/z 331.2266 [M + H]+ (calcd for C21H31O3, 331.2273); GC-MS m/z 330 (M+, 20%), 312 (85%), 297 (100%), 257 (26%), 231 (70%), 214 (55%).
10β-Hydroxy-Δ8-tetrahydrocannabinol (4): yellow oil; [α]25D −78.7 (c 0.59, CHCl3); 1H NMR and 13C NMR, see Tables 1 and 2; HRESIMS m/z 331.2268 [M + H]+ (calcd for C21H31O3, 331.2273); GCMS m/z 330 (M+, 8%), 312 (100%), 297 (98%), 269 (16%), 257 (30%), 231 (68%), 214 (75%).
10α-Hydroxy-Δ9,11-hexahydrocannabinol (5): yellow oil; [α]25D −99.1 (c 0.43, CHCl3); 1H NMR and 13C NMR, see Tables 1 and 2; HRESIMS m/z 331.2266 [M + H]+ (calcd for C21H31O3, 331.2273).
9β,10β-Epoxyhexahydrocannabinol (6): yellow oil; [α]25D −36.6 (c 0.06, CHCl3); 1H NMR and 13C NMR, see Tables 1 and 2; HRESIMS m/z 331.2256 [M + H]+ (calcd for C21H31O3, 331.2273); GC-MS m/z 330 (M+, 28%), 315 (50%), 297 (24%), 271 (100%), 231 (25%), 193 (10%).
11-Acetoxy-Δ9-tetrahydrocannabinolic acid A (7): brown oil; [α]25D −150.0 (c 0.51, CHCl3); IR (neat) νmax 2927, 1734, 1644, 1615, 1565, 1466 cm−1; 1H NMR and 13C NMR, see Tables 1 and 2; HRESIMS m/z 439.2092 [M + Na]+ (calcd for C23H32O6Na, 439.2097).
Cannabiripsol (8): white powder; [α]25D 129 (c 0.09, CHCl3); 1H NMR, consistent with literature values;28 13C NMR data, see Table 2; HRESIMS m/z 349.2393 [M + H]+ (calcd for C21H33O4 349.2379).
CB1 and CB2 Receptor Radioligand Binding Assays
The CB1 and CB2 receptor radioligand binding assays of the isolated compounds were performed according to a previously reported method.33
Animals
Experiments were performed using 8-week-old mice. Male Swiss Webster mice (Harlan, Indianapolis, IN, USA), weighing 24–30 g at the time of testing, were used. The mice were housed in groups of five with a 12 h light/12 h dark cycle. Food and water were provided ad libitum. All mice were selected randomly for each treatment group. Procedures involving animals were performed according to the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) at The University of Mississippi and according to the National Institutes of Health Guide Care and Use of Laboratory Animals (IACUC number 07-018).
Mouse Tetrad Assay
Twenty-four hours prior to testing, animals were acclimated to the experimental setting (ambient temperature, 22− 24 °C, hot-plate chamber, and rectal probe insertion). On the day of testing, preinjection control values for rectal temperature, catalepsy, tail-flick, and hot-plate latencies were determined. Animals were then injected intraperitoneally (ip) with either the vehicle control, Δ9-THC (10–40 mg/kg), or test compound (5–40 mg/kg). Following ip injection, animals were placed individually in activity chambers (San Diego Instruments, San Diego, CA, USA), where the locomotor activity was monitored automatically for 30 min. Total activity was expressed as the total number of interruptions of 16-cell photobeams per chamber. The activity for the last 10 min was recorded for analysis. Animals were then placed on a ring immobility apparatus, and the latency to drop was recorded in seconds with a maximum cutoff of 180 s latency. Rectal temperature was recorded using a digital rectal probe (Physitemp Instruments, Clifton, NJ, USA) and was expressed as the difference between basal and postinjection temperatures. Tail-flick and hot-plate latencies were measured with a maximum tail-flick latency of 15 s and hot-plate latency of 45 s to avoid tissue damage.
Data Analysis
All values were presented as means ± SEM with n = 6–8 animals per group. Antinociception was expressed as the percent maximal effect (% MPE = [(post drug latency − basal latency)/(cutoff latency − basal)] × 100). All data were analyzed using one-way ANOVA followed by a Dunnett’s post hoc test to determine significant differences from the vehicle control at p < 0.05.
Supplementary Material
Acknowledgments
This project was supported by grant no. 5P20RR021929-02 from the National Center for Research Resources and in part by the National Institute on Drug Abuse, contract no. N01DA-5-7746. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. We thank Dr. G. Ma for help with the CHO transfection experiments.
Footnotes
Supporting Information
1H NMR, 13C NMR, and DEPT-135 spectra for the isolated compounds (1–8). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.5b00065.
Notes
The authors declare no competing financial interest.
NOTE ADDED AFTER ASAP PUBLICATION
This paper was published on the Web on May 22, 2015, with errors in the Table of Contents graphic and the Abstract graphic. The corrected version was reposted on May 29, 2015.
References
- 1.ElSohly MA, Slade D. Life Sci. 2005;78:539–548. doi: 10.1016/j.lfs.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed SA, Ross SA, Slade D, Radwan MM, Khan IA, ElSohly MA. Tetrahedron Lett. 2008;49:6050–6053. doi: 10.1016/j.tetlet.2008.07.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed SA, Ross SA, Slade D, Radwan MM, Zulfiqar F, ElSohly MA. J Nat Prod. 2008;71:536–542. doi: 10.1021/np070454a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radwan MM, ElSohly MA, Slade D, Ahmed SA, Wilson L, El-Alfy AT, Khan IA, Ross SA. Phytochemistry. 2008;69:2627–2633. doi: 10.1016/j.phytochem.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radwan MM, Ross SA, Slade D, Ahmed SA, Zulfiqar F, ElSohly MA. Planta Med. 2008;74:267–272. doi: 10.1055/s-2008-1034311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Appendino G, Giana A, Gibbons S, Maffei M, Gnavi G, Grassi G, Sterner O. Nat Prod Commun. 2008;12:1977–1980. [Google Scholar]
- 7.Radwan MM, ElSohly MA, Slade D, Ahmed SA, Khan IA, Ross SA. J Nat Prod. 2009;72:906–911. doi: 10.1021/np900067k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qian S, Cai G, He G, Du FL. Nat Prod Res Dev. 2009;21:784–786. [Google Scholar]
- 9.Cheng L, Kong D, Hu G, Li H. Chem Nat Compd. 2010;46:710–712. [Google Scholar]
- 10.Taglialatela-Scafati O, Pagani A, Scala F, De Petrocellis L, Di Marzo V, Grassi G, Giovanni G, Appendino G. Eur J Org Chem. 2010;11:2067–2072. [Google Scholar]
- 11.Pagani A, Scala F, Chianese G, Grassi G, Appendino G, Taglialatela-Scafati O. Tetrahedron. 2011;67:3369–3373. [Google Scholar]
- 12.Pollastro F, Taglialatela-Scafati O, Allara M, Munoz E, Di Mazo V, De Petrocellis L, Appendino G. J Nat Prod. 2011;74:2019–2022. doi: 10.1021/np200500p. [DOI] [PubMed] [Google Scholar]
- 13.Zulfiqar F, Ross SA, Slade D, Ahmed SA, Radwan MM, Zulfiquar A, Khan IA, ElSohly MA. Tetrahedron Lett. 2012;53:3560–3562. doi: 10.1016/j.tetlet.2012.04.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.ElSohly MA, Gul W. In: Handbook of Cannabis. Pertwee RG, editor. Oxford University Press; Oxford, UK: 2014. pp. 3–22. Chapter 1. [Google Scholar]
- 15.Gaoni Y, Mechoulam R. J Am Chem Soc. 1964;86:1646–1647. [Google Scholar]
- 16.Roberts JD, Gennings C, Shih M. Eur J Pharmacol. 2006;530:54–58. doi: 10.1016/j.ejphar.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 17.Darmani NA, Johnson JC. Eur J Pharmacol. 2004;488:201–212. doi: 10.1016/j.ejphar.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 18.Sofia RD, Kubena RK, Barry H. Psychopharmacologia. 1973;31:121–130. doi: 10.1007/BF00419812. [DOI] [PubMed] [Google Scholar]
- 19.Mannila J, Jaervinen T, Jaervinen K, Tervonen J, Jarho P. Life Sci. 2006;78:1911–1914. doi: 10.1016/j.lfs.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 20.Plange N, Arend KO, Kaup M, Doehmen B, Adams H, Hendricks S, Cordes A, Huth J, Sponsel WE, Remky A. Am J Ophthalmol. 2007;143:173–174. doi: 10.1016/j.ajo.2006.07.053. [DOI] [PubMed] [Google Scholar]
- 21.Parker LA, Kwiatkowska M, Mechoulam R. Physiol Behav. 2006;87:66–71. doi: 10.1016/j.physbeh.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 22.Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 23.Munro S, Thomas KL, Abu-Shaar M. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 24.Barth F. Annu Rep Med Chem. 2005;40:103–118. [Google Scholar]
- 25.Ashton JC, Giass M. Curr Neuropharmacol. 2007;5:73–80. doi: 10.2174/157015907780866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.ElSohly MA, Ross SA, Mehmedic Z, Arafat R, Yi B, Banahan BF. J Forensic Sci. 2002;45:24–30. [PubMed] [Google Scholar]
- 27.Choi YH, Hazekamp A, Peltenburg-Looman AMG, Frederich M, Erkelens C, Lefeber AWM, Verpoorte R. Phytochem Anal. 2004;15:345–354. doi: 10.1002/pca.787. [DOI] [PubMed] [Google Scholar]
- 28.Boeren EG, ElSohly MA, Turner CE. Experientia. 1979;35:1278–1279. doi: 10.1007/BF01963954. [DOI] [PubMed] [Google Scholar]
- 29.Pitt CG, Hauser F, Hawks RL, Sathe S, Wall ME. J Am Chem Soc. 1972;94:8578–8579. doi: 10.1021/ja00779a048. [DOI] [PubMed] [Google Scholar]
- 30.Mechoulam R, Varconi H, Ben-Zvi Z, Edery H, Grunfeld Y. J Am Chem Soc. 1972;94:7930–7931. doi: 10.1021/ja00777a049. [DOI] [PubMed] [Google Scholar]
- 31.Gurny O, Maynard DE, Pitcher RG, Kierstead RW. J Am Chem Soc. 1972;94:7928–7929. doi: 10.1021/ja00777a048. [DOI] [PubMed] [Google Scholar]
- 32.Pitt CG, Fowler MS, Sathe S, Srivastava SC, Williams DL. J Am Chem Soc. 1975;97:3798–3802. doi: 10.1021/ja00846a040. [DOI] [PubMed] [Google Scholar]
- 33.Husni AS, McCurdy CR, Radwan MM, Ahmed SA, Slade D, Ross SA, ElSohly MA, Cutler SJ. Med Chem Res. 2014;23:4295–4300. doi: 10.1007/s00044-014-0972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin BR, Welch SP, Abood M. Adv Pharmacol. 1994;25:341–397. doi: 10.1016/s1054-3589(08)60437-8. [DOI] [PubMed] [Google Scholar]
- 35.Varvel SA, Bridgen DT, Tao Q, Thomas BF, Martin BR, Lichtman AHJ. Pharmacol Exp Ther. 2005;314:329–337. doi: 10.1124/jpet.104.080739. [DOI] [PubMed] [Google Scholar]
- 36.Pertwee RG, Thomas A, Stevenson LA, Ross RA, Varvel SA, Lichtman AH, Martin BR, Razdan RK. Br J Pharmacol. 2007;150:586–594. doi: 10.1038/sj.bjp.0707124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.