

Abstract
Blue cone monochromacy (BCM) is caused by the lack of expression of the normal proteins encoded by the OPN1LW and OPN1MW genes, resulting in the absence of red and green cone sensitivities. We analyzed two cases of BCM in two different families and identified deletion mutations in the locus control region upstream of the two genes. Deletion breakpoints were determined to an accuracy of one base for both cases.
Blue cone monochromacy (BCM; OMIM #303700) is a rare congenital color vision disorder characterized by the absence of cone sensitivity to long (red) and medium (green) wavelengths in the retina.1 Clinical features of BCM include severely impaired color vision and reduced visual acuity (with preservation of only rod and blue cone functions). Photophobia, myopia and congenital nystagmus are also commonly observed.2
The OPN1LW and OPN1MW genes on the X chromosome encode the red and green opsins, respectively, and are thought to be responsible for BCM.3 In the wild-type genomic array of Xq28, the genes are located head-to-tail in a tandem arrangement with a single OPN1LW followed by one or more OPN1MW copies. The actual number of OPN1MW copies varies among individuals. Approximately 50% of Caucasian males have two copies of OPN1MWs (OPN1MW1 and OPN1MW2).1,3,4 The expression of these genes is regulated by the locus control region (LCR), a conserved sequence located upstream of the OPN1LW and OPN1MW gene cluster.5 The genes are encoded by six exons that show a high degree of homology (~98 and 96% identity at the nucleotide and amino acid levels, respectively).1,4
BCM is caused by discrete mutations in each of OPN1LW and OPN1MW genes or a single mutation within the LCR. In 40% of BCM cases, OPN1LW and OPN1MW were inactivated by a deletion in the LCR.6–8 In residual cases, nonhomologous recombination between the two gene loci results in single OPN1LW and/or OPN1LW/OPN1MW hybrid genes carrying deleterious point mutations. The most common genotype in such cases consists of a single OPN1LW/OPN1MW hybrid gene with a c.607T>C (p.C203R) mutation in OPN1LW.8,9
In this study, we performed a fine mutation analysis of the genomic organization at the OPN1LW and OPN1MW gene region in two Japanese families with BCM at a one base-level resolution to further understand the molecular event of the genomic deletion in the LCR.
This study was approved by the Institutional Review Board for Human Genetic and Genome Research at the Hamamatsu University School of Medicine and Nagoya University Graduate School of Medicine. All study procedures adhered to the tenets of the Declaration of Helsinki. Written informed consents were obtained from the patients of all cases before any study procedure or examination was performed.
The pedigree of family 1 is shown in Figure 1, and family 2 has been described previously.10 The characteristics of both families are consistent with an X-linked pattern of inheritance. Patients IDs BCM1 and BCM2 are probands of families 1 and 2, respectively. Patient BCM1, an 18-year-old boy, had complained of low visual acuity since childhood. His visual acuity was 0.4 oculus dexter and 0.4 oculus sinister. Although no remarkable findings were recognized by ophthalmoscopy, BCM was strongly suspected by elaborate color vision testing and full-field electroretinography. Genomic DNA was extracted from the peripheral lymphocytes of patients using standard procedures. To identify disease-causing mutations, we performed a mutation analysis according to the following 3 steps:
PCR amplification and sequencing the amplified products for various regions of the OPN1LW and OPN1MW genes to verify their existence and to detect possible sequence alteration;
‘Junction PCR’ to amplify and sequence the distant regions connected with each other after a possible large deletion for the region in which PCR amplification was unsuccessful;
Further analysis by genomic walking and cloning the unknown genomic fragments when the junction PCR was unsuccessful.
Figure 1.
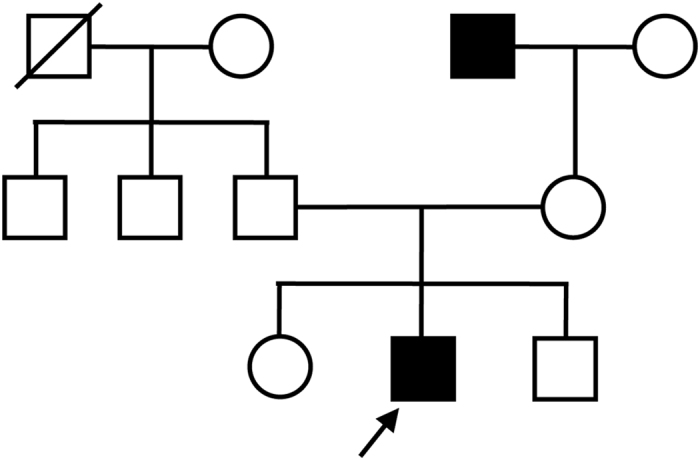
Pedigree of family 1. Square boxes and circles denote male and female members, respectively; black symbols indicate affected individuals; and slashed symbols indicate deceased individuals. The proband is indicated by an arrow.
For step 1, PCR primers were designed to specifically amplify the upstream regions of the OPN1LW gene (accession no. NM020061; Figure 2a,b and Supplementary Table). However, designing specific primers to individually amplify each exon of OPN1LW and OPN1MW (NM_000513 for OPN1MW1 and NM_001048181 for OPN1MW2) was difficult owing to the extremely high homology between these genes. Thus we designed common primers for each of the exons and employed them to co-amplify the exons from the genes. Only 3 exons (exons 2, 4 and 5) were selected because these could be distinguished by sequencing the amplified products (Figure 2a,b and Supplementary Table). A standard protocol11 was used for the direct sequencing. Step 2 was performed in cases when the PCR products were not amplified from patients’ DNA, suggesting that the genomic region concerned did not exist in the patient. After detecting large deletions and narrowing down the region by PCR and sequencing, junction PCR was performed using specific primer pairs designed to span the deletion that would work only when a deletion existed (Figure 2c,d). Step 3 involved PCR-based genomic walking using the GenomeWalker Universal kit (Takara Bio, Otsu, Japan). This kit was used according to the manufacturer’s instructions to determine the unknown DNA sequence.
Figure 2.

Gross determination of the deletion region in two Japanese families with blue cone monochromacy (BCM). (a) Genome structure of human OPN1LW and OPN1MWs. Red and green boxes represent OPN1LW and OPN1MWs exons, respectively; the black box represents the locus control region (LCR). (b) Target positions for examining the existence of each region; solid and broken lines represent unique and coexisting genomic regions, respectively. Each line was drawn along the same scale and position with the upper panel (Figure 2a). Exons 2, 4 and 5 were examined because they differ between OPN1LW and OPN1MWs. Furthermore, only exon 5 could be distinguished between OPN1MW1 and OPN1MW2. Because other exons (exons 1, 3 and 6) had the same sequence in these genes, they were only examined for possible small-scale sequence alterations, including point mutations (Supplementary Table). No such mutations were detected (see text). (c) Summary of PCR analysis of each region. +, PCR product was amplified from patients' DNA and confirmed as OPN1LW or OPN1MW; −, PCR product was not amplified from patients' DNA; #, PCR product was amplified from patients' DNA, but sequences representing OPN1LW exons were absent; ##, PCR product was amplified from patients' DNA, but sequences representing the OPN1MW1 exon was absent. In BCM1, the deletion size was estimated to be ~20 kb (yellow background). In BCM2, the deletion size was estimated to be 41 kb (gray background). The existence of exons 2 and 4 of OPN1MW1 and/or OPN1MW2 as well as exon 5 s of OPN1LW and OPN1MW2 suggested an additional complicated genomic rearrangement (see text). (d) Primer positions for junction PCR. Genomic structure of human OPN1LW. Red and black boxes represent OPN1LW exons and LCR, respectively. Solid arrowheads indicate forward and reverse primers used for junction PCR. The primers had the following sequences: BCM1, 5′- CACATGCGGGTGCCACTACA-3′ and 5′- TCCCTATGGGGAGAGCAGAC-3′; and BCM2, 5′- CTGCCCTGAAGAGAGAGGCT-3′ and 5′- GACCCAGGCATAGGGAGTTG-3′.
In the first step of the analysis of the two BCM cases, small-scale mutations such as a point mutation or a deletion/insertion of a few bases in the OPN1LW and OPN1MWs genes, and their upstream regions (including the LCR) were not detected, but long genomic deletions were suggested in both cases by the failure of PCR amplification using several primer sets (Figure 2c). The approximate deletion sizes were 20 kb in BCM1 and 41 kb in BCM2 (Figure 2c). We therefore proceeded to step 2. The junction PCR successfully identified the deletion breakpoint for BCM1, and the amplified PCR product was sequenced. We located the breakpoints as well as a 16,856-bp deletion and 53-bp insertion ( 5′-CGCTTGCACCCAGGAGGTGGAGGTTGCAGTGAGCCCAGATCGCGCCACTGTAC-3′) (Figure 3a). The proximal boundary of the deletion was 8,899 bp upstream of the OPN1LW translation initiation codon, and the distal boundary was 1,290 bp downstream of OPN1LW exon 2. We propose that this mutation be annotated as ‘c.-8899_IVS2+1290delins53 of OPN1LW’. Interestingly, the proximal boundary of the breakpoint was within an AluSz repeat sequence, and the 53-bp insertion was a partial Alu repeat sequence.
Figure 3.

Determination of the region surrounding the deletion breakpoints in two Japanese families with blue cone monochromacy (BCM). (a) Precise analysis of the deletion–insertion mutation by junction PCR and sequencing in case BCM1. Genomic structure of human OPN1LW. The red and black boxes represent OPN1LW exons and locus control region (LCR), respectively. BCM1 had a 16,856-bp deletion and 53-bp insertion. The proximal and distal boundaries of the deletion were 8,899 bp upstream of the OPN1LW translation initiation codon and within OPN1LW intron 2, respectively. The proximal boundary of the breakpoint was an AluSz repeat sequence, and the 53-bp insertion contained a partial Alu repeat sequence. (b) Precise analysis of the deletion–inversion–insertion mutation by genomic walking in case BCM2. Genomic structure of human OPN1LW and OPN1MW. Red and green boxes represent OPN1LW and OPN1MW exons, respectively; the black box represents the LCR. BCM2 had an 87,682-bp deletion, an inverted re-insertion of 328 bp that is a part of the deleted sequence and a 3-bp insertion. The proximal and distal boundaries of the deletion were 28,144 bp upstream of the OPN1LW initiation codon and 7764 bp downstream of the OPN1MW1 translation termination codon, respectively. In BCM2, exons 2 and 4 of OPN1LW were absent, whereas the presence of exon 5 in OPN1LW and OPN1MW2 were confirmed. It is possible that BCM2 harbored a normal OPN1MW2 as well as a hybrid gene comprising OPN1MW2 exons 1–4 joined to OPN1LW exons 5 and 6. The brackets indicate that this structure is one of possibilities including the order of the hybrid gene and OPN1MW2. (c) Diagram and electropherogram of the region surrounding deletion–inversion–insertion breakpoints in BCM2, which were located within highly repetitive sequences.
In the case of BCM2, the above-described method of determining the deletion region was unsuccessful, and the junction PCR primers failed to amplify a product. We hypothesized that this was due to the presence not only of a deletion but also an insertion of unknown sequence in this region. We therefore proceeded to step 3 and performed genomic walking to identify the unknown insertion sequence. We then isolated a DNA fragment that was rich in highly repetitive sequences, including AluSz (Figure 3b,c). BCM2 harbored a complex mutation with an 87,682-bp deletion, an inverted re-insertion of 328 bp that was a part of the deleted sequence and a 3-bp (GTG) insertion. The proximal and distal boundaries of the deletion were 28,144 bp upstream of the OPN1LW initiation codon and 7,764 bp downstream of the OPN1MW1 translation stop codon, respectively. The 328-bp inversion sequence was located adjacent to and downstream of the breakpoint 25,012–25,339 bp upstream of the OPN1LW translation initiation codon. The 3-bp (GTG) insertion was adjacent to and downstream of the inverted re-insertion. We defined the overall mutation notation as c.-28144_-25340del-25339_-25012inv328insGTG-25011_*7764del84549 of OPN1LW/OPN1MW1 in this study. The proximal and distal breakpoints were within the MER74B and L1M4 repetitive sequences, and the 328-bp inversion sequence included L1M4 and AluSz (Figure 3c). In BCM2, exons 2 and 4 of OPN1LW were absent, whereas exon 5 s of OPN1LW and OPN1MW2 were identified (see Figure 2 legend). This might suggest that BCM2 had normal OPN1MW2 but also a hybrid gene comprising OPN1MW2 exons 1–4 joined to OPN1LW exons 5 and 6. A possible genomic organization of BCM2 is shown in Figure 3b.
Thus, these 2 cases of BCM harbored large deletions including the LCR, but the most common mutation p.C203R was not detected. The two cases differed in terms of size and detailed features of the deletion. Because the LCR and the upstream region of the OPN1LW transcription site are involved in the regulation of OPN1LW and OPN1MW expression,5 deletion of these regions would result in the loss of red and green opsin expression, respectively. We therefore speculate that the cone photoreceptor outer segments in the two families did not express red and green opsins. Both deletions were associated with the AluSz repeat sequence in the two families (Figure 3). It was recently reported that 2 unrelated families with BCM had a complex 3-kb deletion mutation that included the LCR and insertion of an additional aberrant OPN1MW. The deletion–insertion breakpoints were located within 2 Alu repeat sequences (AluSx and AluSx1),12 suggesting the involvement of highly repetitive sequences in deletion events. For the two patients in the present study, segregation analysis was not carried out due to difficulties in collecting samples from the families.
In conclusion, the disease-causing mutation in two Japanese families with BCM was a long deletion that included the LCR. This is the first single base-level mutation analysis of Japanese BCM cases caused by a deletion that includes the LCR.
Acknowledgments
We thank the patients who participated in this study. This work was supported by a Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (B) (no. 17390468 to SM) and Grant-in Aid for Young Scientists (B) (no. 19791261 to CW).
Footnotes
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv)
The authors declare no conflict of interest.
References
- Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS. Molecular genetics of inherited variation in human color vision. Science 1986; 232: 203–210. [DOI] [PubMed] [Google Scholar]
- Berson EL, Sandberg MA, Rosner B, Sullivan PL. Color plates to help identify patients with blue cone monochromatism. Am J Ophthalmol 1983; 95: 741–747. [DOI] [PubMed] [Google Scholar]
- Nathans J, Davenport CM, Maumenee IH, Lewis RA, Hejtmancik JF, Litt M et al. Molecular genetics of human blue cone monochromacy. Science 1989; 245: 831–838. [DOI] [PubMed] [Google Scholar]
- Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science 1986; 232: 193–202. [DOI] [PubMed] [Google Scholar]
- Wang Y, Macke JP, Merbs SL, Zack DJ, Klaunberg B, Bennett J et al. A locus control region adjacent to the human red and green visual pigment genes. Neuron 1992; 9: 429–440. [DOI] [PubMed] [Google Scholar]
- Ayyagari R, Kakuk LE, Coats CL, Bingham EL, Toda Y, Felius J et al. Bilateral macular atrophy in blue cone monochromacy (BCM) with loss of the locus control region (LCR) and part of the red pigment gene. Mol Vis 1999; 5: 13. [PubMed] [Google Scholar]
- Ayyagari R, Kakuk LE, Bingham EL, Szczesny JJ, Kemp J, Toda Y et al. Spectrum of color gene deletions and phenotype in patients with blue cone monochromacy. Hum Genet 2000; 107: 75–82. [DOI] [PubMed] [Google Scholar]
- Nathans J, Maumenee IH, Zrenner E, Sadowski B, Sharpe LT, Lewis RA et al. Genetic heterogeneity among blue-cone monochromats. Am J Hum Genet 1993; 53: 987–1000. [PMC free article] [PubMed] [Google Scholar]
- Gardner JC, Michaelides M, Holder GE, Kanuga N, Webb TR, Mollon JD et al. Blue cone monochromacy: causative mutations and associated phenotypes. Mol Vis 2009; 15: 876–884. [PMC free article] [PubMed] [Google Scholar]
- Terasaki H, Miyake Y. Association of acquired color vision defects in blue cone monochromatism. Jpn J Ophthalmol 1995; 39: 55–59. [PubMed] [Google Scholar]
- Wang C, Nakanishi N, Ohishi K, Hikoya A, Koide K, Sato M et al. Novel RDH5 mutation in family with mother having fundus albipunctatus and three children with retinitis pigmentosa. Ophthalmic Genet 2008; 29: 29–32. [DOI] [PubMed] [Google Scholar]
- Yatsenko SA, Bakos HA, Vitullo K, Kedrov M, Kishore A, Jennings BJ et al. High-resolution microarray analysis unravels complex Xq28 aberrations in patients and carriers affected by X-linked blue cone monochromacy. Clin Genet 2016; 89: 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Data Citations
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.