

Abstract
Objectives:
To investigate the viability and differentiation capacity of dental pulp stem cells (DPSCs) isolated from single donors after two years of cryopreservation.
Methods:
This prospective study was conducted between October 2010 and February 2014 in the Stem Unit, College of Medicine, King Saud University, Riyadh, Saudi Arabia. Seventeen teeth extracted from 11 participants were processed separately to assess the minimum tissue weight needed to yield cells for culturing in vitro. Cell stemness was evaluated before passage 4 using the colony forming unit assay, immunofluorescence staining, and bi-lineage differentiation. Dental pulp stem cells were cryopreserved for 2 years. Post-thaw DPSCs were cultured until senescence and differentiated toward osteogenic, odontogenic, adipogenic, and chondrogenic lineages.
Results:
Viable cells were isolated successfully from 6 of the 11 participants. Three of these 6 cultured cell lines were identified as DPSCs. A minimum of 0.2 g of dental pulp tissue was required for successful isolation of viable cells from a single donor. Post-thaw DPSCs successfully differentiated towards osteogenic, odontogenic, chondrogenic, and adipogenic lineages. The post-thaw DPSCs were viable in vitro up to 70 days before senescence. There was no significant difference between the cells.
Conclusion:
Within the limitations of this investigation, viable cells from dental pulp tissue were isolated successfully from the same donor using a minimum of 2 extracted teeth. Not all isolated cells from harvested dental pulp tissue had the characteristics of DPSCs. Post-thaw DPSCs maintained their multi-lineage differentiation capacity.
Dental pulp is a soft, connective tissue present naturally within the tooth core.1 Dental pulp stem cells (DPSCs) are postnatal cells present in the dental pulp tissue with stemness capacity. Cell stemness is defined as the capacity of undifferentiated cells to undergo an indefinite number of replication and differentiation to specialized cells.2 Dental pulp stem cells have significant potential as a source of adult stem cells for human tissue engineering.3 The regenerative applications of DPSCs include: pulp tissue regeneration as an alternative approach to conventional root canal therapy, bone tissue regeneration in oral maxillofacial surgery and craniofacial anomalies, and as an alternative source for nerve tissue regeneration.4 The first report of DPSC isolation using physical straining of enzymatically processed pulp tissue was published by Gronthos et al.5 Subsequently, several reports of DPSC isolation, characterization, and cryopreservation were published by different investigators worldwide.6-10 However, some questions regarding the clinical practice of DPSC isolation remain unanswered. For example, what is the minimum weight of pulp tissue needed to yield sufficient cells for culturing in vitro? Are DPSCs always present in the dental pulp of extracted teeth? What is the differentiation capacity of DPSCs after cryopreservation? Answering these questions is essential because isolating DPSCs can be laborious, time-consuming, and expensive due to the risk of contamination and the small amount of tissue gained from a single tooth. The objective of the current study was to investigate the viability and differentiation capacity of DPSCs isolated from a single donor after 2 years of cryopreservation.
Methods
This prospective study was approved by the Institutional Ethical Committee, College of Dentistry Research Center, and conducted between October 2010 and February 2014 in the Stem Unit, College of Medicine, King Saud University, Riyadh, Saudi Arabia. The study protocol was in full accordance with the World Medical Association Declaration of Helsinki (2008).
Inclusion criteria were volunteer patients <30 years of age scheduled for tooth extraction, and without a history of medical illness. Exclusion criteria were patients with rampant caries or aggressive periodontitis. A signed written consent form was obtained from all volunteering patients.
Isolation, differentiation, cryopreservation of DPSCs
Each tooth was disinfected by brushing the crown for 30 seconds in 2 mL of chlorhexidine gluconate (Corsodyl®). The tooth was then bathed in saline before it was soaked in Listerine® for 30 seconds. Pulp tissue collection is shown in Figure 1.
Figure 1.
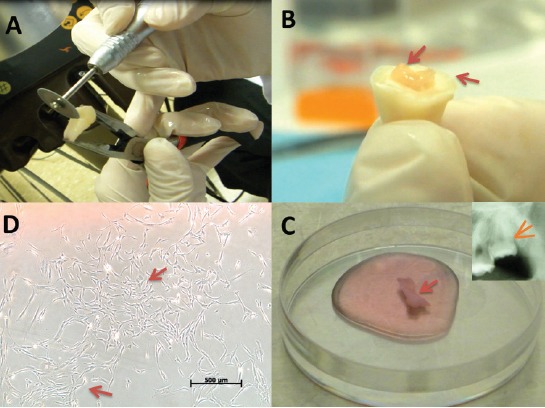
Collecting pulp tissue from extracted teeth. A) Stable finger support while using a diamond disc to create a 360° grove at 2 mm depth under the cemento-enamel junction. B) The crown was separated from the root (arrows) with minimum debris by wedging the chisel in the groove and applying gentle force with a hammer. C) The exposed pulp tissue (arrow) was collected with a hemostat and Endodontic K-files, and placed in 4°C Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 45 mg/L D-glucose, 4 mM L-glutamine, and 110 mg/L sodium pyruvate (Gibco, Loughborough, UK). The culture medium also contained a 10% penicillin-streptomycin solution (Pen-Strep; 10 units penicillin and 10 µg streptomycin per µL, Gibco), Selecting teeth with a large pulp chamber (arrow) ensured the removal of pulp tissue in one piece with minimal debris. D) Dental pulp cells formed visible colonies at day 14 as viewed under an inverted light microscope (arrows).
The weight of the collected samples from each patient was recorded before tissue processing under a laminar flow hood. Then, pulp tissue was minced with a scalpel into cubes <2 mm2 and transferred to a 15 mL centrifuge tube. Enzymatic digestion was performed for 20-45 minutes in a shaking incubator at 37°C using 1 mL of collagenase type 1 (250 units/mg, Gibco) freshly mixed with 1 mL of dispase (5000 caseinolytic units in 100 mL, BD Bioscience, Oxford, UK). The enzymatic digestion was terminated by adding 4 mL of 4°C DMEM when turbidity was evident. Cells were collected by centrifuging at 200 g for 5 minutes at 4°C. The harvested cells and tissue clumps were resuspended in 5 mL of DMEM containing 20% fetal bovine serum (FBS), 1% Pen-Strep, and 1% non-essential amino acids minimum essential medium (Gibco), and cultured in polystyrene 25 cm2 culture flasks with a filtered valve. The culture flasks were incubated at 37°C in 5% CO2. The medium was changed every 4 days. Samples that showed successful cell growth were passaged every 4 to 6 days using 0.25% trypsin-Ethylenediaminetetraacetic acid (Gibco).
Before passage 4, cell characterization and differentiation were carried out using a modification of the protocol published by Vishnubalaji et al.11 The colony forming unit assay was performed in triplicate in 6 cm dishes at a seeding density of 2.5 × 103 cells per dish. Cells were incubated for 2 weeks at 37°C in 5% CO2. Then, the cells were fixed with 1% paraformaldehyde for 10 minutes, washed with phosphate-buffered saline (PBS), and stained with 0.5% crystal violet (Sigma-Aldrich, St. Louis, MO, USA) for 5 minutes. Finally, the cells where washed with PBS to remove excess stain before being evaluated under an inverted light microscope. Aggregates of 40-50 cells were considered a colony.
Antibodies against CD146, CD73, CD29, HLA-DR (phycoerythrin-conjugated mouse anti-human), CD34, CD90, CD45, CD13, CD31 (fluorescein isothiocyanate-conjugated mouse anti-human), CD105, CD14, and CD44 (allophycocyanin-conjugated mouse anti-human) were obtained from BD Biosciences (San Diego, CA, USA). A confluent culture flask from each sample was selected randomly and incubated with the above antibodies and appropriate isotype controls as per the manufacturer’s instructions. Cells were analyzed using a FACSCalibur instrument and CellQuest Pro software version 3.3 (BD Biosciences).
Immunofluorescence staining was performed after culturing cells (2 × 104 cells/cm2) in an 8-chambered slide until they reached 80% confluency. Cells were fixed using acetone/methanol for 10 minutes at 20°C, washed with PBS, and blocked with 3% bovine serum albumin for 30 minutes. Six chambers were incubated for 2 hours at room temperature with a primary antibody against vimentin (BD Biosciences) diluted in PBS (1:100). The remaining 2 chambers were used as controls and filled with PBS. After washing the cells 3 times with PBS, the secondary antibody (goat polyclonal anti-mouse IgG, Abcam, Cambridge, MA, USA) conjugated to fluorescein isothiocyanate (1:4000), was added to all chambers and incubated in the dark for one hour at room temperature. Cells were then washed with PBS, mounted with VectaShield, stained with 4’, 6-diamidino-2-phenylindole (DAPI) as a nuclear dye, and examined using an inverted fluorescence microscope (Leica, ScanScope Console Controller 10.0.01805; Aperio Technologies, Vista, CA, USA).
In vitro osteogenic and adipogenic differentiation was performed in 6-well plates seeded with 0.05 × 106 cells/well. For each differentiation assay, 4 plates were used. Each plate had 3 test wells (one for RNA isolation and 2 for staining) and 3 control wells. Once the cells reached 80% confluency, osteogenic induction medium (DMEM supplemented with 10% FBS, 1% Pen-Strep, 50 µg/mL L-ascorbic acid (Wako Chemicals, Neuss, Germany), 10 mM β-glycerophosphate, 10 nM calcitriol, and 10 nM dexamethasone (Sigma-Aldrich)), or adipogenic induction medium (DMEM supplemented with 10% FBS, 10% horse serum, 1% Pen-Strep, 100 nM dexamethasone, 0.45 mM isobutyl methylxanthine, 3 µg/mL insulin (Sigma-Aldrich), and 1 µM rosiglitazone (BRL49653, Novo Nordisk, Bagsvaerd, Denmark)) were added to the assigned plates.
Osteogenic differentiation was determined by staining osteoblasts for alkaline phosphatase. At the end of the induction periods (days 3, 7, 14, and 21), the cells were fixed with acetone/citrate buffer for 5 minutes at room temperature. Alkaline phosphatase substrate solution (Sigma-Aldrich) was added for one hour at room temperature in the dark. Cells were counterstained immediately with Mayers hematoxylin for 5 minutes and evaluated using an inverted light microscope. Alizarin Red S staining was carried out to evaluate the mineralized cell layer. The differentiated osteogenic cells were fixed in ice-cold ethanol for one hour and then washed twice with PBS. Alizarin Red S stain was applied with gentle agitation for 10 minutes. Then, the cells were washed with PBS 3 times, counterstained with Mayers hematoxylin, and evaluated using an inverted light microscope.
Oil Red O staining was carried out at days 3, 7, 14, and 21 to evaluate adipogenic differentiation. The cells were fixed in 4% paraformaldehyde for 10 minutes at room temperature, and then rinsed with 3% isopropanol. Oil Red-O staining solution was then applied for one hour at room temperature, followed by Mayer’s hematoxylin counterstain. The cells were examined using an inverted light microscope.
Reverse transcription with the real-time quantitative polymerase chain reaction (RT-PCR) was performed to evaluate the relative expression of specific osteogenic and adipogenic genes by the differentiated cells. Ribonucleic acid was isolated from osteogenic and adipogenic plates using a Pure Link RNA isolation kit (Invitrogen). After RNA quantification using a Gene Quant II spectrophotometer, cDNA was synthesized from the isolated RNA according to the manufacturer’s instructions (Transcriptor First Strand cDNA Synthesis Kit, Roche). Real-time quantitative polymerase chain reaction was carried out using the Power SYBR® Green PCR Master Mix kit (Applied Biosystems, Bleiswijk, The Netherlands) with the primer sequences presented in Vishnubalaji et al.11 Cycling parameters included pre-incubation at 95°C for 15 seconds, annealing and extension at 60°C for one minute, and finally a melting curve analysis by raising the temperature to 95°C for 15 seconds followed by cooling at 4°C for 5 minutes.
After characterization of the DPSCs, the remaining cells were cryopreserved for 2 years. Cryovials contained 1 × 106 cells in 1 mL of DMEM with 20% FBS, 1% Pen-Strep, and 10% dimethyl sulfoxide. Vials were cooled stepwise by immediate transfer to a -20°C freezer for 20 minutes, then into a -80°C freezer for 4 days, and finally into liquid nitrogen. After 2 years, cryovials were thawed by adding DMEM medium with gentle aspiration. The collected cells were centrifuged at 200 g for 5 minutes and cultured.
Post-thaw culturing and differentiation of DPSCs
Post-thaw DPSCs from passage 3 were cultured until passage 6. Osteogenic and adipogenic differentiation was then performed as described in the cell characterization section. In vitro odontogenic differentiation was performed as described for osteogenic differentiation except that the induction medium did not include 10 nM calcitriol. The alkaline phosphatase assay and Alizarin Red S staining were carried out to evaluate the mineralized cell layer in osteogenic and odontogenic induction wells. Oil Red O staining was carried out at days 3, 7, 14, and 21 to evaluate adipogenic differentiation. Chondrogenic differentiation was assessed using the pellet culture technique. Sub-confluent cells from passage 6 of post-thaw DPSCs were released by trypsin-EDTA (0.25%, Gibco), counted, and used to generate a micromass culture. Briefly, 0.5 × 106 cells were centrifuged at 200 g for 5 minutes at 4°C in 15 mL polypropylene conical tubes. The resulting pellets were cultured up to 50 days. Control cultures were grown in advanced DMEM/F12 (Gibco) with 1% Pen-Strep. To induce chondrogenic differentiation, control medium was supplemented with 100 µg/µL transforming growth factor-β1 and 0.2 µL/mL dexamethasone. Cultures were carried out in triplicate in a humidified atmosphere containing 5% CO2. The medium was changed every 2 days. Pellets at days 0, 35, and 50 were fixed in 4% formaldehyde in PBS for one hour and embedded in paraffin. Paraffin sections were stained with Masson’s trichrome and Toluidine blue, and analyzed using an inverted light microscope.
Post-thaw DPSCs were cultured until the cells reached senescence. Three 25-cm2 culture flasks were seeded with 0.25 × 106 DPSCs and cultured in the same setting. The cells were passaged every 4 days or when cell confluence reached 75%. Then, cell viability and counts were carried out using a Vi-CELL viability analyzer (Beckman Coulter) for each culture flask. In every passage, new culture flasks were used to seed 0.25 × 106 DPSCs from the previous passage until cells reached senescence.
Statistical comparisons between the cell lines until senescence were performed using a univariate analysis of variance and Tukey HSD, Kruskal-Wallis post-hoc test with Statistical Package for Social Sciences version 22.0 (IBM, Chicago, IL, USA). A p-value of less than 0.05 was considered significant.
Results
The current study included 17 teeth from 11 different individuals. Extracted teeth were disinfected and the pulp tissue was collected between 2 and 72 hours’ post-extraction. In vitro culturing was not successful when the processed sample weighed less than 0.2 gram or when the processing time was greater than 24 hours. Six of 11 samples yielded cells that were cultured in vitro. Three lines out of these 6 (S2, S3, S13) were identified as DPSCs.
Collecting dental pulp tissue from 2 teeth provided approximately 0.2 gram of tissue that initially yielded a few cells appearing between 2 and 8 days of culture. Once the cells reached confluence, passaging of the attached cells resulted in rapid multiplication. The colony forming efficiency was 9-12% at the second passage. At passage 4, every cultured sample yielded 10±2 × 106 cells after 25 ± 7 days in primary culture.
Differentiation of dental pulp stem cells were positive (Figure 2A) for the mesenchymal stem cell markers CD105, CD90, CD73, CD13, CD29, and CD44, and were negative for the hematopoietic and endothelial markers CD34, CD45, CD14, and CD31, as well as for the MHC class II marker HLA-DR.
Figure 2.
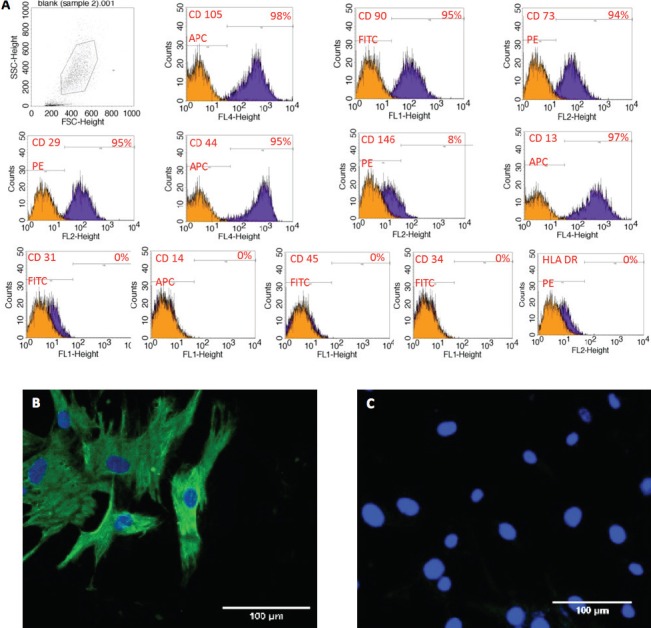
Isolated dental pulp stem cells characterization (DPSCs) showing A) Positive mesenchymal stem markers (CD105, CD90, CD73, CD13, CD29 and CD44) and negative for hematopoietic and endothelial markers (CD34, CD45, CD14, and CD31) as well as for the MHC class II marker human leukocyte antigen - antigen d related. B) Positive immunostaining for 4’, 6-diamidino-2-phenylindole (DAPI) and localization of Vimentin in DPSCs. C) Positive immunostaining for DAPI and negative for localization of Vimentin in control.
Immunocytochemical staining showed that test (DPSCs) and control cells in all chambers had a positive reaction to DAPI, while only DPSC chambers were positive for vimentin (Figures 2B-2C). After osteogenic induction, DPSCs were positive for alkaline phosphatase staining compared with the control. This staining was evident as early as 3 days after induction, with some samples showing a progressive increase in alkaline phosphatase over time (Figures 3A-3B), and others reaching a peak at day 7. The emergence of Alizarin Red S staining appeared later compared to alkaline phosphatase staining. Complete mineralization was achieved by day 14 (Figures 3C-3D), and was maintained up to day 21. Adipogenic induction of DPSCs was mild with few Oil Red O-positive droplets.
Figure 3.
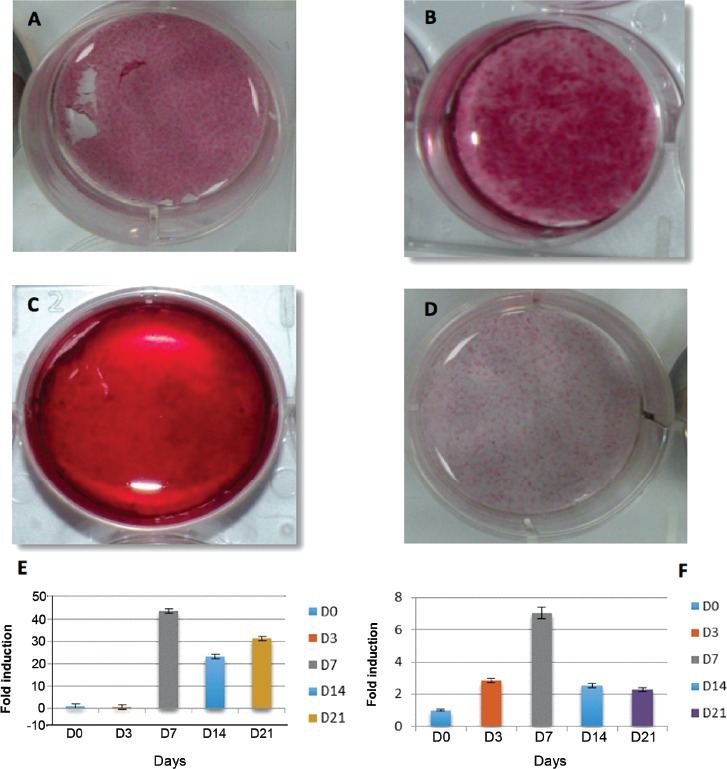
Dental pulp stem cells osteogenic differentiation showing A) day 7, B) day 14 positive ALP staining. C) day 14 positive alizarin red staining, D) control, E) representative graph of RT-polymerase chain reaction of alkaline phosphatase gene expression, and F) RUNX-2 gene expression
Real-time quantitative polymerase chain reaction revealed that osteogenic differentiation resulted in a significant increase in alkaline phosphatase (~45-fold) and RUNX-2 (~7-fold) gene expression, which were highest at day 7 (Figures 3E-3F). Both osteocalcin (~21-fold) and osteopontin (~7-fold) exhibited progressive increases in expression, with the highest amounts seen on day 21. Adipogenic markers showed a similar progressive increase in gene expression. Peak expression was evident on day 21 for adiponectin (~37-fold), PPAR-γ2 (~10-fold), and activated protein-2 (~100-fold).
After 2 years of cryopreservation, thawed DPSCs had 85% viability. In post-thaw DPSCs, the alkaline phosphatase assay showed that osteogenic induction was greatest, followed by odontogenic induction (Figures 4A-4B). Control cells had the lowest alkaline phosphatase activity. Alizarin Red S staining showed more calcified nodules in osteogenic-induced cells, followed by odontogenic-induced cells (Figures 4C-4D). Adipogenic-induced cells were positive for Oil Red O stain as early as 7 days post-induction and maintained in day 21 compared to control (Figures 4E-4F).
Figure 4.
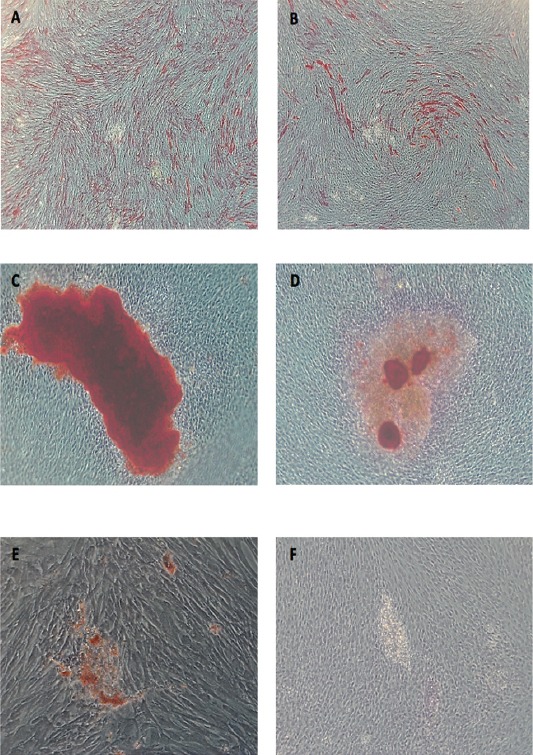
Post-thaw dental pulp stem cells osteogenic, odontogenic, adipogenic differentiation showing A) day 7 ALP activity after osteogenic induction (X5), B) odontogenic induction, C) Day 14 alizarin red S stain (X10) in osteogenic, D) odontogenic induction media, E) day 21 oil red O stained droplet (X20) after adipogenic induction, (F) Control
At days 35 and 50 after induction of chondrogenic differentiation, post-thaw DPSCs demonstrated the formation of essential cartilage matrix proteins, such as proteoglycans, that were stained with Toluidine blue (Figure 5A). Masson’s trichrome was used to detect formation of the extracellular cartilage matrix (Figure 5B). There was no significant difference in cell count (p=0.451), and the days (p=0.053) until senescence between the 3 DPSC lines (Figure 6). Dental pulp stem cells reached senescence in the study condition between 59-70 days.
Figure 5.
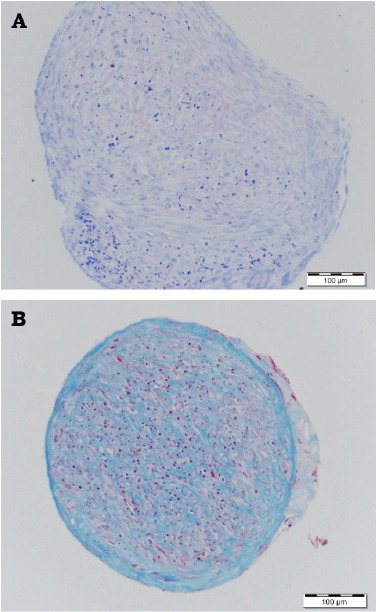
Post-thaw dental pulp stem cells chondrogenic differentiation showing A) Day 35 proteoglycans were positive to Toluidine blue stain, B) Day 50 collagen fibers were positive to Masson’s trichrome stain.
Figure 6.
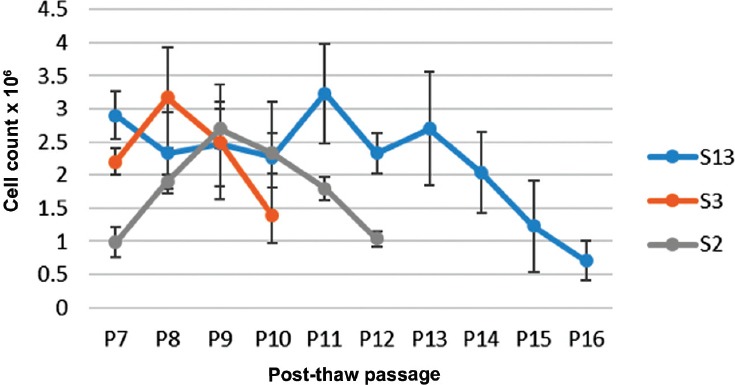
Cell growth curves for 3 dental pulp stem cells line cultured until senescence
Discussion
Isolating DPSCs for dental tissue regeneration research was first reported in 2000 by Gronthos et al5 without details on how the teeth were disinfected. Subsequent studies,12,13 used a disinfection protocol composed of several washes in sterile saline, followed by 1% polyvinylpyrrolidone-iodine, 0.1% sodium thiosulfate, and final immersion in Listerine® antiseptic. Recently, chlorhexidine gel was used in a similar manner.9 In the current study, effective tooth disinfection used a combination of chlorhexidine gluconate and Listerine® mouthwashes, both of which are easily accessible and safe to use.
Removing dental pulp from the enclosed hard dental tissue is challenging. There is a risk of overheating or incorporation of hard tissue debris during cutting. Details on how the pulp tissue was removed were not included in previous reports describing the isolation of DPSCs,9,12-14 except for the detailed technique published in 2011 by Gronthos et al.15 Their technique is similar to the approach used in the current study. Enzymatic digestion was the major obstacle in the current study because prior reports did not indicate the amount of dental pulp tissue used during this step.5,16 In addition, several different enzymes or combinations of enzymes at different concentrations were reported. While Gronthos et al5 used 3 mg/mL of type I collagenase and 4 mg/mL of dispase for 30-45 minutes, others have used various combinations and different time intervals.17-19 Because the aim of our study was to isolate DPSCs from a single donor, we used dental pulp tissue from one or 2 teeth extracted in outpatient clinics to obtain sufficient tissue for processing. The amount of pulp tissue from a single tooth was small. Therefore, adjusting the enzymatic digestion procedure was mandatory. The protocol we developed was designed to digest 0.2 gram of dental pulp tissue, which approximated the amount collected from 2 teeth. Viable cells were successfully cultured from 6 samples; the unsuccessful samples had tissue weights <0.2 gram and no viable cells were observed after tissue digestion. Adjusting the enzymatic digestion step, or using an explant culture, might be alternatives for the successful culturing of DPSCs from a single tooth. Viable cells from pulp tissue were cultured successfully in DMEM containing 20% FBS without the addition of growth factors such as platelet-derived growth factor, transforming growth factor-β, or fibroblast growth factor.19,20 In our study, cell clusters were evident beginning 2-8 days after initial culturing, which was similar to Liu et al.21 Tooth disinfection and culturing in 1% Pen-Strep medium were adequate to prevent contamination.
Differentiation of dental pulp stem cells have been sorted using magnetic beads5,15 and flow cytometry.9,12,22 In the current study, we used flow cytometry and showed that 3 out of 6 samples were positive for mesenchymal stem cell markers and negative for hematopoietic and endothelial cell markers, as well as for a MHC class II marker. The stromal surface markers expressed by our cell lines were in agreement with previous studies.12,23,24 The lack of mesenchymal stem cell markers in the other dental pulp cell lines could be due to the small number of stem cells that could not be identified using the current technique, or due to the lack of stem cells in the samples. This should be investigated in future studies.
Multipotent differentiation potential is a minimal criterion for defining multipotent mesenchymal stromal cells.23 Therefore, osteogenic and adipogenic differentiation capacities were verified using PCR before cryopreservation. The results of the alkaline phosphatase assay and Alizarin Red S stain for osteogenic induction were similar to Ponnaiyan et al.25 The osteogenic differentiated samples showed significant increases in alkaline phosphatase, and the expression of osteopontin, osteocalcin, and RUNX-2, compared to the negative control. This was similar to previous reports.5,26,27 Adipogenic differentiation was induced through the application of dexamethasone-containing media according to the protocol of Vishnubalaji et al.11 Oil Red O staining and the weak expression of adipocyte-specific transcripts were also in agreement with previous studies.24,28,29
There are previous reports on human DPSCs cryopreserved for 7 days,30 3 months,31 and one year.32 Another study reported on preserved rat DPSCs.33 This is the first study showing successful cryopreservation of human DPSCs for 2 years. The viability of the thawed cells was 85%, and the differentiation capacity was not compromised by cryopreservation. Woods et al13 recommended 2 × 106 cells/mL for optimum cryopreservation. However, in our study, we successfully cryopreserved 1 × 106 cells/mL. Distributing DPSCs in more cryovials during cryopreservation provides more samples for future clinical applications.
The differentiation capacity of post-thaw DPSCs was positive to staining tests and similar to staining tests performed on the same cell lines when first isolated 2 years earlier. However, quantitative comparisons34 and PCR analyses are needed to detect any changes in differentiation capacity after long term cryopreservation.
The DPSC lines isolated in the current study were cultured until senescence. Theoretically, cells with stemness capacity should have indefinite self-renewal.2 However, this was not observed during this study. One explanation would be the culturing environment and how DPSCs were plated in vitro.35 This aspect should be investigated in future studies.
This study was limited to establishing a protocol to isolate DPSCs in Saudi Arabia. For that reason, the number of included subjects was limited, the selected teeth were sound with no caries. In addition, the materials and methodology were adopted from protocols used to isolate stem cells from bone marrow and adipose tissue. Therefore, future studies should include a larger number of subjects and carious teeth with a customized technique.
In conclusion, DPSCs are a source of adult stem cells that can be obtained with no associated morbidity/mortality. Isolating DPSCs from a single donor requires adequate pulp tissue and not present in every tissue sample as presented in Table 1.
Table 1.
Study conclusion summarized in synoptical table.

Acknowledgment
We would like to thank the College of Dentistry and the College of Medicine, King Saud University, Riyadh, Saudi Arabia for their support.
Footnotes
Authorship entitlement.
Excerpts from the Uniform Requirements for Manuscripts Submitted to Biomedical Journals updated November 2003. Available from www.icmje.org
The international Committee of Medical Journal Editors has recommended the following criteria for authorship; these criteria are still appropriate for those journals that distinguish authors from other contributors.
Authorship credit should be based on 1) substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data; 2) intellectual content; and 3) final approval of the version to be published. Authors should meet conditions 1, 2, and 3.
Acquisition of funding, collection of data, or general supervision of the research group, alone, does not justify authorship.
An author should be prepared to explain the order in which authors are listed.
References
- 1.La Noce M, Paino F, Spina A, Naddeo P, Montella R, Desiderio V, et al. Dental pulp stem cells: State of the art and suggestions for a true translation of research into therapy. J Dent. 2014;42:761–768. doi: 10.1016/j.jdent.2014.02.018. [DOI] [PubMed] [Google Scholar]
- 2.Cai J, Weiss ML, Rao MS. In search of “stemness”. Exp Hematol. 2004;32:585–598. doi: 10.1016/j.exphem.2004.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tatullo M, Marrelli M, Paduano F. The regenerative medicine in oral and maxillofacial surgery: the most important innovations in the clinical application of mesenchymal stem cells. Int J Med Sci. 2015;12:72–77. doi: 10.7150/ijms.10706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tatullo M, Marrelli M, Shakesheff KM, White LJ. Dental pulp stem cells: function, isolation and applications in regenerative medicine. J Tissue Eng Regen Med. 2015;9:1205–1216. doi: 10.1002/term.1899. [DOI] [PubMed] [Google Scholar]
- 5.Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA. 2000;97:13625–13630. doi: 10.1073/pnas.240309797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SY, Chiang PC, Tsai YH, Tsai SY, Jeng JH, Kawata T, Huang HM. Effects of cryopreservation of intact teeth on the isolated dental pulp stem cells. J Endod. 2010;36:1336–1340. doi: 10.1016/j.joen.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Ferro F, Spelat R, Beltrami AP, Cesselli D, Curcio F. Isolation and characterization of human dental pulp-derived stem cells by using media containing low human serum percentage as clinical grade substitutes for bovine serum. PLoS ONE. 2012;7:e48945. doi: 10.1371/journal.pone.0048945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SY, Huang GW, Shiung JN, Huang YH, Jeng JH, Kuo TF, et al. Magnetic cryopreservation for dental pulp stem cells. Cells Tissues Organs. 2012;196:23–33. doi: 10.1159/000331247. [DOI] [PubMed] [Google Scholar]
- 9.Tirino V, Paino F, De Rosa A, Papaccio G. Identification, isolation, characterization, and banking of human dental pulp stem cells. Methods Mol Biol (Clifton, NJ) 2012;879:443–463. doi: 10.1007/978-1-61779-815-3_26. [DOI] [PubMed] [Google Scholar]
- 10.Hilkens P, Gervois P, Fanton Y, Vanormelingen J, Martens W, Struys T, et al. Effect of isolation methodology on stem cell properties and multilineage differentiation potential of human dental pulp stem cells. Cell Tissue Res. 2013;353:65–78. doi: 10.1007/s00441-013-1630-x. [DOI] [PubMed] [Google Scholar]
- 11.Vishnubalaji R, Al-Nbaheen M, Kadalmani B, Aldahmash A, Ramesh T. Comparative investigation of the differentiation capability of bone-marrow- and adipose-derived mesenchymal stem cells by qualitative and quantitative analysis. Cell Tissue Res. 2012;347:419–427. doi: 10.1007/s00441-011-1306-3. [DOI] [PubMed] [Google Scholar]
- 12.Perry BC, Zhou D, Wu X, Yang FC, Byers MA, Chu TMG, et al. Collection, cryopreservation, and characterization of human dental pulp-derived mesenchymal stem cells for banking and clinical use. Tissue Eng Part C Methods. 2008;14:149–156. doi: 10.1089/ten.tec.2008.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woods EJ, Perry BC, Hockema JJ, Larson L, Zhou D, Goebel WS. Optimized cryopreservation method for human dental pulp-derived stem cells and their tissues of origin for banking and clinical use. Cryobiology. 2009;59:150–157. doi: 10.1016/j.cryobiol.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, et al. Stem cell properties of human dental pulp stem cells. J Dental Res. 2002;81:531–535. doi: 10.1177/154405910208100806. [DOI] [PubMed] [Google Scholar]
- 15.Gronthos S, Arthur A, Bartold PM, Shi S. A method to isolate and culture expand human dental pulp stem cells. Methods Mol Biol. 2011;698:107–121. doi: 10.1007/978-1-60761-999-4_9. [DOI] [PubMed] [Google Scholar]
- 16.Shi S, Gronthos S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J Bone Min Res. 2003;18:696–704. doi: 10.1359/jbmr.2003.18.4.696. [DOI] [PubMed] [Google Scholar]
- 17.Carinci F, Papaccio G, Laino G, Palmieri A, Brunelli G, D’Aquino R, et al. Comparison between genetic portraits of osteoblasts derived from primary cultures and osteoblasts obtained from human pulpar stem cells. J Craniofac Surg. 2008;19:616–625. doi: 10.1097/SCS.0b013e31816aabc8. [DOI] [PubMed] [Google Scholar]
- 18.Patel M, Smith AJ, Sloan AJ, Smith G, Cooper PR. Phenotype and behaviour of dental pulp cells during expansion culture. Arch Oral Biol. 2009;54:898–908. doi: 10.1016/j.archoralbio.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Karbanova J, Soukup T, Suchanek J, Mokry J. Osteogenic differentiation of human dental pulp-derived stem cells under various ex-vivo culture conditions. Acta Medica (Hradec Kralove) 2010;53:79–84. doi: 10.14712/18059694.2016.64. [DOI] [PubMed] [Google Scholar]
- 20.Karaoz E, Demircan PC, Saglam O, Aksoy A, Kaymaz F, Duruksu G. Human dental pulp stem cells demonstrate better neural and epithelial stem cell properties than bone marrow-derived mesenchymal stem cells. Histochem Cell Biol. 2011;136:455–473. doi: 10.1007/s00418-011-0858-3. [DOI] [PubMed] [Google Scholar]
- 21.Liu HC, E LL, Wang DS, Su F, Wu X, Shi ZP, et al. Reconstruction of alveolar bone defects using bone morphogenetic protein 2 mediated rabbit dental pulp stem cells seeded on nano-hydroxyapatite/collagen/poly(L-lactide) Tissue Eng Part A. 2011;17:2417–2433. doi: 10.1089/ten.TEA.2010.0620. [DOI] [PubMed] [Google Scholar]
- 22.Atari M, Barajas M, Hernandez-Alfaro F, Gil C, Fabregat M, Ferres Padro E, Giner L, Casals N. Isolation of pluripotent stem cells from human third molar dental pulp. Histol Histopathol. 2011;26:1057–1070. doi: 10.14670/HH-26.1057. [DOI] [PubMed] [Google Scholar]
- 23.Dominici M, Le Blanc \ K, Mueller I, Slaper-Cortenbach I, Marini F, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 24.Lindroos B, Maenpaa K, Ylikomi T, Oja H, Suuronen R, Miettinen S. Characterisation of human dental stem cells and buccal mucosa fibroblasts. Biochem Biophys Res Commun. 2008;368:329–335. doi: 10.1016/j.bbrc.2008.01.081. [DOI] [PubMed] [Google Scholar]
- 25.Ponnaiyan D, Jegadeesan V. Comparison of phenotype and differentiation marker gene expression profiles in human dental pulp and bone marrow mesenchymal stem cells. Eur J Dentistry. 2014;8:307–313. doi: 10.4103/1305-7456.137631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.d’Aquino R, Graziano A, Sampaolesi M, Laino G, Pirozzi G, De Rosa A, et al. Human postnatal dental pulp cells co-differentiate into osteoblasts and endotheliocytes: a pivotal synergy leading to adult bone tissue formation. Cell Death Differ. 2007;14:1162–1171. doi: 10.1038/sj.cdd.4402121. [DOI] [PubMed] [Google Scholar]
- 27.Laino G, d’Aquino R, Graziano A, Lanza V, Carinci F, Naro F, Pirozzi G, Papaccio G. A new population of human adult dental pulp stem cells: a useful source of living autologous fibrous bone tissue (LAB) J Bone Miner Res. 2005;20:1394–1402. doi: 10.1359/JBMR.050325. [DOI] [PubMed] [Google Scholar]
- 28.Huang GT, Gronthos S, Shi S. Mesenchymal stem cells derived from dental tissues vs. those from other sources: their biology and role in regenerative medicine. J Dental Res. 2009;88:792–806. doi: 10.1177/0022034509340867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koyama N, Okubo Y, Nakao K, Bessho K. Evaluation of pluripotency in human dental pulp cells. J Oral Maxillofac Surg. 2009;67:501–506. doi: 10.1016/j.joms.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Lindemann D, Werle SB, Steffens D, Garcia-Godoy F, Pranke P, Casagrande L. Effects of cryopreservation on the characteristics of dental pulp stem cells of intact deciduous teeth. Arch Oral Biol. 2014;59:970–976. doi: 10.1016/j.archoralbio.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Malekfar A, Valli KS, Kanafi MM, Bhonde RR. Isolation and characterization of human dental pulp stem cells from cryopreserved pulp tissues obtained from teeth with irreversible pulpitis. J Endodon. 2016;42:76–81. doi: 10.1016/j.joen.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Kumar A, Bhattacharyya S, Rattan V. Effect of uncontrolled freezing on biological characteristics of human dental pulp stem cells. Cell Tissue Bank. 2015;16:513–522. doi: 10.1007/s10561-015-9498-5. [DOI] [PubMed] [Google Scholar]
- 33.Davies OG, Smith AJ, Cooper PR, Shelton RM, Scheven BA. The effects of cryopreservation on cells isolated from adipose, bone marrow and dental pulp tissues. Cryobiology. 2014;69:342–347. doi: 10.1016/j.cryobiol.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Ajlan SA, Ashri NY, Aldahmash AM, Alnbaheen MS. Osteogenic differentiation of dental pulp stem cells under the influence of three different materials. BMC Oral Health. 2015;15:132. doi: 10.1186/s12903-015-0113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tatullo M, Marrelli M, Falisi G, Rastelli C, Palmieri F, Gargari M, et al. Mechanical influence of tissue culture plates and extracellular matrix on mesenchymal stem cell behavior: A topical review. Int J Immunopathol Pharmacol. 2016;29:3–8. doi: 10.1177/0394632015617951. [DOI] [PMC free article] [PubMed] [Google Scholar]