

Abstract
Fear-inducing memories can be state dependent, meaning that they can best be retrieved if the brain states at encoding and retrieval are similar. Restricted access to such memories can present a risk for psychiatric disorders and hamper their treatment. To better understand the mechanisms underlying state-dependent fear, we used a mouse model of contextual fear conditioning. We found that heightened activity of hippocampal extrasynaptic GABAA receptors, believed to impair fear and memory, actually enabled their state-dependent encoding and retrieval. This effect required protein kinase C-βII and was influenced by miR-33, a microRNA that regulates several GABA-related proteins. In the extended hippocampal circuit, extrasynaptic GABAA receptors promoted subcortical, but impaired cortical, activation during memory encoding of context fear. Moreover, suppression of retrosplenial cortical activity, which normally impairs retrieval, had an enhancing effect on the retrieval of state-dependent fear. These mechanisms can serve as treatment targets for managing access to state-dependent memories of stressful experiences.
Memories encoded in certain mood-, emotion- or drug-related brain states are most easily retrieved in the same states1. In humans, state-dependent learning has been recognized as a way to organize memories, facilitate decision-making and temporarily avoid negative affect2. In contrast with these generally beneficial effects, it has also been implicated in the nonintegrated encoding of stress-related memories and emotions, placing individuals at risk for a wide variety of psychiatric disorders3,4. State dependency of learning and memory under various psychoactive drugs has been shown in rodent models of reinforcement learning5 and passive avoidance6; however, the molecular and circuit mechanisms of state-dependent learning in general, and fear-related state-dependent learning in particular, remain unknown.
Under normal conditions, fear-provoking memories of stressful experiences are encoded and retrieved by excitatory glutamatergic mechanisms, whereas the inhibitory GABAergic system is thought to impair these processes7. Nevertheless, there is also evidence that GABAA receptor agonists, such as barbiturates, benzodiazepines and alcohol can support state-dependent memory5. Notably, amobarbital, which binds to all GABAA receptors, disinhibits memory retrieval8, whereas diazepam, which predominantly binds to synaptic GABAA receptors9, is ineffective. This suggests that state-dependent learning of stressful experiences is preferentially mediated by extrasynaptic GABAA receptors, which are known to generate tonic inhibition in brain regions important for learning and memory, such as the dentate gyrus of the hippocampus10.
RESULTS
Gaboxadol induces state-dependent fear
To test this hypothesis, we used the specific agonist gaboxadol to increase the activity of extrasynaptic GABAA receptors11. Gaboxadol injected intrahippocampally (i.h.) either before training (Fig. 1a; n = 6 mice per group for the 0.5 μg per hippocampus dose and n = 7 mice per group for all other doses; F 5,35 = 26.798, P < 0.001) or before memory testing (Fig. 1b; n = 7 mice per group for the 0 and 0.125 μg per hippocampus groups and 8 mice per group for 0.25 and 0.5 μg per hippocampus groups; F 3,26 = 20.594, P < 0.001) dose-dependently impaired contextual freezing, an index of learned fear12. These freezing impairments could be interpreted as impaired learning, memory retrieval or fear expression. However, when mice were injected with gaboxadol both before training and testing (G-G group), freezing was indistinguishable from that of vehicle controls (V-V group) and was significantly higher than that of the groups receiving gaboxadol only before training (G-V group) or before the test (V-G group; n = 7 mice per group for V-V, G-V and V-G and 8 mice per group for G-G; F 3,25 = 4.481, P < 0.05; Fig. 1c). This effect was replicated in a within-subject study with mice trained on vehicle or gaboxadol and then tested on or off drug on alternate tests (Fig. 1d; n = 7 mice per group; within-subject effects were F1 = 9.584, P < 0.01 for vehicle and F1 = 9.581, P < 0.01 for gaboxadol). Thus, gaboxadol did not impair memory processes, but instead induced state-dependent contextual fear conditioning. At the lowest dose used to trigger state-dependent fear, gaboxadol did not affect locomotor activity or tone-dependent fear conditioning (Supplementary Fig. 1a,b), consistent with the preferential role of the hippocampus in contextual fear versus cue-dependent learning13,14. Muscarinic cholinergic receptors have also been implicated in state-dependent learning6, but antagonism of these receptors by scopolamine impaired memory without generating state-dependent effects (Supplementary Fig. 2). These findings suggest that state-dependent contextual fear is particularly sensitive to manipulations of GABAergic mechanisms.
Figure 1.
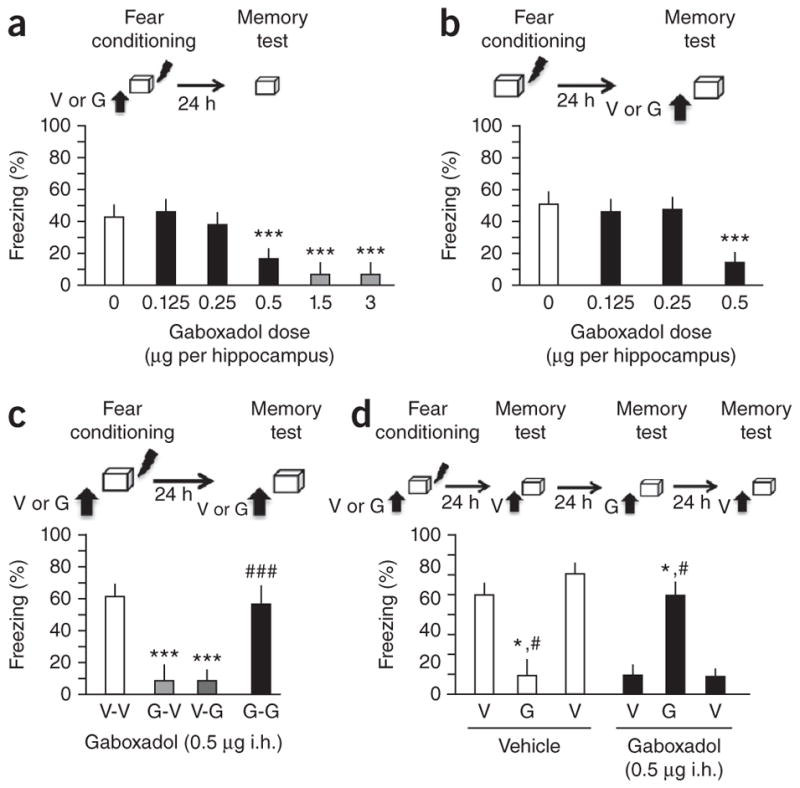
Activation of extrasynaptic GABAA receptors by i.h. injection of gaboxadol induces state-dependent contextual fear. (a) Effect of i.h. injection of vehicle (V) or different doses of gaboxadol (G) 30 min before fear conditioning on freezing at test. The doses inducing side effects (impaired locomotion, tense posture, stereotypic behaviors) are marked red and were not employed in further experiments. (b) Effect of i.h. injection of vehicle or different doses of gaboxadol before the memory test on freezing. (c) Effect of gaboxadol injected i.h. before fear conditioning (G-V), before retrieval (V-G) or both (G-G; left) on freezing. ***P < 0.001 versus vehicle controls; ###P < 0.001 versus G-V or V-G groups (one-way analysis of variance, ANOVA). (d) Mice injected with vehicle before training only froze when tested in the absence of gaboxadol, whereas mice injected with gaboxadol at training only froze when tested in the presence of gaboxadol. *P < 0.01 versus previous test, #P < 0.01 versus following test (repeated measures ANOVA). Error bars represent s.e.m. in all panels.
Gaboxadol mediates state-dependent fear via PKC βIII
GABAA receptor function is closely linked to the activity and phosphorylation of protein kinase C (PKC)15. Of several PKC isoforms, gaboxadol infused before fear conditioning only enhanced the phosphorylation of PKCβII at S660 (Fig. 2a,b; F2,11 = 3.813, P < 0.05). Similarly, PKCβII at S660 was upregulated when gaboxadol was injected both before conditioning and before the memory test or only before the memory test (Fig. 2c, n = 5 hippocampi per group; F12 = 7.29, P < 0.01; full-length blots are presented in Supplementary Fig. 3a,b), suggesting that this isoform is involved in both encoding and retrieval of state-dependent context fear. Accordingly, inhibition of PKCβII before the memory test did not affect freezing in the vehicle control (Fig. 2d; n = 8 mice per group for V-V and V-G, n = 9 mice per group for PKC βII-inhibitor; F2,22 = 0.964, P = 0.399), but it did block retrieval in the gaboxadol-treated group, as shown by reduced freezing when inhibition of PKCβII preceded injection of gaboxadol (Fig. 2d; n = 8 mice per group for V-V and V-G, n = 9 mice per group for PKC δII-inhibitor; F2,22 = 7.375, P < 0.01). Inhibition of PKCδ had no effect on either group, indicating that PKCβII is specifically involved in gaboxadol-mediated retrieval. These findings reveal an important role for PKCβII in retrieval of state-dependent fear and suggest that manipulations of this kinase could modify access to fear-inducing memories.
Figure 2.

Expression of gaboxadol-induced state-dependent fear requires PKCβII signaling. (a) Phosphorylation of different PKC isoforms in hippocampal lysates obtained from vehicle-injected mice, or 1 and 24 h after injection of 0.5 μg per hippocampus of gaboxadol (n = 4 mice per time point). Only PKCβII (S660) levels increased in response to gaboxadol. (b) Representative immunoblots. (c) Phosphorylation of PKCβII (S660) was also increased when gaboxadol was injected before both conditioning and the retrieval test (G-G group) or only before the retrieval test (V-G group) when compared with the V-V control group. Bottom, representative immunoblots. (d) Effect of pre-test infusion of PKCβII (0.25 μg per hippocampus) and PKCδ (0.5 μg per hippocampus) inhibitors on freezing behavior in mice injected with vehicle or gaboxadol before conditioning and PKC inhibitors preceding vehicle or gaboxadol before the memory test. *P < 0.5, ***P < 0.001 when compared with corresponding control groups (one-way ANOVA or t test for c). Error bars represent s.e.m. in all panels.
miR-33 regulates the effects of gaboxadol on state-dependent fear
GABAA receptors regulate the expression of specific microRNAs16,17, which, in turn, may regulate GABAA receptor function18,19. We therefore investigated whether the actions of gaboxadol on tate-dependent fear involve microRNA-mediated mechanisms. Using microarrays, we first compared the microRNA profiles in the hippocampi of naive mice and mice exposed to fear conditioning, and identified 19 differentially expressed microRNAs that were associated with fear conditioning (Supplementary Fig. 4a). Five of these micro-RNAs are predicted to target four or more mRNAs encoding GABAA receptors (Supplementary Fig. 4b), of which miR-33 was the only one whose level changed in response to gaboxadol (Fig. 3a). Contrary to the increase of miR-33 after fear conditioning off gaboxadol, the level of this miRNA significantly decreased 1 and 24 h after fear conditioning on gaboxadol (n = 4 hippocampi per group, F2,9 = 6.189, P < 0.05). To determine whether changes of miR-33 levels are involved in state-dependent fear, we produced lentiviral vectors carrying miR-33 (LV-miR-33), which served to increase the level of miR-33 (Supplementary Fig. 5a–c). To downregulate miR-33, we used miR-33 locked nucleic acid (LNA) inhibitor (miR-33-LNA; Supplementary Fig. 5d–f) and compared its effects to those of scrambled miR LNA (miR-S-LNA) (Supplementary Fig. 6). In control mice injected with a lentivirus carrying scrambled miRNA (LV-SCR), gaboxadol (0.5 μg per hippocampus) induced state-dependent fear (Fig. 3b), as revealed by high freezing levels in the V-V and G-G groups, but not in the V-G and G-V groups (n = 8 mice per group; F3,28 = 18.360, P < 0.001). Overexpression of miR-33 rendered mice insensitive to gaboxadol, as all of the groups showed high levels of freezing (Fig. 3b; n = 8 mice per group; F3,28 = 0.153, P = 0.927). These effects were observed with a threefold average increase of hippocampal miR-33 levels (Fig. 3b; n = 3 hippocampi per group, t5 = 10.297, P < 0.01). Downregulation of miR-33 did not affect freezing in response to this dose of gaboxadol, and all mice froze similarly in both the miR-S-LNA (Fig. 3c; n = 7 mice per group; F3,23 = 12.548, P < 0.001) and miR-33-LNA (Fig. 3c; n = 7 mice per group; F3,23 = 33.989, P < 0.001) groups, despite decreased miR-33 levels (Fig. 3c; n = 4 hippocampi per group, t7 = 8.715, P < 0.01). However, 0.25 μg per hippocampus gaboxadol, a dose that was ineffective in controls (Fig. 3d; n = 7 mice per group; F3,24 = 0.758, P = 0.528), induced state-dependent fear in mice injected with miR-33-LNA (Fig. 3d; n = 7 mice per group; F3,24 = 40.104, P < 0.001) when hippocampal miR-33 levels were decreased (Fig. 3d; n = 4 mice per group; t7 = 9.12, P < 0.001). These findings suggest that miR-33 levels determine the threshold for state-dependent effects mediated by extrasynaptic GABAA receptors and identify a possible mechanism by which microRNAs in general might contribute to the predisposition to develop associated mental disorders.
Figure 3.

miR-33 is downregulated in response to gaboxadol and modulates its effects on state-dependent fear. (a) Of several microRNAs that are differentially regulated by fear conditioning and target multiple GABAR, only miR-33 was significantly affected by gaboxadol (0.5 μg per hippocampus), as revealed by significant downregulation at 1 and 24 h post-conditioning. (b) Freezing in mice injected with 0.5 μg per hippocampus of gaboxadol following i.h. injection of LV-SCR (left) or LV-miR-33 (right). (c) Freezing in mice injected with 0.5 μg per hippocampus of gaboxadol following i.h. injection of miR-S-LNA (left) or miR-33-LNA (right). (d) Freezing in mice injected with 0.25 μg per hippocampus of gaboxadol following i.h. injection of miR-S-LNA (left) or miR-33-LNA (right). *P < 0.05, ***P < 0.001 versus corresponding control groups; ##P < 0.01, ###P < 0.001 versus G-V, V-G or LV-miR-33 + miR-33-LNA groups (one-way ANOVA). Error bars represent s.e.m. in all panels.
miR-33 regulates the levels of GABA-related proteins
Overexpression of miR-33 significantly reduced mRNA expression of several predicted targets related to GABAA function: Gabra4, which encodes the α4 subunit of extrasynaptic GABAA receptors, Gabrb2, which encodes the β2 subunit, and Slc12a5, which encodes the chloride symporter KCC2 (Fig. 4a; n = 4 hippocampi per group; GABRA4: F3,12 = 5.334, P < 0.05; KCC2: F3,12 = 4.782, P < 0.05; GABRB2: F3,12 = 3.662, P < 0.05). miR-33 inhibition reduced the effects of LV-miR-33 on mRNA levels, but did not increase mRNA levels by itself.
Figure 4.

Manipulations of miR-33 alter expression of several GABA-related proteins. (a) Effect of LV-miR-33 and miR-33-LNA on the expression of mRNA encoding GABRA4, KCC2 and GABRB2 and effects by miR-33-LNA. (b) Effect of LV-miR-33 on protein levels of GABRA4, KCC2 and GABRB2 when compared with LV-SCR. (c) Effect of miR-33-LNA on protein levels of GABRA4, KCC2 and GABRB2 when compared with miR-S-LNA. (d) Biotinylation assay revealing binding of miR-33, but not scrambled or mutant miRNAs, to mRNAs encoding GABRB2 and KCC2. (e) Effects of LV-miR-33 and miR-33-LNA on the levels of Synapsin-2 proteins. *P < 0.05, **P < 0.01, ***P < 0.001 versus corresponding control groups; #P < 0.05 versus LV-miR-33 + miR-33-LNA groups (one-way ANOVA). Gabra4a mRNA was not found in the input and was used as a negative control; Syn2ab mRNA was readily detectable in the input, but was not captured by biotinylated miR-33. Error bars represent s.e.m. in all panels.
At the translational level, both LV-miR-33 (Fig. 4b) and miR-33-LNA (Fig. 4c) modulated the levels of GABA-related proteins, and expectedly in opposite directions: LV-miR-33 significantly reduced the level of these proteins (n = 5 hippocampi per group; F4,40 = 22.477, P < 0.01 versus LV-SCR), whereas miR-33-LNA induced their upregulation (F4,40 = 18.25, P < 0.01 versus miR-S-LNA) (Supplementary Fig. 7a–c). The levels of GABA-unrelated proteins relevant for fear conditioning, such as NMDAR subunits or protein kinases, were not affected by LV-miR-33 (Supplementary Fig. 7d,e; NMDAR: F2,24 = 2.116, P = 0.115; kinases: F2,24 = 0.31, P = 0.861). Using a pull-down assay with biotinylated miR-33 or control miRNAs (scrambled miRNA and miRNA with a mutated seed sequence), we validated Gabrb2 (F2,6 = 11.78, P ≪ 0.01) and Kcc2 (F2,6 = 66.9, P < 0.001) mRNAs as targets for miR-33 using an immortalized hippocampal cell line (n = 3 samples per group; Fig. 4d). Because this cell line does not express Gabra4 mRNA, we performed quantitative PCR using Gabra4 as a negative control. Together, these findings suggest that endogenous hippocampal miR-33 mainly affects the translation of these mRNAs into proteins and only disrupts mRNA stability when miR-33 levels are elevated above baseline. This is consistent with previously reported data with other miR-33 targets20. In addition to its direct targets, miR-33 inhibition also affects the levels of other proteins, as revealed by proteomic analysis of pooled hippocampal lysates. Of the nine identified candidate proteins (Supplementary Fig. 8), synapsin-2 (Syn2) is of particular interest because it is a key regulator of asynchronous GABA release, which is thought to generate tonic currents mediated by extrasynaptic GABAA receptors21. We confirmed that Syn2a is not a miR-33 target (Fig. 4d), as Syn2 mRNA was not present in miR-33 pull-downs, despite its presence in the input mRNA (data not shown). Nevertheless, immunoblot analyses of individual hippocampal lysates confirmed that miR-33 manipulations specifically affected the level of Syn2a isoform (n = 5 hippocampi per group; LV-miR-33: t1,8 = 4.21, P < 0.05; miR-33-LNA: t1,8 = 6.54, P < 0.01), but not Syn2b isoform (LV-miR-33: t1,8 = 0.21, P = 0.87; miR-33-LNA: t1,8 = 0.54, P = 0.45) (Fig. 4e). Although significant, the changes of all examined GABA-related proteins were not marked, suggesting that the multitude of affected GABAA-related targets, rather than the magnitude of their changes, contributes the most to the behavioral sensitivity to gaboxadol.
Gaboxadol changes stimulus processing in the external hippocampal circuit
Contextual fear conditioning depends on neuronal plasticity in both the hippocampus and areas receiving direct hippocampal projections22. To establish whether gaboxadol alters plasticity in the extended hippocampal circuit, we compared the levels of early growth factor 1 (EGR-1) and cFos during encoding of context fear in the presence or absence of gaboxadol. These immediate early genes were selected on the basis of their well-documented roles in plasticity relevant for encoding of contextual fear23,24. We first established the main projections from the rostral-dorsal hippocampus by injecting SynaptoTag. This viral vector carries mCherry, which was expressed in the entire neuron, and Syn2-EGFP, which was only expressed at axon terminals (Supplementary Fig. 9a,b). We infused gaboxadol or vehicle through the same cannula 1 month later, performed fear conditioning and then collected brains for immunohistochemical analyses of early growth factor 1 (EGR-1) and cFos responses. Consistent with previous findings25, the most prominent sites of rostral-dorsal hippocampal projections were the lateral septum, retrosplenial cortex (RSC) and entorhinal cortex (EC) (Fig. 5a). Gaboxadol significantly increased the number of EGR-1–positive neurons in the dentate gyrus (F2,8 = 4.802, P < 0.05) and in the lateral septum (F2,8 = 6.879, P < 0.05), the main subcortical projection target of the dorsal hippocampus (Fig. 5b). On the contrary, the number of these neurons was significantly reduced in the cortical targets, the RSC (F2,8 = 11.293, P < 0.01) and EC (F2,8 = 7.046, P < 0.05). The trend of cFos responses to gaboxadol was similar, although a significant difference was only observed for the lateral septum (P < 0.05; Figs. 5c and 6a,b, and Supplementary Fig. 10).
Figure 5.

Gaboxadol modulates EGR-1 responses in the hippocampus and its subcortical and cortical projections. (a) Confocal images showing SynaptoTag (green) and EGR-1 (blue) immunostaining in the lateral septum, hippocampus, RSC and EC. (b) Quantification of EGR-1–positive neurons in hippocampal projections of mice injected i.h. with vehicle without fear conditioning (V, n = 3 mice), vehicle before fear conditioning (V-FC, n = 4 mice) or 0.5 μg per hippocampus of gaboxadol before fear conditioning (G-FC, n = 4 mice). (c) Quantification of cFos-positive neurons in adjacent brain sections. Significant changes were found in the lateral septum, dentate gyrus, RSC cortex and EC cortex. *P < 0.05 versus vehicle, #P < 0.05 and ##P < 0.01 versus V-FC (one-way ANOVA). Error bars represent s.e.m. in all panels.
Figure 6.

Photomicrographs illustrating the effect of gaboxadol on EGR-1 responses in the hippocampus and its subcortical and cortical projections. (a) Outline of hippocampal inputs used for quantification. (b) Micrographs showing average EGR-1 responses in mice of the V, V-FC and G-FC groups. The significant differences were confirmed with an independent replicate with 3 mice per group. We noted some variability in the CA1 response, which showed a trend toward reduced EGR-1 levels in the first experiment, and significantly reduced levels (P < 0.05) in the second experiment.
RSC suppresses gaboxadol-induced state-dependent fear
These changes in immediate early gene responses in the extended hippocampal circuit suggest a shift of information flow from the hippocampus to its subcortical rather than cortical targets during encoding of state-dependent context fear. This is consistent with the view that state-dependent memories are subcortical in nature and are not dependent or even suppressed by cortical activity26. We tested this hypothesis by inactivating RSC, which has a major role in retrieval of context-dependent fear-provoking memory27, during retrieval tests in the presence or absence of gaboxadol. We first infused an adenoviral vector carrying the inhibitory receptor of the designer receptors exclusively activated by designer drugs (DREADD) family, AAV8. hSyn.hM4D(Gi).mCherry, into the RSC and 6 weeks later exposed the mice to fear conditioning on vehicle or gaboxadol (Fig. 7a). The vehicle mice were then tested on vehicle (V-V) and gaboxadol mice on gaboxadol (G-G) when RSC was intact (intraperitoneal (i.p.) injection of vehicle) or inactivated (i.p. injection of the DREADD ligand clozapine-n-oxide, CNO). When RSC was intact, both groups froze similarly, as found in the previous experiments (Fig. 7a; n = 8 mice per group; t14 = 0.17, P = 0.72). However, when RSC was inactivated by CNO, the V-V group froze significantly less, whereas the G-G group froze significantly more, than on the previous test, revealing significant effects of RSC inactivation (F1,14 = 17.46. P < 0.01) and interaction between RSC inactivation and gaboxadol treatment (F1,14 = 19.19, P < 0.01) (Fig. 7b–f). Together, these findings demonstrate that state-dependent memories acquired under gaboxadol are best retrieved when RSC is inactivated. Thus, cortical mechanisms required for the retrieval of normally acquired memories are not required, and even impair retrieval of gaboxadol-induced state-dependent memories.
Figure 7.

Inactivation of RSC at memory retrieval enhances state-dependent fear. (a) Mice from the V-V and G-G groups showed similar freezing levels when RSC activity was intact. Inactivation of RSC with i.p. injection of CNO before the memory test significantly impaired freezing in the V-V group, but enhanced freezing in the G-G group. *P < 0.05 versus V-V (t test), #P < 0.05 and ##P < 0.01 versus corresponding group on a previous test (paired t test). Error bars represent s.e.m. (b) Top, a typical right-field image of a RSC-containing brain slice, from a wild-type mouse that had previously been stereotaxically injected in the RSC with AAV carrying the DREADD and the red fluorescent protein mCherry. All slices were first analyzed for virus spread and only slices of mice with a similar expression pattern of mCherry (n = 4 mice) were used for recordings. Bottom, epifluorescence image showing the expression pattern of infected RSC neurons. (c) Application of CNO to DREADD-expressing RSC neurons reduced their excitability by hyperpolarizing them and increasing the threshold current for firing. Example traces from a whole-cell recording of a DREADD-expressing layer 5 pyramidal neuron in RSC, sampled in current-clamp mode. Under control conditions (gray traces), with the cell at its resting membrane potential, current was injected via the patch pipette in 1-s-long steps (amplitudes of 50, 100 or 150 pA, from bottom to top) to assess the neuron’s suprathreshold excitability. Responses to the same family of current steps were acquired again after bath application of 100 nM CNO (red traces) and showed both a hyperpolarization of the resting membrane potential and a large reduction in the number of action potentials evoked by the step stimuli. (d) Frequency-current relationship for the same neuron. (e) Group analysis of the effect of CNO on the resting potentials of DREADD-expressing RSC neurons, showing a significant hyperpolarization (*P = 0.0039, signed rank test, n = 9 neurons). Gray lines indicate the individual neurons’ pre- and post-drug responses; the symbols connected by the black line represent the population means ± s.e.m. (f) Group analysis of the effect of CNO on rheobase (threshold current amplitude sufficient to generate firing), showing a significant increase (*P = 0.031, signed rank test, n = 9 neurons). Gray lines indicate the individual neurons’ pre- and post-drug responses; the circles connected by the black line represent the population means ± s.e.m.
DISCUSSION
Since its discovery26, state-dependent learning has been demonstrated in studies with humans28,29 and animals5; however, the data have not always been consistent30. Drugs that induce state-dependent operant conditioning or passive avoidance, such as benzodiazepines, NMDAR antagonists or scopolamine31,32, have proved ineffective in fear conditioning33–35. This may be a result of the very narrow dose-range that allows investigation in the absence of side effects, which commonly include changes of locomotor activity and thus confound analyses of freezing behavior. Alternatively, state-dependent regulation of context fear may be restricted to fewer neurobiological mechanisms, such as the hippocampal GABAergic pathway that we identified.
Under control (normal) conditions, fear conditioning critically depends on hippocampal glutamate receptors and cAMP-dependent protein kinase (PKA) signaling36–38. From this perspective, our experiments can be viewed as a comparison of fear responses encoded in two different states: a glutamate receptor/PKA-mediated state in control mice versus an extrasynaptic GABAA receptor/PKCβII-mediated state in gaboxadol-injected mice. These states seem to be separated by an amnestic barrier because, in both groups, retrieval of fear-provoking memory was confined to the state in which fear conditioning occurred. Notably, contextual fear acquired with or without gaboxadol showed many phenomenological similarities, such as contextual specificity, lack of generalization and comparable freezing levels. Nevertheless, the molecular mechanisms underlying fear conditioning in the presence or absence of gaboxadol were different, as revealed by the finding that PKCβII signaling and miR-33 had significant roles in GABAergic mechanisms of context fear, but showed no involvement in controls.
Fear conditioning in the presence and absence of gaboxadol induced opposite changes of the levels miR-33 and its GABA-related targets, which lasted at least up to 24 h post-training. Gabra4, Kcc2 and Gabrb2 mRNA or protein levels changed inversely with the level of miR-33, and these changes were consistent with the direction of behavioral susceptibility to gaboxadol. This is consistent with observations that microRNAs simultaneously target several functionally related mRNAs and proteins39. An unexpected observation was that levels of Syn2a, whose mRNA is not predicted to interact with miR-33, followed this pattern, suggesting that miR-33 indirectly regulates Syn2a through one of its primary targets.
Although some microRNAs are necessary for conditioning and extinction of fear40,41, we found that miR-33 did not affect fear conditioning or anxiety- or depression-like behavior (data not shown) under normal conditions. Instead, it regulated the threshold for GABAA receptor–mediated state-dependent fear. These findings are particularly important in view of the observations that these receptors control brain states ranging from sleep to heightened affect and psychosis42, and that brains of patients suffering from major depression and psychosis show consistent alterations of miR-33 (refs. 43,44) along with a disruption of the glutamatergic and GABAergic balance45. These findings suggest that some brain microRNAs can be predisposing factors for specific brain states and corresponding mental states or disorders.
Extrasynaptic GABAA receptors are especially abundant in interneurons of the dentate gyrus46; thus, an increase of EGR-1 responses in this hippocampal subfield by gaboxadol is most likely a result of disinhibition of the EGR-1 response to fear conditioning. To our surprise, such enhanced activity did not result in an overall enhancement of immediate early gene responses in the hippocampal projections. Instead, EGR-1, and in part cFos responses, were enhanced in subcortical targets of hippocampal inputs, but suppressed in cortical targets. These results experimentally support the conclusions of Girden and Cullar26, who, in their seminal work on state-dependent learning under curare, suggested that conditioning under curare is subcortical in nature and does not require, or is even suppressed by, cortical activity. Accordingly, we found that RSC suppresses the retrieval of state-dependent memories, even though it is required for retrieval of normally acquired memories.
Taken together, our results identify a molecular pathway involving extrasynaptic GABAA receptors, PKCβII signaling and miR-33 as a mediator of state-dependent encoding and retrieval of contextual fear. This pathway enhanced EGR-1 responses in the dentate gyrus of the hippocampus and promoted subcortical while inhibiting cortical processing of context memories. Our evidence for multiple mechanisms of fear has important implications for the treatment of patients experiencing fear and anxiety comorbid with other mental disorders47, as, despite similar manifestations, these symptoms may require disorder-specific therapies.
ONLINE METHODS
Animals
9-week-old male C57BL/6N mice were obtained from a commercial supplier (Harlan), individually housed on a 12-h light/dark cycle (lights on at 7 a.m.), and allowed ad libitum access to food and water. All procedures were approved by Northwestern University’s Animal Care and Use Committee in compliance with US National Institutes of Health standards. The individual experiments were not performed on littermates, so we did not apply randomization procedures, but all behavioral tests and immunohistochemical analyses were performed by experimentalists who were unaware of the treatments. During training, blinding was performed so that a laboratory member not involved in the experiments would prepare and color code the solution. In addition, the experimenter performing the tests was not aware of the numbering code.
Surgery and cannulation
Double guided cannulas (Plastic One) were implanted in the dorsal hippocampus or RSC as described previously27. Mice were anesthetized with 1.2% tribromoethanol (vol/vol, Avertin) and implanted with bilateral 26-gauge cannulas using a stereotaxic apparatus. Stereotaxic coordinates for the dorsal hippocampus were 1.7 mm posterior, ±1.0 mm lateral and 2.0 mm ventral to bregma, according to the mouse brain atlas48.
Pharmacological treatments
Drugs were injected i.h. at a volume of 0.25 μl per side at a rate of 0.15 μl min−1 or i.p. at a volume of 0.2 ml. Extrasynaptic GABAA receptors were activated by gaboxadol (varying doses, indicated in corresponding figures, dissolved in 0.9% saline; vol/vol, Sigma-Aldrich), PKCβII was inhibited by PKCβII peptide inhibitor I trifluoroacetate salt (0.25 μg per hippocampus, dissolved in artificial cerebrospinal fluid; Sigma-Aldrich) and PKCδ was inhibited by myristoyl (N-terminal) SFNSYELGSL peptide (0.5 μg per hippocampus, dissolved in artificial cerebrospinal fluid; GenScript).
labeling of hippocampal projections
Adeno-associated viral (AAV) vector containing SynaptoTag (0.5 μl) was injected into the dorsal hippocampus25. The vector components were arranged sequentially downstream from left ITR of AAV2: synapsin promoter, mCherry, IRES, Syb2-EGFP fusion, WPRE, hGH poly-A sequence, right ITR. The construct was used to generate AAV vectors as described25. 1 month after the injection of the AAV vector, mice were anesthetized with an i.p. injection of 240 mg per kg of body weight of Avertin and transcardially perfused with ice-cold 4% paraformaldehyde (vol/vol) in phosphate buffer (pH 7.4, 150 ml per mouse). Brains were post-fixed for 48 h in the same fixative and then immersed for 24 h each in 10, 20 and 30% sucrose (vol/vol) in phosphate buffer. The tissue was frozen in liquid nitrogen, and 50-μm coronal sections were used for visualization of mCherry and EGFP fluorescence by confocal laser-scanning microscope (Olympus Fluoview FV10i) at 40×.
Inactivation of the RSC using DREADD
The viral vector carrying a construCt coding for the inhibitory DREADD (AAV8.hSyn.hM4D(Gi)-mCherry, University of North Carolina Vector Core, 1012 vp ml−1) was bilaterally infused into the dorsal RSC 1.7 mm posterior, ±0.4 mm lateral, and 1.0 mm ventral to bregma1. Infusions were performed using an automatic microinfusion pump (CMA Microdialysis) connected to a Hamilton microsyringe. The viral vectors were infused in a volume of 0.4 μl per site over 5 min. 6 weeks later the animals were exposed to fear conditioning with or without gaboxadol. To inactivate the RSC, CNO was injected i.p. at dose of 2 mg per kg 40 min before the indicated memory test. After the experiments, tissue was collected and virus spread established during slice electrophysiology (Fig. 7) or immunohistochemically using anti-mCherry antibodies (1:1,500; Abcam, Ab167453; data not shown).
Fear conditioning
Contextual fear conditioning was performed in an automated system (TSE Systems) as previously described27. Briefly, mice were exposed for 3 min to a novel context, followed by a foot shock (2 s, 0.7 mA, constant current). 24 h later, mice were tested for memory retrieval. Testing consisted of 3 min in the conditioning context, during which freezing was measured every 10 s. In control experiments, mice were exposed to a novel context (3 min), followed by a tone (30 s, 74 dB SPL, 10 kHz) and a foot shock (2 s, 0.7 mA, constant current) to induce tone-dependent fear learning. During testing for fear to the tone, freezing was scored every 5 s in a novel context during a 30-s tone presentation. Freezing was expressed as a percentage of the total number of observations during which the mice were motionless.
miRNA microarray analysis
Mice were subjected to fear conditioning as described49. 24 h after the last session of fear conditioning, mice were killed by cervical dislocation, and dorsal hippocampi were excised and frozen in liquid nitrogen. Total RNA was extracted using miRCURY RNA Isolation Kit-Tissue (Exiqon) and sent to Exiqon for microRNA analysis. Two independent hippocampal samples for each naive and fear-conditioned mice were analyzed in triplicate (a total of 12 samples) by microRNA microarrays.
Proteomic analysis
Mice were injected i.h. with 0.25 μl per side of either 40 μM miR-33-LNA or scrambled control (miR-S-LNA). 1 week later, mice were killed by cervical dislocation, and dorsal hippocampi were excised, resuspended in a modified radioimmunoprecipitation (RIPA) buffer (50 mM Tris-HCl pH 7.4, 1% NP-40 (vol/vol), 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail (Boehringer Manheim), 1 mM Na3VO4 and 1 mM NaF), incubated for 15 min on ice, and centrifuged for 15 min (15,000g) at 4 °C. Samples were sent to Kendrick Labs, where they were analyzed by computerized comparisons of two-dimensional gel patterns for differences with the aid of SameSpots software v4.0 (Nonlinear Dynamics).
Western blot
Dorsal hippocampi were collected around the tips of the hippocampal cannulas. Tissue was lysed in modified RIPA buffer, incubated 15 min on ice, and centrifuged for 15 min (15,000g) at 4 °C. Samples were subjected to SDS-PAGE (10 μg per well) and transferred to PVDF membrane (Millipore). Membranes were blocked with I-block (Tropix), incubated with primary antibody overnight at 4 °C, and with secondary antibody for 1 h at 20–22 °C. Bands were detected using alkaline phosphatase chemiluminescence. Primary antibodies used were against β-tubulin (1:4,000, T4026, Sigma), KCC2 (1:2,000, MABN88, Millipore), GABRA4 (1:1,500, sc-20917, Santa Cruz), GABRB2 (1:1,500, ab8340, Abcam), pPKCα/β II (1:800, T638/641), pPKCβ II (1:800, S660), pPKCθ (1:800, T538), pPKCδ/θ (1:800, S638/676) (PKC Sampler kit #9921, Cell Signaling), synapsin-2 (1:1,000, NB 120-13258, Novus Biologicals), NR1 (1:6,000, sc-9058, Santa Cruz), NR2A (1:1,000, 07-632, Millipore), NR2B (1:2,000, ab65783, Abcam), PKA (1:20,000, ab76238, Abcam), CaMKII (1:6,000, C6974, Sigma), Erk-1/2 (1:4,000, M5670, Sigma). All antibodies gave bands at the predicted molecular sizes and were validated by preadsorption experiments with immunogenic peptides used for their production. We also validated the Erk and NR2A antibodies with tissue of knockout mice (data not shown).
Biotinylation assay
Mouse mHippoE-2 cells (Cedarlane Labs) were maintained in 1× DMEM with 10% fetal bovine serum (FBS, vol/vol), 25 mM glucose and 1% penicillin/streptomycin (vol/vol), and maintained at 37 °C with 5% CO2. Biotinylated miRNA mimics (miRIDIAN mimics, Dharmacon) used were miR-33 (GUGCAUUGUAGUUGCAUUGCA-biotin), scrambled miR-33 in which the miR-33 sequence was scrambled (GGCUCGAUGUUCGAAUAUUGU-biotin), and miR-33mut in which the seed sequence of miR-33 was replaced with the seed sequence of cel-miR-67, a commonly used control miRNA, as it does not target known mammalian mRNAs (GGCUCAUUUAGUUGCAUUGCA-biotin). Biotinylation assay was performed as previously described50. 1 d before transfection, 106 cells were seeded onto 10-cm tissue culture dishes (four dishes for each experimental group). The next day cells were transfected with 50 pmol of biotinylated miRNA per dish using RNAiMAX transfection reagent (Life Technologies). 24 h later cells were lysed, lysates from the four replicate dishes combined and biotinylated miRNAs pulled-down using Dynabeads MyOne Streptavidin C1 (Life Technologies). A portion of the cell lysate was saved as input sample. RNAs obtained in pull-down and input were purified using RNA Easy kit (Quiagen), and concentration measured using Nanodrop (Thermo Scientific). RNA amounts were quantified by real-time PCR. The enrichment ratio of the control-normalized pull-down RNA to the control-normalized input levels was calculated and expressed relative to miR-33 group, as described previously50,51.
Real-time PCR
Dorsal hippocampi were collected around the tips of the hippocampal cannulas. Total RNA was extracted using miRCURY RNA Isolation Kit-Tissue (Exiqon). Reverse transcription was performed on 20 ng of total RNA using Universal cDNA Synthesis Kit (Exiqon) for the analysis of microRNAs; on 100 ng of total RNA using First Strand cDNA Synthesis Kit (Applied Biosystems) for the analysis of mRNAs; or on 80 ng of total RNA using First Strand cDNA Synthesis Kit (Applied Biosystems) for the biotinylation assay. Real-time PCR analysis was performed on an Applied Biosystems 7300 instrument using SYBR green detection system (Applied Biosystems) and primers specific for miR-33 (Exiqon), miR-124 (Exiqon), miR-381-5p (Exiqon), miR-136-5p (Exiqon), miR-144-3p (Exiqon), miR-494-3p (Exiqon), GABRA4 (Qiagen), GABRB2 (Qiagen), KCC2 (Qiagen) or synapsin 2A (Qiagen).
Immunohistochemistry
Mice were anesthetized with an i.p. injection of 240 mg per kg of Avertin and transcardially perfused with ice-cold 4% paraformaldehyde in phosphate buffer (pH 7.4, 150 ml per mouse). Brains were postfixed for 48 h in the same fixative and then immersed for 24 h each in 10, 20 and 30 sucrose in phosphate buffer. Tissue was frozen in liquid nitrogen and 50-μm coronal sections were used for free-floating immunohistochemistry with primary antibodies against cFos (1:4,000, sc-52, Santa Cruz) or EGR-1 (1:8,000, sc-110, Santa Cruz) as described previously52. The antibodies were validated with tissue from cFos and Egr1 knockout mice obtained from M. Xu (University of Chicago) and W. Tourtellotte (Northwestern University), respectively (data not shown). Immunostaining with cFOS and EGR-1 was visualized with coumarin (TSA systems; excitation 402 nm, emission 443 nm). Slices were mounted in Vectashield (Vector) and observed with a confocal laser-scanning microscope (Olympus Fluoview FV10i) at 40×. Areas of synapsin immunostaining (green) were identified, marked, and superimposed on cFos or EGR-1 images. Quantification was performed using the ImageJ software (US National Institutes of Health) by an experimenter unaware of the experimental conditions. We captured single- or double-labeled images to accurately mark the areas in these regions that received hippocampal input (as revealed by green synapsin immunolabeling), and then quantified the number of EGR-1 and cFos positive–neurons. The individual samples were then decoded, and statistical analyses was performed.
miRNA inhibition
LNA microRNA inhibitors (Exiqon) were dissolved in water to 400 μM, heated at 65 °C for 10 min, cooled on ice, and diluted in artificial cerebrospinal fluid (ACSF) to a final working concentration of 40 μM. Scrambled LNA, which does not bind to any cellular microRNA, was used as a control. Mice were injected i.h. with 0.5 μl of the inhibitors.
miRNA overexpression
Plasmids (pMIRNA1/pCDH encoding miR-33) or scrambled miRNA driven by the CMV promoter were obtained from C. Fernández-Hernando. The plasmid was packaged into lentiviral particles at the Northwestern University Genomic Core Facility. For in vivo overexpression, lentiviral particles were injected i.h. at 0.25 μl per side, and allowed to incubate for 30 d before any experiments were performed. After the experiments tissue was collected and virus spread established immunohistochemically using mouse monoclonal anti-green fluorescent protein (GFP) antibodies (1:5,000; Abcam, Ab 1218). For in vitro miR-33 overexpression, B35 neuroblastoma cells (40% confluent) were treated with miR-33 lentiviral particles in the presence of 7 μg/ml polybrene. Cells were maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin (Cellgro), with occasional medium exchange. 8 d after treatment with lentiviral particles, total cellular RNA was extracted and processed for real-time PCR analysis.
Electrophysiology
Coronal brain slices (thickness, 0.3 mm) were prepared from mice injected into the RSC with DREADD virus (AAV8.hSyn.hM4D (Gi)-mCherry, University of North Carolina Vector Core). Whole-cell current-clamp recordings were performed as previously described53. Briefly, slices were transferred to the recording chamber of an upright microscope, and perfused with oxygenated artificial cerebrospinal fluid (ACSF) (in mM: 127 mM NaCl, 25 mM NaHCO3, 25 mM D-glucose, 2.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2 and 1.25 mM NaH2PO4), containing NBQX (10 μM), CPP (5 μM), and gabazine (20 μM), at maintained at 32 °C by a feedback-controlled in-line heater. Neurons expressing mCherry were visualized with bright-field and epifluorescence optics, and patched with borosilicate pipette (4–6 MΩ) filled with potassium internal solution (in mM: 128 potassium methanesulfonate, 10 HEPES, 10 phosphocreatine, 4 MgCl2, 4 ATP, 0.4 GTP, 3 ascorbate, 1 EGTA, 1 QX-314, pH 7.25, 290–295 mOsm). To determine rheobase, a family of current step stimuli (1-s duration, ranging in amplitude from −200 to 700 pA in 50-pA increments) was injected, with the cell at the resting membrane potential. The bath solution was switched from plain ACSF to ACSF containing CNO (100 nM), and after 10 min the responses to current steps were sampled again. Recordings with series resistance >25 MΩ were excluded from analysis. Signals were amplified with an Axon Multiclamp 700B (Molecular Devices), sampled at 40 KHz, filtered at 4 KHz. Data acquisition and hardware settings were controlled by Ephus software54. Group data were compared using the non-parametric signed rank test (signrank function in Matlab), with significance defined as P < 0.05. Because the experimental design was a within-slice treatment with all slices overexpressing DREADD, the data collection could not be randomized or blinded.
Statistics
Statistical analyses were performed using SPSS software. One-way ANOVA was followed by Tukey’s test for post hoc comparisons of three or more experimental groups (only when ANOVA was significant) or Student’s t tests for comparison of two experimental groups. Homogeneity of variance was confirmed with Levene’s test for equality of variances. All comparisons were conducted using two-tailed tests and the P value for all cases was set to <0.05 for significant differences. Group sizes were determined using power analysis assuming a moderate effect size of 0.5.
A Supplementary methods checklist is available.
Supplementary Material
Acknowledgments
We thank W. Xu and T.C. Sudhof (Stanford University) for providing us with the SynaptoTag viral vector, C. Fernandez-Hernando (Yale School of Medicine) for providing pMIRNA1/pCDH plasmids encoding miR-33 and scrambled miRNA, and the Genomic Core (Northwestern University) for generating lentiviral vectors containing miR-33 and scrambled constructs. We also thank B. Frick and F. Kassam for their help with the behavioral experiments. This work was supported by US National Institutes of Health grants NIH/NIMH MH078064 (J.R.), NIH/NINDS NS061963 and NS087479 (G.M.G.S.), and a Ken and Ruth Davee Award for Innovative Investigations in Mood Disorders, (J.R. and V.J.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
V.J. and J.R. designed the experiments, analyzed the data and wrote the manuscript. V.J. performed the experiments. K.A.C. and K.L. performed some of the behavioral and biochemical experiments. H.J.C. and A.L.G. helped with the biochemical experiments. N.Y. and G.M.G.S. performed the electrophysiological recordings.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Pompilio L, Kacelnik A, Behmer ST. State-dependent learned valuation drives choice in an invertebrate. Science. 2006;311:1613–1615. doi: 10.1126/science.1123924. [DOI] [PubMed] [Google Scholar]
- 2.Reus VI, Weingartner H, Post RM. Clinical implications of state-dependent learning. Am J Psychiatry. 1979;136:927–931. doi: 10.1176/ajp.136.7.927. [DOI] [PubMed] [Google Scholar]
- 3.Silberman EK, Putnam FW, Weingartner H, Braun BG, Post RM. Dissociative states in multiple personality disorder: a quantitative study. Psychiatry Res. 1985;15:253–260. doi: 10.1016/0165-1781(85)90062-9. [DOI] [PubMed] [Google Scholar]
- 4.Spiegel D, Hunt T, Dondershine HE. Dissociation and hypnotizability in posttraumatic stress disorder. Am J Psychiatry. 1988;145:301–305. doi: 10.1176/ajp.145.3.301. [DOI] [PubMed] [Google Scholar]
- 5.Overton DA. Historical context of state-dependent learning and discriminative drug effects. Behav Pharmacol. 1991;2:253–264. [PubMed] [Google Scholar]
- 6.Piri M, Rostampour M, Nasehi M, Zarrindast MR. Blockade of the dorsal hippocampal dopamine D1 receptors inhibits the scopolamine-induced state-dependent learning in rats. Neuroscience. 2013;252:460–467. doi: 10.1016/j.neuroscience.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Möhler H. Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors. J Neurochem. 2007;102:1–12. doi: 10.1111/j.1471-4159.2007.04454.x. [DOI] [PubMed] [Google Scholar]
- 8.Stein L, Berger BD. Paradoxical fear-increasing effects of tranquilizers: evidence of repression of memory in the rat. Science. 1969;166:253–256. doi: 10.1126/science.166.3902.253. [DOI] [PubMed] [Google Scholar]
- 9.Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mortensen M, Patel B, Smart TG. GABA potency at GABA(A) receptors found in synaptic and extrasynaptic zones. Front Cell Neurosci. 2011;6:1. doi: 10.3389/fncel.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandra D, et al. GABAA receptor alpha 4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc Natl Acad Sci USA. 2006;103:15230–15235. doi: 10.1073/pnas.0604304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanchard RJ, Blanchard DC. Crouching as an index of fear. J Comp Physiol Psychol. 1969;67:370–375. doi: 10.1037/h0026779. [DOI] [PubMed] [Google Scholar]
- 13.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 14.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 15.Song M, Messing RO. Protein kinase C regulation of GABAA receptors. Cell Mol Life Sci. 2005;62:119–127. doi: 10.1007/s00018-004-4339-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sathyan P, Golden HB, Miranda RC. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J Neurosci. 2007;27:8546–8557. doi: 10.1523/JNEUROSCI.1269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sengupta JN, et al. MicroRNA-mediated GABAAalpha-1 receptor subunit down-regulation in adult spinal cord following neonatal cystitis-induced chronic visceral pain in rats. Pain. 2013;154:59–70. doi: 10.1016/j.pain.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao C, et al. Computational prediction of MicroRNAs targeting GABA receptors and experimental verification of miR-181, miR-216 and miR-203 targets in GABA-A receptor. BMC Res Notes. 2012;5:91. doi: 10.1186/1756-0500-5-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barmack NH, Qian Z, Yakhnitsa V. Long-term climbing fiber activity induces transcription of microRNAs in cerebellar Purkinje cells. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130508. doi: 10.1098/rstb.2013.0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rayner KJ, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medrihan L, Ferrea E, Greco B, Baldelli P, Benfenati F. Asynchronous GABA release is a key determinant of tonic inhibition and controls neuronal excitability: a study in the Synapsin II−/− mouse. Cereb Cortex. 2014 Jun 24; doi: 10.1093/cercor/bhu141. published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orsini CA, Kim JH, Knapska E, Maren S. Hippocampal and prefrontal projections to the basal amygdala mediate contextual regulation of fear after extinction. J Neurosci. 2011;31:17269–17277. doi: 10.1523/JNEUROSCI.4095-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radulovic J, Kammermeier J, Spiess J. Relationship between fos production and classical fear conditioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci. 1998;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bozon B, Davis S, Laroche S. A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron. 2003;40:695–701. doi: 10.1016/s0896-6273(03)00674-3. [DOI] [PubMed] [Google Scholar]
- 25.Xu W, Sudhof TC. A neural circuit for memory specificity and generalization. Science. 2013;339:1290–1295. doi: 10.1126/science.1229534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Girden E, Culler E. Conditioned responses in curarized striate muscle in dogs. J Comp Psychol. 1937;23:261–274. [Google Scholar]
- 27.Corcoran KA, et al. NMDA receptors in retrosplenial cortex are necessary for retrieval of recent and remote context fear memory. J Neurosci. 2011;31:11655–11659. doi: 10.1523/JNEUROSCI.2107-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodwin DW, Powell B, Bremer D, Hoine H, Stern J. Alcohol and recall: state-dependent effects in man. Science. 1969;163:1358–1360. doi: 10.1126/science.163.3873.1358. [DOI] [PubMed] [Google Scholar]
- 29.Bustamante JA, Jordan A, Vila M, Gonzalez A, Insua A. State dependent learning in humans. Physiol Behav. 1970;5:793–796. doi: 10.1016/0031-9384(70)90281-7. [DOI] [PubMed] [Google Scholar]
- 30.Weafer J, Gallo DA, de Wit H. Amphetamine fails to alter cued recollection of emotional images: study of encoding, retrieval and state-dependency. PLoS ONE. 2014;9:e90423. doi: 10.1371/journal.pone.0090423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koek W. Drug-induced state-dependent learning: review of an operant procedure in rats. Behav Pharmacol. 2011;22:430–440. doi: 10.1097/FBP.0b013e328348ed3b. [DOI] [PubMed] [Google Scholar]
- 32.Colpaert FC, Koek W, Bruins Slot LA. Evidence that mnesic states govern normal and disordered memory. Behav Pharmacol. 2001;12:575–589. doi: 10.1097/00008877-200112000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Maren S, Aharonov G, Stote DL, Fanselow MS. N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 34.Anagnostaras SG, Maren S, Fanselow MS. Scopolamine selectively disrupts the acquisition of contextual fear conditioning in rats. Neurobiol Learn Mem. 1995;64:191–194. doi: 10.1006/nlme.1995.0001. [DOI] [PubMed] [Google Scholar]
- 35.Davis M. Diazepam and flurazepam: effects on conditioned fear as measured with the potentiated startle paradigm. Psychopharmacology (Berl) 1979;62:1–7. doi: 10.1007/BF00426027. [DOI] [PubMed] [Google Scholar]
- 36.Gao C, et al. IQGAP1 regulates NR2A signaling, spine density and cognitive processes. J Neurosci. 2011;31:8533–8542. doi: 10.1523/JNEUROSCI.1300-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Impey S, et al. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- 38.Abel T, et al. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 39.Merchan F, Boualem A, Crespi M, Frugier F. Plant polycistronic precursors containing non-homologous microRNAs target transcripts encoding functionally related proteins. Genome Biol. 2009;10:R136. doi: 10.1186/gb-2009-10-12-r136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin Q, et al. The brain-specific microRNA miR-128b regulates the formation of fear-extinction memory. Nat Neurosci. 2011;14:1115–1117. doi: 10.1038/nn.2891. [DOI] [PubMed] [Google Scholar]
- 41.Griggs EM, Young EJ, Rumbaugh G, Miller CA. MicroRNA-182 regulates amygdala-dependent memory formation. J Neurosci. 2013;33:1734–1740. doi: 10.1523/JNEUROSCI.2873-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brickley SG, Mody I. Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron. 2012;73:23–34. doi: 10.1016/j.neuron.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreau MP, Bruse SE, David-Rus R, Buyske S, Brzustowicz LM. Altered microRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol Psychiatry. 2011;69:188–193. doi: 10.1016/j.biopsych.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banigan MG, et al. Differential expression of exosomal microRNAs in prefrontal cortices of schizophrenia and bipolar disorder patients. PLoS ONE. 2013;8:e48814. doi: 10.1371/journal.pone.0048814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Penzes P, Buonanno A, Passafaro M, Sala C, Sweet RA. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J Neurochem. 2013;126:165–182. doi: 10.1111/jnc.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milenkovic I, et al. The parvalbumin-positive interneurons in the mouse dentate gyrus express GABAA receptor subunits alpha1, beta2, and delta along their extrasynaptic cell membrane. Neuroscience. 2013;254:80–96. doi: 10.1016/j.neuroscience.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 47.Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Franklin K, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Elsevier; 2004. [Google Scholar]
- 49.Huh KH, et al. Hippocampal Erk mechanisms linking prediction error to fear extinction: roles of shock expectancy and contextual aversive valence. Learn Mem. 2009;16:273–278. doi: 10.1101/lm.1240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ørom UA, Lund AH. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods. 2007;43:162–165. doi: 10.1016/j.ymeth.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Lal A, et al. Capture of microRNA-bound mRNAs identifies the tumor suppressor miR-34a as a regulator of growth factor signaling. PLoS Genet. 2011;7:e1002363. doi: 10.1371/journal.pgen.1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guzmán YF, et al. Fear-enhancing effects of septal oxytocin receptors. Nat Neurosci. 2013;16:1185–1187. doi: 10.1038/nn.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamawaki N, Borges K, Suter BA, Harris KD, Shepherd GM. A genuine layer 4 in motor cortex with prototypical synaptic circuit connectivity. Elife. 2014;3:e05422. doi: 10.7554/eLife.05422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suter BA, et al. Ephus: multipurpose data acquisition software for neuroscience experiments. Front Neural Circuits. 2010;4:100. doi: 10.3389/fncir.2010.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.