

Abstract
Extracellular superoxide dismutase (SOD3) gene transfer to tissue damage results in increased healing, increased cell proliferation, decreased apoptosis, and decreased inflammatory cell infiltration. At molecular level, in vivo SOD3 overexpression reduces superoxide anion (O2 −) concentration and increases mitogen kinase activation suggesting that SOD3 could have life-supporting characteristics. The hypothesis is further strengthened by the observations showing significantly increased mortality in conditional knockout mice. However, in cancer SOD3 has been shown to either increase or decrease cell proliferation and survival depending on the model system used, indicating that SOD3-derived growth mechanisms are not completely understood. In this paper, the author reviews the main discoveries in SOD3-dependent growth regulation and signal transduction.
1. Introduction
Extracellular superoxide dismutase (EC-SOD, SOD3) [1, 2], similar to cytosolic CuZn-SOD (SOD1) [3] and mitochondrial MnSOD (SOD2) [4, 5], catalyzes the dismutation of superoxide anion (O2 −) into hydrogen peroxide (H2O2) (in this review reactive oxygen species refer to O2 − and H2O2), which to date is the only reported physiological function of the enzyme. Thus, the cellular effects of SOD enzyme activity are caused by changes in the local concentrations of O2 − and H2O2, which are second messengers in signal transduction that have an impact on growth capacity and the transformation of primary cells. Although the enzymes have a significant therapeutic potential their delivery to injury site is challenging due to limitations in gene transfer efficiency. Hence researchers have developed SOD mimics that function similarly with SOD enzymes regulating redox balance with consequent impact on growth, differentiation, and death [6–10]. The importance of local regulation of reactive oxygen species (ROS) by SOD3 has been highlighted by our previous studies of local and systemic delivery of sod3 via adenovirus to sites of cardiovascular injury: both gene transfer methods increase plasma SOD activity, but only the local gene delivery demonstrates a therapeutic response [11]. The data is supported by observations reporting that Arg-213-Gly mutation at C-terminal end of SOD3 reduces the affinity of the enzyme to heparan sulphate proteoglycans of endothelial cells thus increasing plasma SOD3 concentration by 10-fold [12, 13]. The mice carrying Arg-213-Gly mutation have tissue level changes, such as increased neutrophil mediated inflammation, cellular degeneration and premature aging, abnormal gait, and reduced lifetime that may be result of increased neutrophil ROS production [14]. Based on the abovementioned data decreased SOD3 content at cell membranes impairs life-supporting cellular functions. Notably, H2O2 can have toxic effects on cellular functions at high concentrations, thus suggesting a need to regulate ROS production in the tissue environment. Indeed, a number of reports have demonstrated tight regulation of SOD3 expression at the transcriptional, posttranscriptional, and posttranslational levels [12, 15–23]. This regulation is influenced by various factors, most importantly by the level of O2 − substrate and the reaction end product H2O2 [23–25].
2. Therapeutic Effects of SOD3 Overexpression
One of the first milestones in SOD3 research was the discovery of the tissue-protective nature of the enzyme in cardiovascular models. The earliest observations reported reduced cardiovascular damage by recombinant SOD3 administration [26–30]; these observations were confirmed by a series of gene transfer studies [11, 24, 31–39] and later reviewed in [40–44]. Characteristically, treatment of cardiovascular tissues with SOD3 reduces the extent of the damage, increases the healing process, improves cardiac function, reduces the remodeling of vasculature, attenuates apoptosis, inhibits inflammatory and smooth muscle cell migration, and increases cell proliferation and endothelial cell layer recovery. The role of SOD3 in neoangiogenesis is less clear. We have reported increased endothelialization and reduced macrophage and smooth muscle cell migration with consequent long-term inhibition of neointima formation in rabbit denudation and in rabbit in-stent models [11, 38], suggesting a role for the enzyme in vascular cell proliferation and inflammatory cell migration. We have further demonstrated, using rat hind limb injury model, SOD3-dependent increases in tissue injury recovery that were mediated by activation of mitogen signal transduction with consequent increased satellite cell proliferation in muscles [24]; by activation of antiapoptotic signaling that involved increased extracellular signal regulated kinase 1/2 (ERK1/2), protein kinase B (AKT), and forkhead box O3a (FOXO3a) activation [39]; and by reduction of macrophage-specific inflammation, which was correlated with reduced expression of the inflammatory cytokines tumor necrosis factor α (TNFα), interleukin 1α (IL1α), interleukin 6 (IL6), macrophage inflammatory protein 2 (MIP2), and monocyte chemotactic protein 1 (MCP-1) and the adhesion molecules vascular adhesion molecule (VCAM), intercellular adhesion molecule (ICAM), P-selectin, and E-selectin [36]. Although we did not observe increased neoangiogenesis by overexpressing SOD3, another recent study performed in SOD3 knockout mice suggested defective vessel formation in the absence of the enzyme. The authors demonstrated that SOD3 does not directly promote vascular endothelial growth factor receptor (VEGFR) activation but it is able to enhance the ability of VEGF ligand to phosphorylate VEGF-R [45]. Thus, in vivo data suggest that SOD3 expression activates growth-promoting, antiapoptotic, and anti-inflammatory signal transduction pathways in cardiovascular models (Figure 1).
Figure 1.
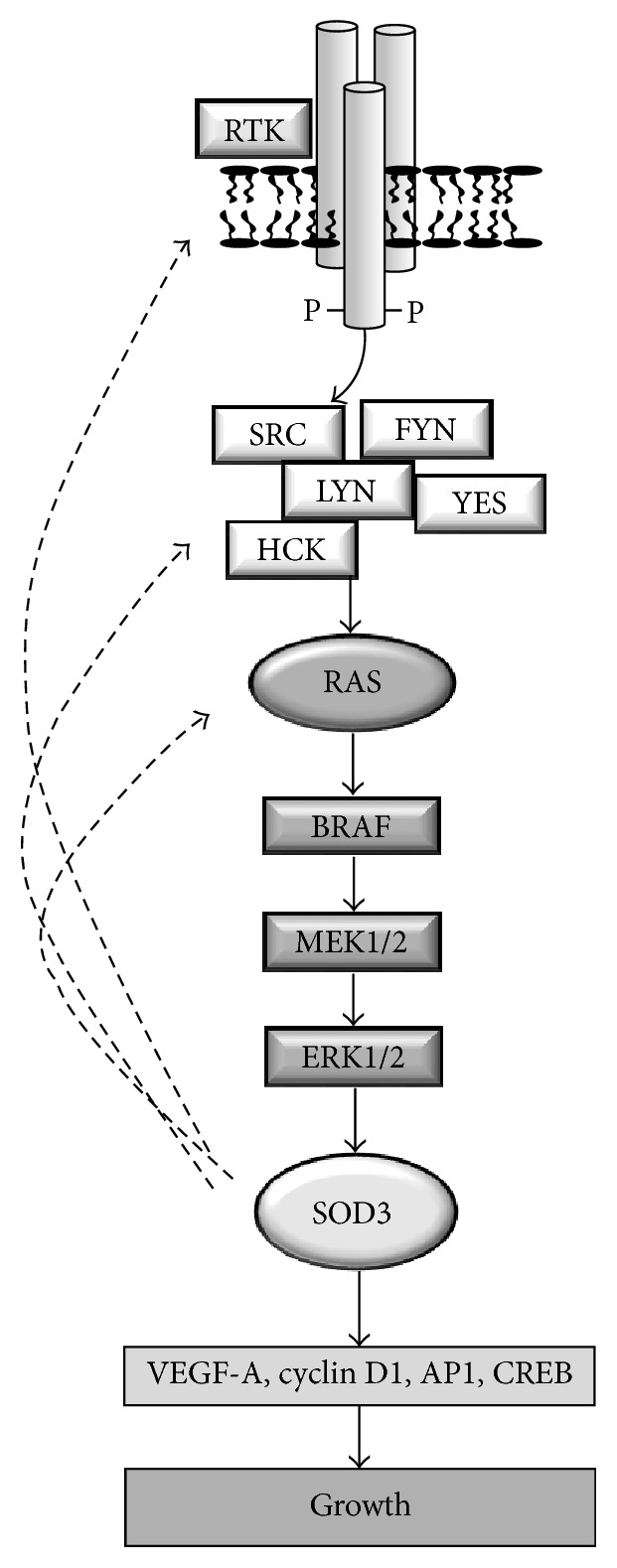
Suggested positive feedback loop in SOD3 signal transduction. Phosphorylation of RTKs activates the cell membrane associated SRC proto-oncogene family members that contribute to RAS GTP loading and stimulation of mitogenic signal transduction to BRAF, MEK1/2, and ERK1/2 kinases. In vitro transient transfection of RAS, BRAF, MEK1/2, and ERK1/2 increases both SOD3 mRNA and protein expression hence suggesting mitogen pathway induced SOD3 synthesis. SOD3 production results in increased synthesis of growth promoters, such as VEGF and cyclin D1, and increased activation of activator protein 1 (AP1) and cAMP response element-binding protein (CREB). Importantly, SOD3 activates cell surface receptor tyrosine kinases (RTKs), increases phosphorylation of SRC family members, and regulates the GTP loading to small GTPases, such as RAS.
The function of SOD3 in lung models has been investigated using SOD3 null and transgenic mice. The earliest observations suggested that SOD3 null mice had a significantly shortened life span and experienced death associated with lung edema under conditions of hyperoxia [46]. These observations were confirmed in conditional knockout mice that showed reduced survival associated with disorders resembling adult respiratory distress syndrome, such as thickening of alveolar septa, increased inflammation, hemorrhage, and loss of patent alveoli [47]. Hence, the lung model data support results obtained from cardiovascular damage models, suggesting survival-supporting and growth-promoting roles for SOD3 in the tissue environment.
The most dramatic prosurvival effect of SOD3 has been observed in total body irradiation (TBI) studies. In one study, intravenous administration of 0.5 × 106 mesenchymal stem cells (MSCs) previously transduced with SOD3-expressing adenovirus multiplicity of infection (MOI) 2000 resulted in a 90% survival rate 35 days after 9 Gy TBI without hematopoietic stem cell (HSC) transfusion, whereas 90% of control animals died [48]. These data were confirmed in a study of mice receiving 5.81 Gy TBI, which showed a similar survival rate [49]. In this study, in the absence of HSC transplantation, the transfusion of MSCs (1 × 106) transduced with SOD3-expressing adenovirus (MOI 50) resulted in a 90% 30-day survival rate compared to a 20% survival rate in control animals. Blood value analysis 10 days after TBI demonstrated that there were eightfold higher white blood cell counts (1.1 × 108 in controls versus 8.9 × 108 in the SOD3 group), 40-fold higher platelets values (2.4 × 109 in controls versus 97 × 109 in the SOD3 group), and significantly increased hemoglobin levels (105 g/L in controls versus 128 g/L in the SOD3 group) in SOD3-treated animals compared to controls [49]. Although the authors concluded that the increased survival was caused by significantly decreased apoptosis in SOD3-treated animals, another possible survival mechanism could be increased cycling of primitive HSCs with consequent hematopoietic cell differentiation. The data provided by Gan and coworkers suggested that the gene expression of members (i.e., p53, p21, and p16) of the p53-mediated growth arrest pathway was reduced in MSC-SOD3-transplanted animals [49], supporting the hypothesis that increased SOD3-driven mitogen stimulus in the bone marrow together with reduced apoptosis might explain the increased survival after TBI observed in SOD3-treated animals. Previous studies have suggested a common bone marrow niche and homing site for HSCs and MSCs [50, 51], thus indicating that SOD3-treated MSCs could have a paracrine effect on quiescent HSCs, inducing them to proliferate and to differentiate by directly affecting primitive progenitor cell cycling or via erythropoietin signaling [52]. Hence, the in vivo data observed from various animal models suggest that SOD3 maintains normal tissue homeostasis by promoting cell survival and proliferation.
3. Hydrogen Peroxide Action in Signal Transduction
Hydrogen peroxide regulates a number of cellular functions, such as cell proliferation, differentiation, migration, and survival. The first evidence that H2O2 could function as a second messenger came from studies demonstrating increased H2O2 production in association with increased platelet-derived growth factor (PDGF), epithelial growth factor (EGF), and vascular endothelial growth factor (VEGF) receptor tyrosine kinase phosphorylation with simultaneously reduced protein tyrosine phosphatase (PTP) activity [53–55]. In general, ROS are able to affect cell signaling by two mechanisms: (1) by inactivating PTPs, thereby increasing tyrosine kinase phosphorylation, and (2) by directly oxidizing tyrosine kinase receptors, causing their phosphorylation [56, 57].
H2O2 and O2 ∙− are known to be involved in the initiation of tumorigenesis and in malignant transformation [58]. In addition to increasing cell proliferation, survival, and migration, H2O2 activates SRC family proto-oncogenes, which regulate vascular development and vascular permeability [59], the latter of which is an early step in tumor stroma development [60]. The role of H2O2-derived signaling in the later phase of tumor development allows cancer cell survival in hypoxic environments by maintaining activation of the AKT pathway, with a consequent increased expression of hypoxia inducible factor 1α [61].
4. SOD3 Expression in Tumorigenesis
Previous data have suggested that there is a correlation between increased sod3 mRNA production and increased growth of benign tumors [62], indicating a role for SOD3 in early tumorigenesis. In vitro studies have supported this conclusion by demonstrating that moderate overexpression of SOD3 stimulates mouse primary embryonic fibroblast (MEF) cell proliferation, mimicking the RAS oncogene response in primary cells [63] and further corroborating the close relationship of SOD3 expression and cellular growth. Consistent with these results, sod3 mRNA synthesis is upregulated at low RAS activation levels; however, sod3 mRNA expression, which negatively correlates with mir21 expression, is strongly downregulated when the RAS activation level increases to ≥10-fold relative to parental cells [23]. In contrast to the case in benign growth, SOD3 expression is progressively downregulated in a number of cancers and cancer cell lines [62, 64–67], correlating with the RAS activation level [23], which suggests that sod3 could be a prognostic differentiation marker. Silencing of the SOD3 gene can be divided into reversible immediate events and stable late events. Immediate events following RAS activation occur via SOD3 self-regulation through small RAS GTPase regulatory genes, mir21 upregulation, and p38 MAPK phosphorylation [23, 68–72], whereas late regulatory events consist of DNA methylation and histone acetylation [15, 16, 72–74]. The correlation of decreased SOD3 expression with increased malignancy has led to the hypothesis that SOD3 could function as a tumor suppressor that must be silenced to allow the progression of carcinogenesis [66]. Although the hypothesis is feasible based on conventional tumor suppressor gene silencing mechanisms, the mechanisms of how reduced SOD3 expression could increase transformed cell proliferation have not been fully elucidated.
5. SOD3 as a Growth Promoter in Tumorigenesis
As mentioned above, SOD3 has been shown to promote normal primary cell proliferation in various model systems [11, 24, 33, 34, 38, 63]. The close connection of SOD3 to growth-associated signal transduction was demonstrated in a recent microarray functional KEGG and GO pathway analysis suggesting that the highest number of SOD3-affected genes was in the MAPK signaling (254 genes, p < 0.02) and endothelial cell proliferation pathways (33 genes, p < 0.018). Other significantly affected pathways included various cancer-associated signal transduction and cell proliferation pathways [75]. We have previously shown that RAS-BRAF-MEK1/2-ERK1/2, a major signal transduction pathway in cancer, activates SOD3 mRNA expression and enzyme activity in vitro and in vivo, which then increases GTP loading to RAS [24]. These data suggest the existence of a positive feedback loop that maintains the mitogen pathway in a phosphorylated state, inducing growth-supporting and antiapoptotic signal transduction pathways in injured tissues [24, 39] (Figure 1). We have further shown that thyroid stimulating hormone (TSH), cAMP-PKA, and PLC-Ca2+ increase sod3 production in thyroid cells, demonstrating that SOD3 in the thyroid contributes to cell proliferation and differentiation [62]. The role of SOD3 in growth promotion was further strengthened by data indicating that expression of SOD3 induces the activation of AP-1, c-Jun, and CRE promoter regions; increased expression of FOXO3a and FOXQ1 transcription factors; and increased expression of the cell cycle proteins cyclin D1, cell division cycle 25A (CDC25A), and proliferating cell nuclear antigen (PCNA) [24, 39, 63]. Importantly, H2O2 treatment of cells has been shown to stimulate SOD3 mRNA synthesis and mimic SOD3 function in cells that were treated with N-acetylcysteine (NAC) and diphenyleneiodonium (DPI), suggesting substrate specific regulation of the enzyme [23, 24, 75].
Signal transduction studies have suggested that cell membrane bound SOD3 increases the phosphorylation of the cell membrane tyrosine kinase (RTK) receptors, epidermal growth factor receptor (EGFR), erb-b2 receptor tyrosine kinase 2 (ERBB2), receptor-like tyrosine kinase (RYK), anaplastic lymphoma kinase (ALK), Fms-like tyrosine kinase 3 (FLT3), Ephrin A10 (EPHA10), and VEGF-R [45, 67, 75]; cell membrane-associated signaling molecules, such as the SRC proto-oncogene family members HCK, FYN, SRC, YES, and LYN; and cytoplasmic signaling molecules, including AKT, glycogen synthase kinase 3 (GSK3), and β-catenin [75]. Hence, SOD3 overexpression activates two main growth-related signal transduction pathways, RAS-ERK1/2 and β-catenin cascades (Figure 2).
Figure 2.

Suggested model for dose-dependent effect of SOD3 on RAS activation and β-catenin cellular localization. Moderately increased SOD3 expression at cell membranes promotes cell membrane bound RAS GTP loading by activating GEF expression and by inhibiting GAP and GDI synthesis causing increased RAS-ERK1/2 signaling. Robustly increased SOD3 expression inhibits RAS GTP loading by inhibiting GEF expression and by activating GAP and GDI synthesis causing decreased RAS-ERK1/2 signaling. Moderately increased SOD3 expression promotes AKT and GSK3 phosphorylation and β-catenin nuclear entry, whereas robustly increased SOD3 expression arrests β-catenin to cytoplasm by increasing the expression of WWTR1, SNAI2, and AXIN2. Note that both moderate and robust SOD3 expressions increase the phosphorylation of RTKs, SRCs, AKT, and GSK3. SOD3 dose-dependent signal transduction regulation occurs at the level of small GTPases and β-catenin cytoplasm-nuclear localization.
Interestingly, recent studies have indicated that reducing the expression of SOD3 to physiological levels can increase the growth of transformed malignant cells in vitro and in vivo. Based on in vivo data, VEGF-C-driven SOD3 expression increases the tumorigenesis and metastasis of xenografted mammary cells [67]. Additionally, knockdown of VEGF-C in mammary cancer cell lines significantly reduces the expression of SOD3, tumor formation, and metastasis of the cells, whereas restoration of SOD3 expression in VEGF-C knockdown cells to the levels of control cells with carcinogenic characteristics partly recovers the aggressiveness of the cells, increasing both primary tumor growth and metastasis [67]. The growth-promoting effects of SOD3 are further supported by studies in cancer cell lines harboring decreased endogenous SOD3 expression; transfection of SOD3 into these cell lines results in in vitro and in vivo growth selection of cells, favoring those with modestly increased SOD3 levels. In vitro transfection of high SOD3 concentrations into cancer cells followed by mixed population long-term culture results in apoptosis of cells with high supraphysiological concentrations of SOD3 plasmid, whereas cells that contained moderately increased SOD3 expression compared to control cancer cells took over the culture due to their increased proliferation capacity. In vivo studies with xenografted luciferase-marked cells support this observation, showing an initial decrease in tumorigenesis and in luciferase signal in tumors derived from SOD3-transfected cancer cells containing a strong increase in SOD3 mRNA expression. The initial decreased growth phase was followed by faster tumor development and in vivo selection of cells, which contained moderately increased SOD3 mRNA expression levels compared to control cell-derived tumors [63]. Thus, SOD3, by affecting local ROS concentrations, might have progrowth characteristics in early tumorigenesis as a mediator of the RAS oncogene and, in certain cellular environments, may work in coordination with other growth factors that stimulate cancer cell proliferation.
6. SOD3 as a Growth Suppressor in Cancer
Various studies have demonstrated cancer growth suppression caused by supraphysiological SOD3 overexpression in vitro and in vivo. In general, these studies have been performed using cells transfected with SOD3 at high concentrations or using cells transduced with adenovirus expressing SOD3 [63, 75–79], which induces strong mRNA production, thus reaching supraphysiological SOD3 and H2O2 levels for three to four days [80]. High expression of SOD3 results in DNA damage and activation of the DNA damage response (DDR), including phosphorylation of histone H2AX, phosphorylation of checkpoint kinase1/2 (CHK1/2), phosphorylation of p53, increased production of p21, and consequent growth arrest and apoptosis [63]. Additionally, supraphysiological SOD3 expression in anaplastic cancer cells activates AKT-GSK3-β-catenin signaling but prevents β-catenin nuclear transfer by increasing the gene expression of WW domain-containing transcription regulator protein 1 (WWTR1), snail homolog 2 (SNAI2), and axis inhibition protein 2 (AXIN2) [75], which are responsible for β-catenin cytoplasmic arrest, binding, and degradation, respectively [81–83].
At the tissue level, supraphysiological SOD3 overexpression correlates with reduced oxidative stress marker 4-hydroxynonenal staining in xenografted tumors and decreased intracellular dihydroethidium staining in cancer cells transduced with adenovirus expressing SOD3 [77, 78]. Functionally, supraphysiological overexpression of SOD3 inhibits the nuclear localization of NF-κB, reduces VEGF-A expression, decreases cell proliferation, inhibits tumor growth, decreases metastasis (suggesting a reduction of in vivo cancer cell migration), and increases apoptosis [76–78]. Furthermore, SOD3 has been shown to affect hypoxia inducible factor HIF-1α signaling, which enables vascular growth, thus regulating tumor progression. supraphysiological SOD3 overexpression by adenovirus (MOI 50–100) decreases HIF-1α levels by inducing degradation, whereas virus doses of MOI 25 or less have minor or no impact on HIF-1α levels [84].
7. SOD3 Affects Growth in a Dose-Dependent Manner
SOD3 overexpression studies have demonstrated a dual role for the enzyme in growth regulation depending on the expression level of the enzyme: rescued or moderately increased SOD3 expression promotes cell proliferation, whereas supraphysiological overexpression of the enzyme causes growth arrest and apoptosis [63, 67, 75, 77, 78]. Notably, moderately increased SOD3 levels stimulate cell proliferation, mimicking the function of the RAS oncogene in primary cultures and causing mitogenic burst followed by growth arrest-related senescence, immortalization of primary cells, and even transformation of the cells together with additional changes in cellular signaling [63, 85–87]. Interestingly, our data have suggested the existence of SOD3 dose-dependent regulation of downstream signal transduction at the level of RAS small GTPases [24, 75]. A moderate twofold increase in SOD3 activity in tissues markedly increases RAS GTP loading and downstream growth signaling [24, 75], whereas robust supraphysiological SOD3 overexpression decreases RAS, RAC, RHO, and CDC42 activation by regulating the gene expression of regulators of these small GTPases. Mechanistically, moderate SOD3 expression increases the mRNA expression of guanine nucleotide exchange factors (GEFs) responsible for GTP loading to GTPases and decreases the expression of GTPase activating proteins (GAPs) and guanine nucleotide dissociation inhibitors, which are responsible for maintaining GDP-RAS association and inhibiting localization of GTPases to cell membranes, where they are activated [88–90]. Robust supraphysiological expression of SOD3, which has been shown to reduce primary tumor growth metastasis and cancer cell proliferation, reduces the expression of GEFs and increases the expression of GAPs and GDIs, resulting in inhibition of the activation of downstream ERK1/2 kinases [75] (Figure 2). Hence, modification of the gene expression of regulators of these small GTPases is a cornerstone in SOD3-derived control of the RAS-ERK1/2 mitogen pathway activation and cellular growth. It is important to note that, in line with the effect of SOD3 on cellular functions, the effect of H2O2 is concentration dependent. Low physiological (<0.7 μM) concentration induces growth, whereas concentrations above 50 μM induce DNA damage and senescence [91]. Therefore, SOD3-driven signal transduction resembles H2O2-activated signaling.
8. Conclusions
The role of SOD3 in tumorigenesis is only partly solved. Recent studies have suggested that SOD3 has dose-dependent effects on primary tumor growth and metastasis activity that, however, may depend on the ability of different kinds of tumor cells to detoxify ROS differently. Thus, several avenues of research must still be pursued. Most importantly, the in vitro results that showed that moderate SOD3 mRNA overexpression induces primary cell immortalization and transformation should be repeated using in vivo model systems, and the mechanism of this effect should be further elucidated. Second, increased SOD3 mRNA expression correlates with increased benign growth and decreased SOD3 mRNA expression correlates with increased malignant progression, thus creating a dilemma regarding the signal transduction differences between primary and transformed cells. The presented hypothesis suggesting increased aggressiveness of cancer cells caused by decreased SOD3 mRNA expression requires a mechanistic explanation. Furthermore, as strong expression of SOD3 mRNA induces apoptosis and death of cancer cells, it would be of great interest to determine if SOD3 gene regulation in certain cellular conditions allows supraphysiological expression of the enzyme, causing cellular death, thus suggesting tumor suppressor characteristics for SOD3. Although the function of SOD3 as a regulator of cellular growth has been well established by a number of studies, the enzyme itself might not be a suitable cancer drug or druggable target molecule. Rather, SOD3-related signal transduction studies might indicate mediators of tumor progression, which could then be useful targets for preclinical and clinical studies.
Acknowledgments
Work has been supported by Ministry of Health, SDN 5x1000 2012, and by Fondazione SDN Decision no. RC2010-M-0001.
Competing Interests
The author has no conflict of interests.
References
- 1.Marklund S. A novel superoxide dismutase of high molecular weight from bovine liver. Acta Chemica Scandinavica. 1973;27(4):1458–1460. doi: 10.3891/acta.chem.scand.27-1458. [DOI] [PubMed] [Google Scholar]
- 2.Marklund S. L. Human copper-containing superoxide dismutase of high molecular weight. Proceedings of the National Academy of Sciences of the United States of America. 1982;79(24 I):7634–7638. doi: 10.1073/pnas.79.24.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCord J. M., Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) The Journal of Biological Chemistry. 1969;244(22):6049–6055. [PubMed] [Google Scholar]
- 4.Scrutton M. C. Purification and some properties of a protein containing bound manganese (avimanganin) Biochemistry. 1971;10(21):3897–3905. doi: 10.1021/bi00797a016. [DOI] [PubMed] [Google Scholar]
- 5.Weisiger R. A., Fridovich I. Superoxide dismutase. Organelle specificity. The Journal of Biological Chemistry. 1973;248(10):3582–3592. [PubMed] [Google Scholar]
- 6.Nagano T., Hirano T., Hirobe M. Superoxide dismutase mimics based on iron in vivo. The Journal of Biological Chemistry. 1989;264(16):9243–9249. [PubMed] [Google Scholar]
- 7.Athar M., Iqbal M., Giri U. Novel copper superoxide dismutase mimics and damage mediated by O2.−. Nutrition. 1995;11(5, supplement):559–563. [PubMed] [Google Scholar]
- 8.Batinic-Haberle I., Rajic Z., Tovmasyan A., et al. Diverse functions of cationic Mn(III) N-substituted pyridylporphyrins, recognized as SOD mimics. Free Radical Biology and Medicine. 2011;51(5):1035–1053. doi: 10.1016/j.freeradbiomed.2011.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miriyala S., Spasojevic I., Tovmasyan A., et al. Manganese superoxide dismutase, MnSOD and its mimics. Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease. 2012;1822(5):794–814. doi: 10.1016/j.bbadis.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batinic-Haberle I., Tovmasyan A., Roberts E. R. H., Vujaskovic Z., Leong K. W., Spasojevic I. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxidants & Redox Signaling. 2014;20(15):2372–2415. doi: 10.1089/ars.2012.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laukkanen M. O., Kivelä A., Rissanen T., et al. Adenovirus-mediated extracellular superoxide dismutase gene therapy reduces neointima formation in balloon-denuded rabbit aorta. Circulation. 2002;106(15):1999–2003. doi: 10.1161/01.CIR.0000031331.05368.9D. [DOI] [PubMed] [Google Scholar]
- 12.Sandström J., Nilsson P., Karlsson K., Marklund S. L. 10-Fold increase in human plasma extracellular superoxide dismutase content caused by a mutation in heparin-binding domain. The Journal of Biological Chemistry. 1994;269(29):19163–19166. [PubMed] [Google Scholar]
- 13.Adachi T., Yamada H., Yamada Y., et al. Substitution of glycine for arginine-213 in extracellular-superoxide dismutase impairs affinity for heparin and endothelial cell surface. Biochemical Journal. 1996;313(1):235–239. doi: 10.1042/bj3130235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwon M.-J., Lee K.-Y., Lee H.-W., Kim J.-H., Kim T.-Y. SOD3 variant, R213G, altered SOD3 function, leading to ROS-mediated inflammation and damage in multiple organs of premature aging mice. Antioxidants & Redox Signaling. 2015;23(12):985–999. doi: 10.1089/ars.2014.6035. [DOI] [PubMed] [Google Scholar]
- 15.Zelko I. N., Stepp M. W., Vorst A. L., Folz R. J. Histone acetylation regulates the cell-specific and interferon-γ-inducible expression of extracellular superoxide dismutase in human pulmonary arteries. American Journal of Respiratory Cell and Molecular Biology. 2011;45(5):953–961. doi: 10.1165/rcmb.2011-0012oc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zelko I. N., Mueller M. R., Folz R. J. CpG methylation attenuates Sp1 and Sp3 binding to the human extracellular superoxide dismutase promoter and regulates its cell-specific expression. Free Radical Biology and Medicine. 2010;48(7):895–904. doi: 10.1016/j.freeradbiomed.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laukkanen M. O., Lehtolainen P., Turunen P., et al. Rabbit extracellular superoxide dismutase: expression and effect on LDL oxidation. Gene. 2000;254(1-2):173–179. doi: 10.1016/s0378-1119(00)00272-9. [DOI] [PubMed] [Google Scholar]
- 18.Enghild J. J., Thøgersen I. B., Oury T. D., Valnickova Z., Højrup P., Crapo J. D. The heparin-binding domain of extracellular superoxide dismutase is proteolytically processed intracellularly during biosynthesis. The Journal of Biological Chemistry. 1999;274(21):14818–14822. doi: 10.1074/jbc.274.21.14818. [DOI] [PubMed] [Google Scholar]
- 19.Laukkanen M. O., Leppänen P., Turunen P., Porkkala-Sarataho E., Salonen J. T., Ylä-Herttuala S. Gene transfer of extracellular superoxide dismutase to atherosclerotic mice. Antioxidants & Redox Signaling. 2001;3(3):397–402. doi: 10.1089/15230860152409040. [DOI] [PubMed] [Google Scholar]
- 20.Petersen S. V., Oury T. D., Valnickova Z., et al. The dual nature of human extracellular superoxide dismutase: one sequence and two structures. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(24):13875–13880. doi: 10.1073/pnas.2436143100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karlsson K., Edlund A., Sandstrom J., Marklund S. L. Proteolytic modification of the heparin-binding affinity of extracellular superoxide dismutase. Biochemical Journal. 1993;290(2):623–626. doi: 10.1042/bj2900623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandström J., Carlsson L., Marklund S. L., Edlund T. The heparin-binding domain of extracellular superoxide dismutase C and formation of variants with reduced heparin affinity. The Journal of Biological Chemistry. 1992;267(25):18205–18209. [PubMed] [Google Scholar]
- 23.Cammarota F., de Vita G., Salvatore M., Laukkanen M. O. Ras oncogene-mediated progressive silencing of extracellular superoxide dismutase in tumorigenesis. BioMed Research International. 2015;2015:13. doi: 10.1155/2015/780409.780409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laurila J. P., Castellone M. D., Curcio A., et al. Extracellular superoxide dismutase is a growth regulatory mediator of tissue injury recovery. Molecular Therapy. 2009;17(3):448–454. doi: 10.1038/mt.2008.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gottfredsen R. H., Larsen U. G., Enghild J. J., Petersen S. V. Hydrogen peroxide induce modifications of human extracellular superoxide dismutase that results in enzyme inhibition. Redox Biology. 2013;1(1):24–31. doi: 10.1016/j.redox.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sjoquist P.-O., Carlsson L., Jonason G., Marklund S. L., Abrahamsson T. Cardioprotective effects of recombinant human extracellular-superoxide dismutase type C in rat isolated heart subjected to ischemia and reperfusion. Journal of Cardiovascular Pharmacology. 1991;17(4):678–683. doi: 10.1097/00005344-199104000-00023. [DOI] [PubMed] [Google Scholar]
- 27.Wahlund G., Marklund S. L., Sjöquist P.-O. Extracellular-superoxide dismutase type C (EC-SOD C) reduces myocardial damage in rats subjected to coronary occlusion and 24 hours of reperfusion. Free Radical Research Communications. 1992;17(1):41–47. doi: 10.3109/10715769209061087. [DOI] [PubMed] [Google Scholar]
- 28.Abrahamsson T., Brandt U., Marklund S. L., Sjoqvist P.-O. Vascular bound recombinant extracellular superoxide dismutase type C protects against the detrimental effects of superoxide radicals on endothelium-dependent arterial relaxation. Circulation Research. 1992;70(2):264–271. doi: 10.1161/01.RES.70.2.264. [DOI] [PubMed] [Google Scholar]
- 29.Sjoquist P.-O., Marklund S. L. Endothelium bound extracellular superoxide dismutase type C reduces damage in reperfused ischaemic rat hearts. Cardiovascular Research. 1992;26(4):347–350. doi: 10.1093/cvr/26.4.347. [DOI] [PubMed] [Google Scholar]
- 30.Leite P. F., Danilovic A., Moriel P., et al. Sustained decrease in superoxide dismutase activity underlies constrictive remodeling after balloon injury in rabbits. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(12):2197–2202. doi: 10.1161/01.atv.0000093980.46838.41. [DOI] [PubMed] [Google Scholar]
- 31.Chen E. P., Bittner H. B., Davis R. D., Folz R. J., Van Trigt P. Extracellular superoxide dismutase transgene overexpression preserves postischemic myocardial function in isolated murine hearts. Circulation. 1996;94(9, supplement II):412–417. [PubMed] [Google Scholar]
- 32.Li Q., Bolli R., Qiu Y., Tang X.-L., Guo Y., French B. A. Gene therapy with extracellular superoxide dismutase protects conscious rabbits against myocardial infarction. Circulation. 2001;103(14):1893–1898. doi: 10.1161/01.CIR.103.14.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozumi K., Tasaki H., Takatsu H., et al. Extracellular superoxide dismutase overexpression reduces cuff-induced arterial neointimal formation. Atherosclerosis. 2005;181(1):55–62. doi: 10.1016/j.atherosclerosis.2005.01.051. [DOI] [PubMed] [Google Scholar]
- 34.Qian Z., Haessler M., Lemos J. A., et al. Targeting vascular injury using Hantavirus-pseudotyped lentiviral vectors. Molecular Therapy. 2006;13(4):694–704. doi: 10.1016/j.ymthe.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 35.Kamezaki F., Tasaki H., Yamashita K., et al. Gene transfer of extracellular superoxide dismutase ameliorates pulmonary hypertension in rats. American Journal of Respiratory and Critical Care Medicine. 2008;177(2):219–226. doi: 10.1164/rccm.200702-264OC. [DOI] [PubMed] [Google Scholar]
- 36.Laurila J. P., Laatikainen L. E., Castellone M. D., Laukkanen M. O. SOD3 reduces inflammatory cell migration by regulating adhesion molecule and cytokine expression. PLoS ONE. 2009;4(6) doi: 10.1371/journal.pone.0005786.e5786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He S.-Q., Zhang Y.-H., Venugopal S. K., et al. Delivery of antioxidative enzyme genes protects against ischemia/reperfusion-induced liver injury in mice. Liver Transplantation. 2006;12(12):1869–1879. doi: 10.1002/lt.21001. [DOI] [PubMed] [Google Scholar]
- 38.Bräsen J. H., Leppänen O., Inkala M., et al. Extracellular superoxide dismutase accelerates endothelial recovery and inhibits in-stent restenosis in stented atherosclerotic Watanabe heritable hyperlipidemic rabbit aorta. Journal of the American College of Cardiology. 2007;50(23):2249–2253. doi: 10.1016/j.jacc.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 39.Laatikainen L. E., Incoronato M., Castellone M. D., Laurila J. P., Santoro M., Laukkanen M. O. SOD3 decreases ischemic injury derived apoptosis through phosphorylation of Erk1/2, Akt, and Foxo3a. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0024456.e24456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fattman C. L., Schaefer L. M., Oury T. D. Extracellular superoxide dismutase in biology and medicine. Free Radical Biology and Medicine. 2003;35(3):236–256. doi: 10.1016/S0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- 41.Fukai T., Folz R. J., Landmesser U., Harrison D. G. Extracellular superoxide dismutase and cardiovascular disease. Cardiovascular Research. 2002;55(2):239–249. doi: 10.1016/S0008-6363(02)00328-0. [DOI] [PubMed] [Google Scholar]
- 42.Zelko I. N., Mariani T. J., Folz R. J. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radical Biology and Medicine. 2002;33(3):337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 43.Fukai T., Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxidants & Redox Signaling. 2011;15(6):1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin Z., Reszka K. J., Fukai T., Weintraub N. L. Extracellular superoxide dismutase (ecSOD) in vascular biology: an update on exogenous gene transfer and endogenous regulators of ecSOD. Translational Research. 2008;151(2):68–78. doi: 10.1016/j.trsl.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oshikawa J., Urao N., Kim H. W., et al. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS ONE. 2010;5(4) doi: 10.1371/journal.pone.0010189.e10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carlsson L. M., Jonsson J., Edlund T., Marklund S. L. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(14):6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gongora M. C., Lob H. E., Landmesser U., et al. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. The American Journal of Pathology. 2008;173(4):915–926. doi: 10.2353/ajpath.2008.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdel-Mageed A. S., Senagore A. J., Pietryga D. W., et al. Intravenous administration of mesenchymal stem cells genetically modified with extracellular superoxide dismutase improves survival in irradiated mice. Blood. 2009;113(5):1201–1203. doi: 10.1182/blood-2008-07-170936. [DOI] [PubMed] [Google Scholar]
- 49.Gan J., Meng F., Zhou X., et al. Hematopoietic recovery of acute radiation syndrome by human superoxide dismutase-expressing umbilical cord mesenchymal stromal cells. Cytotherapy. 2015;17(4):403–417. doi: 10.1016/j.jcyt.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 50.Ahmadbeigi N., Soleimani M., Vasei M., et al. Isolation, characterization, and transplantation of bone marrow-derived cell components with hematopoietic stem cell niche properties. Stem Cells and Development. 2013;22(23):3052–3061. doi: 10.1089/scd.2013.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Méndez-Ferrer S., Michurina T. V., Ferraro F., et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suliman H. B., Ali M., Piantadosi C. A. Superoxide dismutase-3 promotes full expression of the EPO response to hypoxia. Blood. 2004;104(1):43–50. doi: 10.1182/blood-2003-07-2240. [DOI] [PubMed] [Google Scholar]
- 53.Sundaresan M., Yu Z.-X., Ferrans V. J., Irani K., Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270(5234):296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 54.Lennicke C., Rahn J., Lichtenfels R., Wessjohann L. A., Seliger B. Hydrogen peroxide—production, fate and role in redox signaling of tumor cells. Cell Communication and Signaling. 2015;13, article 39 doi: 10.1186/s12964-015-0118-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bae Y. S., Kang S. W., Seo M. S., et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. The Journal of Biological Chemistry. 1997;272(1):217–221. doi: 10.1074/jbc.272.1.217. [DOI] [PubMed] [Google Scholar]
- 56.Denu J. M., Tanner K. G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37(16):5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 57.Paulsen C. E., Truong T. H., Garcia F. J., et al. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nature Chemical Biology. 2012;8(1):57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mitsushita J., Lambeth J. D., Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Research. 2004;64(10):3580–3585. doi: 10.1158/0008-5472.CAN-03-3909. [DOI] [PubMed] [Google Scholar]
- 59.Nakamura K., Hori T., Sato N., Sugie K., Kawakami T., Yodoi J. Redox regulation of a src family protein tyrosine kinase p56lck in T cells. Oncogene. 1993;8(11):3133–3139. [PubMed] [Google Scholar]
- 60.Cammarota F., Laukkanen M. O. Mesenchymal stem/stromal cells in stromal evolution and cancer progression. Stem Cells International. 2016;2016:11. doi: 10.1155/2016/4824573.4824573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fruehauf J. P., Meyskens F. L., Jr. Reactive oxygen species: a breath of life or death? Clinical Cancer Research. 2007;13(3):789–794. doi: 10.1158/1078-0432.ccr-06-2082. [DOI] [PubMed] [Google Scholar]
- 62.Laatikainen L. E., Castellone M. D., Hebrant A., et al. Extracellular superoxide dismutase is a thyroid differentiation marker down-regulated in cancer. Endocrine-Related Cancer. 2010;17(3):785–796. doi: 10.1677/ERC-10-0021. [DOI] [PubMed] [Google Scholar]
- 63.Castellone M. D., Langella A., Cantara S., et al. Extracellular superoxide dismutase induces mouse embryonic fibroblast proliferative burst, growth arrest, immortalization, and consequent in vivo tumorigenesis. Antioxidants & Redox Signaling. 2014;21(10):1460–1474. doi: 10.1089/ars.2013.5475. [DOI] [PubMed] [Google Scholar]
- 64.Svensk A.-M., Soini Y., Pääkkö P., Hirvikoski P., Kinnula V. L. Differential expression of superoxide dismutases in lung cancer. American Journal of Clinical Pathology. 2004;122(3):395–404. doi: 10.1309/A45Q-HB0Q-RRX6-CT9A. [DOI] [PubMed] [Google Scholar]
- 65.Yokoe H., Nomura H., Yamano Y., et al. Alteration of extracellular superoxide dismutase expression is associated with an aggressive phenotype of oral squamous-cell carcinoma. Experimental and Therapeutic Medicine. 2010;1(4):585–590. doi: 10.3892/etm_00000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O'Leary B. R., Fath M. A., Bellizzi A. M., et al. Loss of SOD3 (EcSOD) expression promotes an aggressive phenotype in human pancreatic ductal adenocarcinoma. Clinical Cancer Research. 2015;21(7):1741–1751. doi: 10.1158/1078-0432.CCR-14-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang C. A., Harrell J. C., Iwanaga R., Jedlicka P., Ford H. L. Vascular endothelial growth factor C promotes breast cancer progression via a novel antioxidant mechanism that involves regulation of superoxide dismutase 3. Breast Cancer Research. 2014;16(5, article 462) doi: 10.1186/s13058-014-0462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang X., Ng W.-L., Wang P., et al. MicroRNA-21 modulates the levels of reactive oxygen species by targeting SOD3 and TNFα . Cancer Research. 2012;72(18):4707–4713. doi: 10.1158/0008-5472.can-12-0639. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 69.Fukai T., Siegfried M. R., Ushio-Fukai M., Cheng Y., Kojda G., Harrison D. G. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. The Journal of Clinical Investigation. 2000;105(11):631–1639. doi: 10.1172/JCI9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adachi T., Toishi T., Takashima E., Hara H. Infliximab neutralizes the suppressive effect of TNF-α on expression of extracellular-superoxide dismutase in vitro. Biological and Pharmaceutical Bulletin. 2006;29(10):2095–2098. doi: 10.1248/bpb.29.2095. [DOI] [PubMed] [Google Scholar]
- 71.Kamiya T., Hara H., Yamada H., Imai H., Inagaki N., Adachi T. Cobalt chloride decreases EC-SOD expression through intracellular ROS generation and p38-MAPK pathways in COS7 cells. Free Radical Research. 2008;42(11-12):949–956. doi: 10.1080/10715760802566566. [DOI] [PubMed] [Google Scholar]
- 72.Laukkanen M. O., Mannermaa S., Hiltunen M. O., et al. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(9):2171–2178. doi: 10.1161/01.ATV.19.9.2171. [DOI] [PubMed] [Google Scholar]
- 73.Teoh-Fitzgerald M. L., Fitzgerald M. P., Zhong W., Askeland R. W., Domann F. E. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene. 2014;33(3):358–368. doi: 10.1038/onc.2012.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kamiya T., Machiura M., Makino J., Hara H., Hozumi I., Adachi T. Epigenetic regulation of extracellular-superoxide dismutase in human monocytes. Free Radical Biology & Medicine. 2013;61:197–205. doi: 10.1016/j.freeradbiomed.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 75.Laukkanen M. O., Cammarota F., Esposito T., Salvatore M., Castellone M. D. Extracellular superoxide dismutase regulates the expression of small GTPase regulatory proteins GEFs, GAPs, and GDI. PLoS ONE. 2015;10(3) doi: 10.1371/journal.pone.0121441.e0121441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tanaka M., Kogawa K., Nakamura K., et al. Anti-metastatic gene therapy utilizing subcutaneous inoculation of EC-SOD gene transduced autologous fibroblast suppressed lung metastasis of Meth-A cells and 3LL cells in mice. Gene Therapy. 2001;8(2):149–156. doi: 10.1038/sj.gt.3301362. [DOI] [PubMed] [Google Scholar]
- 77.Wheeler M. D., Smutney O. M., Samulski R. J. Secretion of extracellular superoxide dismutase from muscle transduced with recombinant adenovirus inhibits the growth of B16 melanomas in mice. Molecular Cancer Research. 2003;1(12):871–881. [PubMed] [Google Scholar]
- 78.Teoh M. L. T., Sun W., Smith B. J., Oberley L. W., Cullen J. J. Modulation of reactive oxygen species in pancreatic cancer. Clinical Cancer Research. 2007;13(24):7441–7450. doi: 10.1158/1078-0432.CCR-07-0851. [DOI] [PubMed] [Google Scholar]
- 79.Teoh M. L. T., Fitzgerald M. P., Oberley L. W., Domann F. E. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Research. 2009;69(15):6355–6363. doi: 10.1158/0008-5472.CAN-09-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Laukkanen M. O., Leppanen P., Turunen P., Tuomisto T., Naarala J., Yla-Herttuala S. EC-SOD gene therapy reduces paracetamol-induced liver damage in mice. Journal of Gene Medicine. 2001;3(4):321–325. doi: 10.1002/jgm.194. [DOI] [PubMed] [Google Scholar]
- 81.Matsuoka S., Huang M., Elledge S. J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282(5395):1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 82.D'Uva G., Bertoni S., Lauriola M., et al. Beta-catenin/HuR post-transcriptional machinery governs cancer stem cell features in response to hypoxia. PLoS ONE. 2013;8(11) doi: 10.1371/journal.pone.0080742.e80742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leung J. Y., Kolligs F. T., Wu R., et al. Activation of AXIN2 expression by β-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. The Journal of Biological Chemistry. 2002;277(24):21657–21665. doi: 10.1074/jbc.m200139200. [DOI] [PubMed] [Google Scholar]
- 84.Sibenaller Z. A., Welsh J. L., Du C., et al. Extracellular superoxide dismutase suppresses hypoxia-inducible factor-1α in pancreatic cancer. Free Radical Biology & Medicine. 2014;69:357–366. doi: 10.1016/j.freeradbiomed.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sarkisian C. J., Keister B. A., Stairs D. B., Boxer R. B., Moody S. E., Chodosh L. A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nature Cell Biology. 2007;9(5):493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 86.Di Micco R., Fumagalli M., d'Adda di Fagagna F. Breaking news: high-speed race ends in arrest—how oncogenes induce senescence. Trends in Cell Biology. 2007;17(11):529–536. doi: 10.1016/j.tcb.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 87.Bartek J., Bartkova J., Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26(56):7773–7779. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 88.Tcherkezian J., Lamarche-Vane N. Current knowledge of the large RhoGAP family of proteins. Biology of the Cell. 2007;99(2):67–86. doi: 10.1042/BC20060086. [DOI] [PubMed] [Google Scholar]
- 89.Bos J. L., Rehmann H., Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129(5):865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 90.DerMardirossian C., Bokoch G. M. GDIs: central regulatory molecules in Rho GTPase activation. Trends in Cell Biology. 2005;15(7):356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 91.Song Y., Driessens N., Costa M., et al. Roles of hydrogen peroxide in thyroid physiology and disease. Journal of Clinical Endocrinology & Metabolism. 2007;92(10):3764–3773. doi: 10.1210/jc.2007-0660. [DOI] [PubMed] [Google Scholar]