

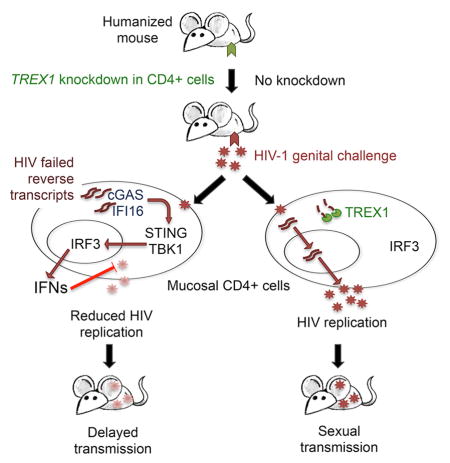
Summary
Despite their antiviral effect, the in vivo effect of interferons on HIV transmission is difficult to predict, because interferons also activate and recruit HIV-susceptible cells to sites of infection. HIV does not normally induce Type I interferons in infected cells, but does if TREX1 is knocked down. Here we investigated the effect of topical TREX1 knockdown and local interferon production on HIV transmission in human cervicovaginal explants and humanized mice. In explants in which TREX1 was knocked down, HIV induced interferons, which blocked infection. In humanized mice, even though TREX1 knockdown increased infiltrating immune cells, it delayed viral replication for 3–4 weeks. Similarly intravaginal application of Type I interferons the day before HIV infection induced interferon responsive genes, reduced inflammation and decreased viral replication. However, intravenous interferon enhanced inflammation and infection. Thus, in models of human sexual transmission, a localized interferon response inhibits HIV transmission, but systemic interferons do not.
Graphical abstract
Introduction
Most viruses trigger expression of Type I interferons (IFN) when pattern recognition receptors detect viral nucleic acids (Coccia et al., 1994). IFNs orchestrate comprehensive antiviral gene expression programs within infected cells and promote innate and acquired antiviral immune responses by enhancing antigen recognition, lymphocyte activation, and recruitment of immune cells to infection sites. Acute infection with the human immunodeficiency virus (HIV), however, does not induce antiviral IFNs in the CD4+ T cells and macrophages that are productively infected (Goldfeld et al., 1991; Unterholzner and Bowie, 2008; Yan et al., 2010). HIV evades immune surveillance at multiple stages of the viral life cycle. During viral entry, TLR RNA sensors do not recognize HIV genomic RNA, because most virions bypass endosomes where these sensors are located. After fusion, genomic RNA is shielded within the viral capsid from cytosolic RNA receptors. HIV reverse transcripts are bound at both ends to HIV integrase, which is predicted to interfere with recognition by cGAS, the cytosolic DNA sensor. However, HIV reverse transcriptase (RT) also generates incomplete reverse transcripts that are not bound to integrase. These can trigger the cGAS-STING-IRF3 pathway of IFN induction if they are not digested by TREX1, a ubiquitous cytosolic 3′–5′ exonuclease (Cai et al., 2014; Gao et al., 2013; Sun et al., 2013; Yan et al., 2009; Yan et al., 2010). When TREX1 is knocked down or knocked out, in vitro HIV infection triggers Type I IFN expression in infected cells that inhibits viral replication. Type I IFNs can also be induced by mutating the HIV capsid or depleting host cofactors with which it interacts, and by knocking down SAMHD1 (Lahaye et al., 2013; Rasaiyaah et al. 2013; Zhang et al., 2014).
Although IFNs have a strong and unequivocal antiviral effect for most viruses, because HIV infects immune cells, the net effect of IFNs on HIV is more complicated. Type I IFNs block both early and late stages of the HIV life cycle (Agy et al., 1995; Coccia et al., 1994; Shirazi and Pitha, 1992). Although Type I IFNs also induce the expression of all known HIV restriction factors, including the APOBECs, SAMHD1 and tetherin, Type I IFNs can potentially enhance HIV infection by promoting T cell activation and recruiting HIV-susceptible cells to the site of infection. Therefore, although in vitro experiments show that TREX1 deficiency inhibits HIV replication, the in vivo consequences of TREX1 knockdown are difficult to predict.
The antiviral effects of Type I IFNs prompted researchers to evaluate their administration as a treatment of HIV infection in the early days of the HIV epidemic. Early studies showed clinical improvement (Hübner et al., 2007; Judge et al., 2005). These promising results were replicated by two randomized control studies, which demonstrated that IFNα treatment significantly reduced viral loads in chronically infected patients (Jackson et al., 2006; Saba et al., 2010). However, subsequent clinical trials did not demonstrate similar therapeutic benefit (Fitzgerald-Bocarsly and Jacobs, 2010; Lehmann et al., 2010; Swiecki and Colonna, 2010). Researchers abandoned IFN-based therapies when HAART became widely available in the mid-1990s (Saba et al., 2010; Wu and KewalRamani, 2006). IFNs, like other cytokines, are meant to act locally at the site of infection, and are probably most effective at controlling viral infection when they are produced at high concentrations where the infection begins. The high concentrations required for therapeutic benefit when exogenous IFN is administered lead to systemic side effects, including fever, neutropenia and depression. The equivocal outcomes of systemic IFN treatment may have been due, in part, to the nonspecific generalized immune activation that accompanies sustained systemic IFN administration. Knocking down TREX1 does not induce IFNs in uninfected cells (Yan et al., 2010), providing a means to localize IFN production to infected cells and evaluate whether IFNs produced in infected cells provide a net protective effect.
Here we knockdown TREX1 using CD4-aptamer-siRNA chimeras (CD4-AsiCs) in CD4+ cells in human cervicovaginal explants and in the genital tract of female humanized mice to evaluate the effect on HIV transmission of localized IFN production in infected cells. We previously showed that CD4-AsiCs, which are composed of a CD4-targeting aptamer covalently linked to the passenger strand of an siRNA and then annealed to the active strand, cause specific gene knockdown in CD4+ T cells and monocytes/macrophages without toxicity, cell activation or innate immune off-target effects (Wheeler et al., 2011; Wheeler et al., 2013). Gene knockdown in tissue lasts for almost two weeks (Collins et al., 2000; Wheeler et al., 2011). Topical application of CD4-AsiCs designed to knockdown CCR5 and/or HIV gag and vif block vaginal transmission in humanized mice.
Because studying early events in the sexual transmission of HIV in humans is difficult, our understanding of sexual transmission of the virus relies heavily on studies in macaques challenged with cell-free simian immunodeficiency virus (SIV) (Arfi et al., 2008; Bergamaschi and Pancino, 2010; d’Ettorre et al., 2014; Miller et al., 2005; Piguet and Steinman, 2007). In this model, SIV first infects and expands in CD4+ T cells in the genital mucosa before spreading to myeloid cells (Haase, 2010; Miller et al., 2005; Zhang et al., 1999). Infection is contained within the genital tract for about a week before disseminating to regional lymph nodes and systemically. This provides a “window of opportunity” for interventions to prevent HIV from establishing a foothold. Genital infection stimulates a pro-inflammatory cytokine cascade, which recruits activated immune cells to the genital mucosa (Caux et al., 2000; Dieu-Nosjean et al., 1999; Miller et al., 2005), which then promotes viral replication and spreading, both locally and systemically. Plasmacytoid dendritic cells (pDCs), the primary producers of Type I IFNs, are recruited to the genital mucosa, but IFN production is delayed (Klatt et al., 2014). Thus, because SIV does not induce IFNs in infected cells and IFN production by recruited cells in the genital tract is delayed, SIV does not have to cope with the antiviral effects of host IFNs while it is establishing the infection.
A recent study investigated the effect of manipulating Type I IFNs on transmission of repeated SIV rectal challenges in rhesus macaques (Sandler et al., 2014). Blocking the Type I IFN receptor increased viral replication and AIDS progression, while administration of IFN-α2a around the time of challenge blocked transmission. However, continued exposure to exogenous IFNs actually caused desensitization to its antiviral effect, increased the viral setpoint, and accelerated disease progression. This work highlights the potential protective role of Type I IFNs, but also suggests that the in vivo effects of IFNs are complex.
Immune responses can differ between human and nonhuman primates, since immune genes continue to coevolve with pathogens, and SIV differs in important ways from HIV. In particular, the SIV viral protein Vpx promotes degradation of host restriction factor SAMHD1, which limits HIV infection in resting T cells and myeloid cells, thereby enabling SIV to replicate in cell populations that HIV cannot. Given the complexity of the effects of IFNs, which activate components of the immune system capable of both propagating and inhibiting HIV (Boasso et al., 2008; Fitzgerald-Bocarsly and Jacobs, 2010; Hardy et al., 2009; Poli et al., 1994), it is important to study the role of IFNs in HIV transmission in human systems. In particular, the exact role IFNs play in acute HIV infection, the timing of their induction, and the effects they exert on their most immediate downstream targets during the earliest stages of viral transmission remain poorly understood. Here we use two HIV-human transmission model systems, polarized human cervicovaginal explants and humanized mice, to investigate the effect of inducing Type I IFNs in infected cells by TREX1 knockdown to assess the effects of endogenous IFNs on HIV female genital transmission. These models both have limitations. The explants are only viable for 10 to 14 days (Collins et al., 2000; Wheeler et al., 2011) and do not take into account recruitment of immune cells to the tissue. Although human myeloid and lymphoid cells are present in the female genital tract of humanized mice, they are less abundant than in human tissues, and their lymph nodes are often undeveloped or absent (Olesen et al., 2011). Moreover, humanized mice have chronic immune activation from graft vs host responses. Although there is good cross-reactivity between human and mouse cytokines and chemokines, signaling may not replicate precisely what occurs in humans. Nonetheless, these imperfect models are as close as we can get to human transmission.
Here we show that CD4-AsiCs knockdown TREX1 expression by 75–95% in human cervicovaginal tissue and the female genital tract of humanized mice. Knocking down TREX1 increased expression of Type I IFNs and interferon stimulated genes (ISGs) in HIV-exposed, but not uninfected, human tissue and in humanized mice in the first 12–24 hours after exposure, but did not cause upregulation of inflammatory cytokines, such as IL1β, IL-6, and IL-8. Intravaginal (IVAG) IFN administration had a similar effect. In contrast, exogenous IFNs, given intravenously (IV) to humanized mice, induced ISGs as well as proinflammatory cytokine gene expression, even in the absence of HIV infection. In tissue explants, TREX1 knockdown suppressed HIV infection, and suppression was largely mediated by Type I IFNs, since it was strongly inhibited by neutralizing antibodies to Type I IFNs. In humanized mice, a robust Type I IFN response decreased viral replication during the first 48 hours after exposure, despite recruitment of immune cells to the genital mucosa. Importantly, TREX1 knockdown in the genital tract delayed HIV infection for ~3–4 weeks, suggesting that IFN induction in infected cells suppresses local viral replication.
Results
TREX1 knockdown in CD4+ cells in vitro and in human cervicovaginal tissue explants
CD4-AsiCs were designed to knockdown TREX1 in CD4+ cells using the CD4 aptamer and siRNA linkage, previously shown to knockdown CCR5 and viral genes selectively in CD4+ T cells and macrophages in cervicovaginal tissue explants and the genital tract of humanized mice (Wheeler et al., 2011; Wheeler et al., 2013). The aptamer directs the chimeric RNA selectively into CD4+ cells, where Dicer cleaves the AsiC to separate the aptamer from the siRNA. A chimeric RNA encoding the CD4 aptamer linked at its 3′-end to the sense strand of one of two TREX1 siRNAs was in vitro transcribed using 2′-fluoropyrimidines to enhance stability and minimize off-target effects (Figure 1a–b). The in vitro transcribed RNA was then annealed to the antisense siRNA strand. To evaluate gene knockdown, PBMCs were incubated for 48 h with 0.25–4 μM TREX1 CD4-AsiCs and analyzed by qRT-PCR for TREX1 mRNA (Figure 1c–d). TREX1 knockdown occurred specifically in CD4+, but not CD8+ T cells. The best knockdown occurred using 4 μM AsiC. ‘Sequence a’, which knocked down TREX1 by ~50% even at the lowest concentration and by ~90% at the highest concentration, was more effective than ‘sequence b’ and was used for subsequent experiments, unless otherwise noted. TREX1 knockdown in primary human CD4+ T cells also significantly reduced TREX1 protein and the proportion of cells that stained above background for TREX1 from 83% to 7%, as assessed by flow cytometry (Figure S1).
Figure 1. CD4-AsiCs knockdown TREX1 in CD4+ cells in vitro and in polarized cervicovaginal explants.

(a, b) Two TREX1 CD4-AsiCs (sequence a, a; sequence b, b) (c, d) CD4+ (red) and CD8+ (blue) T cells were treated and analyzed for TREX1 gene knockdown by qRT-PCR relative to GAPDH. (f, g) CD4+CD14+ macrophages, CD4+ T cells, and CD19+ B cells, sorted from polarized cervicovaginal explants treated with TREX1 CD4-AsiCs, or medium, were analyzed for TREX1 knockdown by qRT-PCR. Graphs show mean ± SEM from 3 independent experiments (*p<0.05, **p<0.005, ***p<0.0005, by Student’s t-test, relative to mock-treated control samples). The change in TREX1 in B cells was not reproducible. See also Figure S1.
Type I IFN signaling leads to phosphorylation and nuclear translocation of the IRF3 transcription factor. To investigate whether IFN signaling is activated after HIV infection in cells knocked down for TREX1, we assessed IRF3 localization by imaging flow cytometry in primary human MDMs and CD4+ T cells (Figure S2 a, b). TREX1 knockdown did not significantly change IRF3 localization in uninfected cells. However, as expected, IRF3 translocated to the nucleus after HIV infection in cells knocked down with TREX1 siRNA compared to cells transfected with a non-targeting negative control siRNA (MDMs, p<0.01; CD4+ T cells, p<0.0001). To determine whether IRF3 activation is mediated by cGAS and IFI16, DNA sensors that recognize HIV reverse transcripts (Gao et al., 2013; Monroe et al., 2013), we co-transfected CD4+ T-cells with TREX1 siRNA alone or together with cGAS or IFI16 siRNAs. siRNA targeting the DNA sensor AIM2 or the RNA sensor RIG-I, which are not required for the induction of type I IFN by HIV in TREX1 knock-out cells (Yan et al., 2010), were used as controls. Knockdown of cGAS or IFI16, but not AIM2 or DDX58, the gene encoding RIG-I, significantly and strongly inhibited IRF3 nuclear translocation in response to HIV infection in TREX1 knocked down cells (Figure S2 f, h, j). Thus TREX1 knockdown induces IFN pathway activation and IRF3 nuclear translocation in response to HIV that is dependent on DNA sensing by both cGAS and IFI16, like recognition of HSV-1 and Listeria monocytogenes DNA (Orzalli et al., 2015; Hansen et al., 2014).
We next evaluated TREX1 gene knockdown in human cervicovaginal explants. TREX1 CD4-AsiCs (1 and 4 μM) were applied twice at an interval of 24 h to the epithelial surface of polarized human cervicovaginal tissue. Four days after the second treatment, tissues were digested to single cell suspensions, which were sorted into CD4+/CD3+ T cells, CD4+/CD14+ macrophages and CD19+ B cell subsets and analyzed for TREX1 mRNA by qRT-PCR (Figure 1f, g). TREX1 was knocked down by 80–95% in CD4+ T cells and macrophages, but not in B cells. Both concentrations led to comparable knockdown. We previously showed that CD4-AsiCs targeting other genes do not activate an innate immune IFN response on their own (Wheeler et al., 2011). However, because IFN induction may be sequence dependent, we evaluated whether TREX1-specific CD4-AsiCs induce Type I IFNs. We treated cervical explants with TREX1 CD4-AsiCs in the absence of HIV infection and measured expression levels of type I IFN mRNAs in the tissue at the expected peak response time (6 hr) by sensitive qRT-PCR assay (Figure S3 a). As expected, TREX1 CD4-AsiCs did not induce an IFN response, whereas Poly(I:C), used as a positive control, did.
TREX1 knockdown inhibits HIV replication and induces IFN-β in polarized human cervicovaginal explants
TREX1 knockdown inhibits HIV expression in vitro by up-regulating Type I IFNs (Yan et al., 2010). To evaluate whether knocking down TREX1 also inhibits HIV infection in human tissue, we used a previously validated polarized human cervicovaginal explant model of HIV transmission and infection. The observed p24 Ag production in infected explants measured active viral replication in the explants, rather than carryover of virus in the infectious inoculum, since p24 Ag levels did not increase in tissues that were infected in the presence of the HIV inhibitors, AZT or nevirapine (Figure S3 b). To evaluate the effect of TREX1 knockdown in the explant system, we measured viral replication in cervicovaginal tissue from 3 healthy donors infected after they were treated with medium or CD4-AsiCs against TREX1 or, as positive control, a cocktail against CCR5, gag, and vif. Explants were treated 3 and 2 d prior to challenge with HIVBaL and viral replication was monitored by measuring viral p24 antigen in the lower transwell chamber (Figure 2a–c). As previously demonstrated, the CCR5 and antiviral gene cocktail completely prevented productive HIV infection. CD4-AsiCs encoding either TREX1 siRNA sequence inhibited HIV infection, but ‘sequence a’ performed better, almost completely suppressing p24 Ag production. Although HIV infection in the absence of gene knockdown did not lead to detectable IFNβ release until day 9 of culture, when it was barely above background, the infected tissues that were treated with TREX1 AsiCs produced IFNβ that was detected in the first measurement on day 3 (Figure 2d). Tissues treated with the more effective ‘sequence a’ AsiC generated more IFNβ. Tissues treated with the CD4-AsiC cocktail that blocked infection did not release IFNβ. To evaluate the importance of Type I IFNs in inhibiting HIV transmission in the tissue, blocking mAbs to IFNα and IFNβ were added to the culture medium 24 h before infection (Figure 2e–g). The blocking antibodies had no effect on HIV production in explants that were knocked down for CCR5 and the viral genes, which was not surprising since they did not generate Type I IFNs. Antibody treatment blunted the amount of IFNβ protein detected in the culture supernatants and increased HIV infection in TREX1 AsiC-treated explants. However, blocking IFNα and IFNβ in TREX1 AsiC-treated tissues did not completely restore HIV infection to the level observed in tissues not subjected to knockdown. Thus, TREX1 knockdown suppressed HIV infection in cervicovaginal tissues, largely via induction of Type I IFNs.
Figure 2. CD4-AsiC knockdown of TREX1 inhibits HIV replication in polarized human cervicovaginal explants partly by inducing Type I IFNs.

(a–d) Polarized explants (n=8) from 3 donors treated with PBS (blue) or twice pretreated with 4 μM CD4-AsiCs targeting TREX1 (siRNA sequence a, light green; siRNA sequence b, dark green) or CCR5, gag, and vif (cocktail, red) before HIV-1BaL challenge. Data for uninfected control cultures are shown in yellow. HIV infection analyzed by p24 Ag ELISA (b, c) and IFN-β protein measured by ELISA (d). Blocking monoclonal antibodies (mAbs) against IFN-α and IFN-β were applied 24 h prior to, and at the time of HIV challenge (e–g). Graphs show mean ± SEM (*p<0.05, **p<0.005, ***p<0.0005, by Student’s t test, relative to uninfected control). See also Figure S2, S3, S4.
Recently transmitted HIV-1 viruses, termed transmitted/founder (T/F) viruses, are relatively resistant to suppression by Type I IFNs (Parrish, et al., 2013). To determine whether TREX1 knockdown could inhibit infection with T/F HIV-1 virus, we compared the effect of TREX1 knockdown on replication of a T/F virus relative to HIVBaL in human cervical explants (Figure S4), using treatment with the CD4 aptamer on its own as a control. T/F HIV replication was significantly inhibited by treatment with TREX1 CD4-AsiCs. However T/F virus was less strongly suppressed than HIVBaL.
Exposure of monocyte-derived macrophages (MDMs) to Type I IFN inhibits HIV replication when administered just before or after infection
To prepare for in vivo experiments with IFNs, we first defined the dose dependence and timing of IFN protection from HIV infection in vitro. MDMs were incubated for 24 h with different amounts of a recombinant form of IFN, engineered to bind multiple IFN receptors with high affinity (rIFN), prior to infection with 3 doses of HIVBaL (Figure S5). Adding 10,000 IU rIFN in 0.3 ml culture medium suppressed viral replication in MDMs by >95% for even the highest viral challenge. Treatment up to 24 h prior to or 6 h after viral challenge completely prevented viral replication at the highest rIFN concentration, but adding the recombinant protein 6 h before infection was more effective than 24 h before infection at the lower doses. When rIFN was added 2 d before or 1 d after viral challenge, there was little protection.
TREX1 knockdown IVAG or topical rIFN upregulate ISGs and inhibit early HIV replication in humanized BLT mice
To investigate the in vivo effect of TREX1 expression on HIV transmission, we treated humanized BLT mice IVAG with PBS or 40 pmol of TREX1 CD4-AsiCs on 2 consecutive days. Half of the mice were not exposed to HIV and half were challenged the following day with HIV1JR-CSF using a viral dose that reproducibly infects all control mice (n=6) (Wheeler et al., 2011; Wheeler et al., 2013) (Figure 3a). Additional groups of mice (n=6) were treated with IV rIFN (104 IU) or IVAG rIFN (2×103 IU) the day before viral challenge. One hour post-rIFN administration, we measured rIFN levels in the serum. After IV injection, rIFN was detected in the serum at a level of 43.5 ± 0.57 IU/ml (n=3), but it was not detectable in the serum after IVAG application (data not shown). Mice were sacrificed 16 h after viral challenge, and cervicovaginal tissue was digested into single cell suspensions. Systemic IFN significantly increased the number of human CD45+ hematopoietic cells in the female genital tract, irrespective of HIV infection, while TREX1 knockdown and topical IVAG IFN increased human infiltrating cells in the genital tract only following HIV infection (Figure 3b). Gene expression was examined in human CD45+ hematopoietic cells, human CD4+ cells, and in the total mixed cell population isolated from the vaginal mucosa by qRT-PCR. TREX1 CD4-AsiCs strongly knocked down TREX1 mRNA (by ~95%) in genital tract CD4+ cells in both HIV uninfected and infected mice (Figure 3c). Both topical and systemic IFNs significantly increased CCR5 and IFN-responsive TREX1 and APOBEC3G and other ISG mRNAs in tissue CD4+ cells (Figure 3b, e–i, k). In the absence of HIV infection, TREX1 knockdown did not significantly change expression of inflammatory cytokines, CCR5, Type I and II IFNs, or ISGs, including SAMHD1 and APOBEC3G (Figure 3e–j).
Figure 3. TREX1 knockdown or topical IFN upregulate ISGs and inhibit HIV transmission in humanized mice.

(a) Schematic. BLT mice (n=6 per treatment group) were treated twice IVAG with PBS (blue) or 40 pmol TREX1 CD4-AsiCs (green) or once with rIFN 104 IU IV (dark red) or 2×103 IU IVAG (light red). Half of the mice in each group (n=3) were challenged 24 hr after the last treatment with HIVJR-CSF, and all mice were sacrificed 16 h after that. Cervicovaginal tissue was digested to a single cell suspension, and sorted into human CD45+ and CD4+ cell populations. (b) Flow cytometry analysis of human CD45+ cells in the vaginal tissue of each group of mice. (c–i) RNA from sorted cells or total tissue was analyzed for mRNA levels by qRT-PCR. TREX1 (c) and HIV gag (d) mRNA in vaginal CD4+ cells and CCR5 expression in vaginal CD45+ cells (e) are shown. (f–k) mRNA levels for human inflammatory cytokines TNFA (f) and IL1B (g), HIV restriction factors SAMHD1 (h) and APOBEC3G (i, A3G) and a panel of IFNs and ISGs (j, k) in vaginal tissue are shown. Human gene mRNA levels, relative to GAPDH mRNA, were normalized to the value in the HIV-uninfected, PBS control. gag RNA, which was not detected (ND) in uninfected samples, was normalized to GAPDH mRNA. Shown are the mean ± SEM for each group. (*, p<0.05 by 2-sided Student’s t-test, relative to PBS control). The graphs represent data from one experiment with BLT-mice prepared with human hematopoietic stem cells from one donor. See also Figure S5.
After HIV challenge, HIV gag mRNA was detected above background in control and IFN-treated BLT mouse genital tissue, but was not detected in mice treated with TREX1 AsiCs at this early time point (Figure 3d). Thus TREX1 knockdown was protective. TREX1 knockdown, and to a greater extent topical and systemic IFNs, also increased CCR5 mRNA in the genital tissue, likely due to infiltration of activated immune cells and activation of tissue resident cells (Figure 3b, e). With exposure to HIV, TREX1 knockdown led to significantly increased Type I and Type II IFNs and ISG mRNAs, including the mRNAs for the HIV restriction factor ISGs, SAMHD1 and APOBEC3G (Figure 3h, i, k). TREX1 knockdown did not have a consistent effect on HIV-induced inflammatory gene expression – it significantly increased TNFα, reduced IL-1β and IL-8, and did not change IL-6 mRNA expression. Thus, early after HIV infection, TREX1 knockdown in CD4+ cells in the female genital tract reduced HIV load and induced IFNs and antiviral gene expression, but at the same time increased recruitment of activated CCR5+ immune cells, expanding the numbers of susceptible cells in the tissue.
Topical, but not systemic, IFN significantly suppressed early HIV replication, assessed by measuring gag mRNA 16 hr after infection, compared to control mice, but inhibition was less effective than TREX1 knockdown, which suppressed HIV to undetectable levels (Figure 3d). Systemic rIFN more potently induced ISG expression than TREX1 knockdown, but also enhanced tissue recruitment of CCR5+ hematopoietic cells and caused more pro-inflammatory gene expression. The recruitment of activated HIV-susceptible cells and induction of inflammation in the genital tract after systemic IFN administration may have canceled the antiviral effects of ISGs on HIV replication.
TREX1 CD4-AsiCs upregulate gene expression of Type I IFNs and ISGs in the vaginal tissue of HIV-exposed humanized BLT mice
We next evaluated the effect of TREX1 knockdown on gene expression in the female genital tract of humanized mice 24 and 48 h after viral challenge (Figure 4a). Mice treated IVAG with TREX1 AsiCs were compared to mice treated IVAG with PBS as negative control, or lipopolysaccharide (LPS), which induces both Type I IFNs and inflammatory cytokines, as positive control. Each group was treated on 2 consecutive days and challenged with HIV IVAG on the following day. Blood and cervicovaginal tissue were harvested 24 and 48 h later, single cell suspensions were prepared, and human CD4+, human CD45+, and total cell populations were analyzed for mRNA expression by qRT-PCR (Figure 4c–i). IVAG LPS, as expected, increased the numbers of human CD45+ cells in the vaginal tissue (Figure 4b). TREX1 AsiCs knocked down TREX1 in CD4+ cells in the genital tract by 90% at 24 h and 75% at 48 h post challenge, while IVAG LPS significantly increased the expression of IFN-responsive TREX1 in CD4+ cells in vaginal tissue at both time points, and in the blood significantly at 48 h (Figure 4c). HIV infection in genital tract CD4+ cells was suppressed to near background levels by TREX1 knockdown at both time points, while LPS pretreatment enhanced gag mRNA expression in the genital tract. (Figure 4d). Unexpectedly, HIV gag mRNA was detected in blood CD4+ cells of some LPS-treated mice at 48 hr, suggesting that infection may not be restricted to the genital tract in the setting of genital inflammation. However, because gag detection in blood cells was just above background, this finding needs to be interpreted with caution. CCR5 mRNA, an indicator of T cell activation, was significantly decreased in vaginal and blood CD4+ cells in TREX1 AsiC-treated mice, while it was significantly increased in LPS-treated mice in both compartments compared to mock treated, HIV-infected mice (Figure 4e). Knockdown of TREX1 increased the expression of Type I IFNs and ISGs and decreased expression of proinflammatory cytokines in tissue and blood, while LPS pretreatment increased both inflammation and the IFN response (Figure 4f–i). Thus, TREX1 knockdown in genital CD4 cells, which enhanced antiviral IFN gene expression and suppressed inflammation, blocked HIV infection at 24 h and significantly controlled it to near background levels at 48 h. Generalized innate immune activation by LPS administration in the genital tract enhanced HIV replication locally and accelerated dissemination.
Figure 4. TREX1 CD4-AsiCs upregulate Type I IFNs and ISGs and inhibit HIV transmission to BLT mice.

(a) Schematic. BLT mice (n=6 per treatment group) were treated twice with IVAG PBS (blue), LPS (red), or 40 pmol TREX1 CD4-AsiCs (green). All mice were infected with HIVJR-CSF. Blood and cervicovaginal tissues were obtained at sacrifice 24 h and 48 h (n=3 per treatment group) after viral challenge. PBMCs or genital tract single cell suspensions were either not sorted or sorted into human CD45+ and human CD4+ cell populations for analysis as indicated. (b) Flow cytometry analysis of human CD45+ cells in the tissue (left) and blood (right). (c–e) mRNA levels relative to human GAPDH were analyzed by qRT-PCR for human TREX1 (c), HIV gag (d) and human CCR5 (e) using RNA extracted from CD4+ cells in genital tract (left) and blood (right). (f–i) mRNA levels were analyzed by qRT-PCR on RNA extracted from genital tract single cell suspensions or PBMCs as indicated for the following human genes - TNFA (f), IL1B (g), and a panel of IFNs, inflammatory cytokines, and ISGs (h, i). Human gene mRNA levels were normalized to the value in the HIV-uninfected, PBS control. gag RNA was normalized to GAPDH. ND, not detected. All graphs show mean ± SEM (*, p<0.05 relative to PBS-treated control mice, determined using Student t-test). Graphs represent data from one experiment using BLT-mice prepared with human hematopoietic stem cells from one donor. See also Figure S5.
TREX1 CD4-AsiCs delay, but do not prevent, transmission of HIV to humanized BLT mice
To evaluate the net antiviral effect of TREX1 knockdown, we compared HIV infection and CD4+ T cell depletion in humanized mice treated IVAG with TREX1 AsiCs with mock-treated mice and mice treated with the AsiC cocktail against CCR5 and viral genes that blocks viral transmission (Wheeler et al., 2011; Wheeler et al., 2013) (Figure 5a). (The control group data were previously published in (Wheeler et al., 2011), but the TREX1 knockdown data, obtained at the same time, were not previously published.) All mock treated mice became infected and showed profound CD4+ T cell depletion (Figure 5b–e). Viremia was first detected 3–4 weeks post challenge and CD4 counts began to drop within 2 weeks and CD4+ T cells were severely depleted in all mice by 8 weeks. Mice treated with the cocktail were not infected – they had undetectable viremia, assessed by HIV p24 Ag and HIV gag mRNA, and no change in CD4+ T cell counts. Mice treated with TREX1 AsiCs initially looked like they were protected, but they all developed detectable viremia by 7 weeks. CD4+ T cell depletion only became significant after 9 weeks. Thus viral production was delayed for about a month by knocking down TREX1 prior to viral challenge compared to control mice. Despite the delay in infection, the viral setpoint in TREX1 AsiC-treated mice (~105 copies/ml blood) and extent of CD4+ T cell depletion at 12 weeks, when the experiment was terminated, were indistinguishable from those in control mice.
Figure 5. TREX1 CD4-AsiCs delay HIV transmission to BLT mice.

(a) Experimental design. Mice (n=4 per treatment group) were treated according to the indicated dosing schedules (right) with PBS (blue, b), a cocktail of CD4-AsiCs targeting CCR5, gag and vif, using a regimen that previously blocked HIV transmission (red, c) (Wheeler et al., 2011), or TREX1 CD4-AsiCs (green, d). 40 pmol of each CD4-AsiC was administered IVAG twice according to the dosing schedule. Mice were assessed for HIV RNA by qRT-PCR (left), plasma HIV p24-Ag by ELISA (middle), and circulating CD4+ T cell count by flow cytometry (right). The dashed line represents the threshold of detection of the assay. No CD4 analysis was performed at week 4. (e) shows average ± SEM for each group (*p<0.05; ***p<0.005, ***p<0.0005, by 1-way ANOVA, relative to PBS control). The graphs represent data from one of two independent experiments with different batches of BLT-mice prepared with human hematopoietic stem cells from two different donors.
Discussion
Here we demonstrate that TREX1 knockdown using CD4-AsiCs induces expression and secretion of Type I IFNs and ISGs in HIV-infected cells that inhibits HIV transmission in human tissue explants and in humanized mice for several weeks when administered prior to viral challenge. Protection from HIV replication in tissue explants was largely abrogated by IFNα/β blocking antibodies, suggesting that protection is mediated by Type I IFN production in infected cells. The residual protection may have been due to incomplete blockade of all Type I IFNs. We also cannot exclude the possibility that some antiviral effects of TREX1 knockdown are IFN-independent. Indeed ISGs can be induced in the absence of TREX1 independently of IFNs (Hasan et al., 2013). Within the first 2 days after HIV challenge, HIV replication was not detected in the genital tract of mice treated with TREX1 CD4-AsiCs and was suppressed in mice given rIFN IVAG, suggesting that local IFNs strongly inhibit HIV transmission in vivo. This was despite evidence of significant immune cell infiltration in the genital mucosa and CD4+ T cell activation to express CCR5, the HIV coreceptor used during sexual transmission. In contrast, IV administration of rIFN using five times the IVAG dose, which induced local IFN and ISG gene expression in the genital mucosa, but less robustly than IVAG administration, did not inhibit HIV replication at this early timepoint. Unlike treatment with IVAG IFN or TREX1-AsiCs, IV IFN activated proinflammatory cytokine expression (TNFα, IL-1β, IL-6 and IL-8), which likely counteracted the protective effects provided by rIFN.
In some humanized mice (but not those treated with TREX1 CD4-AsiCs), we detected HIV gag RNA at very low levels in CD4+ blood cells as early as 1 and 2 days following IVAG challenge, accompanied by suggestions of systemic immune activation by qRT-PCR analysis of CCR5 and cytokine gene expression in CD4+ blood cells (Figure 4d). Importantly, at these early times, systemic dissemination was exacerbated by IVAG pretreatment with LPS, which caused both local and systemic IFN induction and immune activation. A similar increase in sexual transmission was observed when rhesus macaques treated IVAG with other TLR agonists were challenged with SIV (Wang et al., 2005). In that case as well, both IFNs and inflammatory cytokines were induced in the genital tract. However infected cells were not detected in the blood of mice treated IVAG with CD4-AsiCs against TREX1. These data suggest that in humanized mice, HIV may not be contained within the genital tract for the first week after exposure. Nonetheless, we did not detect plasma viremia by RT-PCR or p24 Ag assay until 3 weeks after infection (Figure 5). Because the level of gag mRNA in circulating CD4+ T cells was just above background, the measured gag mRNA in circulating CD4+ T cells within the first few days after infection should be interpreted with caution and needs to be confirmed. The discrepancy between RT-PCR analysis of CD4+ T cells and plasma viremia assays suggests that RT-PCR analysis of circulating CD4+ T cells may be a more sensitive way to detect systemic dissemination than amplification of viral RNA in circulating virions in the serum. Early dissemination might be the consequence of chronic low levels of immune activation from graft vs host responses that are not completely suppressed in the NOD/SCID/Il2rg−/− (NSG) mice. Their chronic immune activation may mean that conclusions about transmission in humanized mice may not reflect transmission in women, who have no underlying immune activation or vaginal coinfection. However, these mice may be a good model for women with vaginal infections, such as bacterial vaginosis or trichomonas infection, who are more vulnerable to becoming infected. If these mouse findings are confirmed, they suggest that it is worth investigating whether early viral dissemination might occur in women with ongoing vaginal or systemic infection.
Although TREX1 knockdown suppressed HIV infection early on, HIV was transmitted with a delay in viral kinetics of ~3–4 weeks. In these experiments TREX1 CD4-AsiCs were administered one and two days before viral challenge. In an earlier study (Wheeler et al., 2013), CCR5 knockdown in humanized mice using CD4-AsiCs provided complete protection from transmission when HIV challenge occurred within a day of AsiC application, but although gene knockdown in the tissue persisted for ~2 weeks, protection was incomplete when HIV challenge was delayed for 4 or 6 days. The lack of complete protection after delayed challenge was attributed to influx of HIV-susceptible cells, not exposed to AsiCs, into the genital mucosa. Similarly, in this study, HIV might have persisted in the tissue (possibly within myeloid cells that do not necessarily replicate the virus, but efficiently transmit it to T cells (Arfi et al., 2008; Bergamaschi and Pancino, 2010; Kumar et al., 2014; Piguet and Steinman, 2007)) and then replicated in tissue resident CD4+ cells in which TREX1 knockdown had waned or in recruited CD4+ T cells that were not present at the time of knockdown. Although the knockdown of TREX1 only delayed the progression of HIV infection, but did not inhibit it completely, the suppression of local HIV replication in the genital tract might reduce person-to-person transmission. Future experiments in which TREX1 AsiCs are administered both before and after HIV challenge should address this question. By measuring viral DNA species and integration of the provirus within different human cell subtypes in the genital tract of humanized mice, we may be able to determine the cell types in which the virus persists during TREX1 knockdown. The viral setpoint and CD4 depletion in TREX1 knocked down mice eventually reached the same levels as in mock-treated mice. This surprising finding suggests that the viral setpoint is not determined by the original viral replicative burst, but by the complex interaction of the virus with the host immune system. The conservation of the viral setpoint here may be related to the well-known and poorly understood clinical finding that viral levels return to the pretreatment setpoint when antiretroviral drugs are halted. What determines the viral setpoint in any setting is not well understood.
The in vitro data presented here suggest that there is a narrow temporal window during which IFNs can effectively control infection - the ~30 hours around the time of exposure. Our results echo those found in SIV infection in macaques, in which IFN only effectively blocks infection within a 3-day period (Unterholzner and Bowie, 2008). This may be one of the reasons that chronic IFN therapy showed inconsistent results in clinical trials and ultimately did not appreciably improve patient outcomes (Fitzgerald-Bocarsly and Jacobs, 2010; Jackson et al., 2006; Lehmann et al., 2010; Saba et al., 2010; Swiecki and Colonna, 2010). In addition to timing, location also seems to play a critical role. IFNs at the site of transmission provided protection, but after systemic IFN treatment, protection was lost, likely due to a shift in balance between protective and harmful IFN-triggered downstream events. Our finding in humanized mice differs from results in macaques, where IV PEGylated-IFN begun before SIV challenge and continued for 4 weeks provided protection from rectal challenge (Unterholzner and Bowie, 2008). Multiple variables could account for these differing observations including differences between species, site of infection, virus, and dose and type of rIFN.
This study is the first application to use in vivo gene knockdown as a tool to explore the earliest stages of HIV disease pathogenesis and the role IFNs play in transmission. Knockdown of individual host genes in all the cells that HIV infects using CD4-AsiCs provides a straightforward method to explore the role of individual host genes in HIV transmission. Preliminary results suggest that the CD4-aptamer also recognizes macaque CD4 (data not shown). Thus, this tool could also be used to study the role of host genes in SIV transmission in macaques, where gene knockout is not easy. We have recently found that EpCAM-AsiCs given subcutaneously can silence gene expression in EpCAM+ cells distributed at distal sites throughout the mouse (Gilboa-Geffen et al., 2015). Preliminary studies of subcutaneous injection of CD4-AsiCs in humanized mice also show strong knockdown (~80%) in CD4+ T cells in local lymph nodes, as well as in the spleen and distal lymph nodes. These findings suggest that this powerful tool could be used to study the influence of individual host genes not only on transmission, but also on disease progression and pathogenesis.
Experimental procedures
CD4-AsiC synthesis
CD4-AsiCs were synthesized using the primer sequences given in Table S1 using in vitro transcription as described (Davis et al., 1998; McNamara et al., 2006; Wheeler et al., 2011). Sequences for the conjugated siRNAs are shown in Table S2.
Human cervical polarized tissue explants
Human cervical tissue was obtained with BCH and HMS Human Investigational Review Board approval from healthy donors undergoing hysterectomy for benign conditions and was prepared as previously described (Wheeler et al., 2011; Wheeler et al., 2013).
BLT mouse experiments
Animal work was approved by the Animal Care and Use Committees of Massachusetts General Hospital, Boston Children’s Hospital (BCH) and Harvard Medical School (HMS). All in vivo experiments were performed using progesterone-treated, ketamine/xylazine anesthetized NOD/SCID/IL2rg−/− (NSG) female mice, bearing humanized bone marrow following reconstitution with CD34+ cells from human fetal liver and surgical human thymic grafts (“BLT mice”), prepared by the MGH Humanized Mouse Program as previously described (Brainard et al., 2009; Kumar et al., 2008; Wheeler et al., 2011; Wheeler et al., 2013). Uniform HIV infection in challenge experiments was obtained by requiring high levels of human immune reconstitution using previously described criteria (Wheeler et al., 2011; Wheeler et al., 2013). These criteria included a minimum of 25% of human CD45+ cells in the blood, of which at least half are lymphocytes (and at least 40% of these are T cells). The absolute number of human T cells was required to be at least 200/100 μL of blood. BLT mice were treated with PBS or 80 pmols of CD4-AsiCs in PBS by atraumatic application to the vaginal mucosa in 15 μL according to the indicated schedule. For infections, HIVJR-CSF (105 TCID50) diluted in 10 μL PBS was applied atraumatically to the vaginal mucosa. Mice were kept supine for 5 min after each application. For some experiments 1 μg/mL of LPS (List Biological Laboratories, Inc.) was applied IVAG. For other experiments, rIFN (R&D Systems) was administered either by tail vein injection (10,000 IU in 50 μL PBS) or by IVAG administration (2,000 IU in 10 μL PBS). For all post-treatment analysis, both the vaginal tissue and peripheral blood were harvested and processed as previously described (Wheeler et al., 2011; Wheeler et al., 2013). Single cell suspensions were stained using 1/100 dilutions of hCD45 and hCD4 antibodies (BioLegend), and sorted by FACS for gene expression analysis.
Additional experimental methods are described in Supplemental Information.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (AI102816 and AI090671, JL; AI078897, AMT, ADL), the Harvard CFAR (AI060354, AMT, ADL) and Ragon Institute (JL, AMT, ADL) and fellowships from the Cancer Research Institute, the Adelstein Fund, the Harvard Medical School MD/PhD Program and the Point Foundation (LAW), and the Harvard CFAR (RT). We thank T. Allen and T. Dudek and the Virology Core of the Ragon Institute for providing viral stocks for in vivo studies and E. Oliva, M. Miri, and A. Bodo, MGH Surgical Pathology, and L. Yang, BIDMC, for tissue specimens.
Footnotes
Supplemental Information includes five Supplemental Figures, three Supplemental Tables, Supplemental Experimental Procedures and References.
Author contributions JL, LAW and RT designed the research plan and wrote the manuscript. LAW and RT performed experiments with assistance of VV, NB, XL, BB, LO, SM, and SR. ADL and AMT supervised and helped design the humanized mouse experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agy MB, Acker RL, Sherbert CH, Katze MG. Interferon treatment inhibits virus replication in HIV-1- and SIV-infected CD4+ T-cell lines by distinct mechanisms: evidence for decreased stability and aberrant processing of HIV-1 proteins. Virology. 1995;214:379–386. doi: 10.1006/viro.1995.0047. [DOI] [PubMed] [Google Scholar]
- Arfi V, Riviere L, Jarrosson-Wuilleme L, Goujon C, Rigal D, Darlix JL, Cimarelli A. Characterization of the early steps of infection of primary blood monocytes by human immunodeficiency virus type 1. J Virol. 2008;82:6557–6565. doi: 10.1128/JVI.02321-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi A, Pancino G. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology. 2010;7:31. doi: 10.1186/1742-4690-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boasso A, Hardy AW, Anderson SA, Dolan MJ, Shearer GM. HIV-induced type I interferon and tryptophan catabolism drive T cell dysfunction despite phenotypic activation. PLoS One. 2008;3:e2961. doi: 10.1371/journal.pone.0002961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brainard DM, Seung E, Frahm N, Cariappa A, Bailey CC, Hart WK, Shin HS, Brooks SF, Knight HL, Eichbaum Q, et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J Virol. 2009;83:7305–7321. doi: 10.1128/JVI.02207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Caux C, Ait-Yahia S, Chemin K, de Bouteiller O, Dieu-Nosjean MC, Homey B, Massacrier C, Vanbervliet B, Zlotnik A, Vicari A. Dendritic cell biology and regulation of dendritic cell trafficking by chemokines. Springer Semin Immunopathol. 2000;22:345–369. doi: 10.1007/s002810000053. [DOI] [PubMed] [Google Scholar]
- Coccia EM, Krust B, Hovanessian AG. Specific inhibition of viral protein synthesis in HIV-infected cells in response to interferon treatment. J Biol Chem. 1994;269:23087–23094. [PubMed] [Google Scholar]
- Collins KB, Patterson BK, Naus GJ, Landers DV, Gupta P. Development of an in vitro organ culture model to study transmission of HIV-1 in the female genital tract. Nat Med. 2000;6:475–479. doi: 10.1038/74743. [DOI] [PubMed] [Google Scholar]
- d’Ettorre G, Ceccarelli G, Giustini N, Mastroianni CM, Silvestri G, Vullo V. Taming HIV-related inflammation with physical activity: a matter of timing. AIDS Res Hum Retroviruses. 2014;30:936–944. doi: 10.1089/aid.2014.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KA, Lin Y, Abrams B, Jayasena SD. Staining of cell surface human CD4 with 2′-F-pyrimidine-containing RNA aptamers for flow cytometry. Nucleic Acids Res. 1998;26:3915–3924. doi: 10.1093/nar/26.17.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieu-Nosjean MC, Vicari A, Lebecque S, Caux C. Regulation of dendritic cell trafficking: a process that involves the participation of selective chemokines. J Leukoc Biol. 1999;66:252–262. doi: 10.1002/jlb.66.2.252. [DOI] [PubMed] [Google Scholar]
- Fitzgerald-Bocarsly P, Jacobs ES. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. J Leukoc Biol. 2010;87:609–620. doi: 10.1189/jlb.0909635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa-Geffen A, Hamar P, Le MTN, Wheeler LA, Trifonova R, Petrocca F, Wittrup A, Lieberman J. Gene Knockdown by EpCAM Aptamer-siRNA Chimeras Suppresses Epithelial Breast Cancers and Their Tumor-Initiating Cells. Mol Cancer Ther. 2015;14:2279–2291. doi: 10.1158/1535-7163.MCT-15-0201-T. [DOI] [PubMed] [Google Scholar]
- Goldfeld AE, Birch-Limberger K, Schooley RT, Walker BD. HIV-1 infection does not induce tumor necrosis factor-alpha or interferon-beta gene transcription. J Acquir Immune Defic Syndr. 1991;4:41–47. [PubMed] [Google Scholar]
- Haase AT. Targeting early infection to prevent HIV-1 mucosal transmission. Nature. 2010;464:217–223. doi: 10.1038/nature08757. [DOI] [PubMed] [Google Scholar]
- Hansen K, Prabakaran T, Laustsen A, Jørgensen SE, Rahbæk SH, Jensen SB, Nielsen R, Leber JH, Decker T, Horan KA, et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014;33(15):1654–66. doi: 10.15252/embj.201488029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy GA, Sieg SF, Rodriguez B, Jiang W, Asaad R, Lederman MM, Harding CV. Desensitization to type I interferon in HIV-1 infection correlates with markers of immune activation and disease progression. Blood. 2009;113:5497–5505. doi: 10.1182/blood-2008-11-190231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, Yan N. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol. 2013;14:61–71. doi: 10.1038/ni.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J Virol. 2007;81:12596–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Burchard J, Leake D, Reynolds A, Schelter J, Guo J, Johnson JM, Lim L, Karpilow J, Nichols K, et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA. 2006;12:1197–1205. doi: 10.1261/rna.30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- Klatt NR, Bosinger SE, Peck M, Richert-Spuhler LE, Heigele A, Gile JP, Patel N, Taaffe J, Julg B, Camerini D, et al. Limited HIV infection of central memory and stem cell memory CD4+ T cells is associated with lack of progression in viremic individuals. PLoS Pathog. 2014;10:e1004345. doi: 10.1371/journal.ppat.1004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Abbas W, Herbein G. HIV-1 latency in monocytes/macrophages. Viruses. 2014;6:1837–1860. doi: 10.3390/v6041837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Ban HS, Kim SS, Wu H, Pearson T, Greiner DL, Laouar A, Yao J, Haridas V, Habiro K, et al. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008;134:577–586. doi: 10.1016/j.cell.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, El Marjou A, Lacabaratz C, Lelievre JD, Manel N. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Lehmann C, Lafferty M, Garzino-Demo A, Jung N, Hartmann P, Fatkenheuer G, Wolf JS, van Lunzen J, Romerio F. Plasmacytoid dendritic cells accumulate and secrete interferon alpha in lymph nodes of HIV-1 patients. PLoS One. 2010;5:e11110. doi: 10.1371/journal.pone.0011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- Miller CJ, Li Q, Abel K, Kim EY, Ma ZM, Wietgrefe S, La Franco-Scheuch L, Compton L, Duan L, Shore MD, et al. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J Virol. 2005;79:9217–9227. doi: 10.1128/JVI.79.14.9217-9227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. IFI16 DNA Sensor Is Required for Death of Lymphoid CD4 T-cells Abortively Infected with HIV. Science. 2014;343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen R, Wahl A, Denton PW, Garcia JV. Immune reconstitution of the female reproductive tract of humanized BLT mice and their susceptibility to human immunodeficiency virus infection. J Reprod Immunol. 2011;88:195–203. doi: 10.1016/j.jri.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A. 2015;112:E1773–81. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, et al. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci USA. 2013;110:6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piguet V, Steinman RM. The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol. 2007;28:503–510. doi: 10.1016/j.it.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G, Biswas P, Fauci AS. Interferons in the pathogenesis and treatment of human immunodeficiency virus infection. Antiviral Res. 1994;24:221–233. doi: 10.1016/0166-3542(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba E, Grivel JC, Vanpouille C, Brichacek B, Fitzgerald W, Margolis L, Lisco A. HIV-1 sexual transmission: early events of HIV-1 infection of human cervico-vaginal tissue in an optimized ex vivo model. Mucosal Immunol. 2010;3:280–290. doi: 10.1038/mi.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, Levin D, Wijeyesinghe S, Makamdop KN, Del Prete GQ, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511:601–605. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirazi Y, Pitha PM. Alpha interferon inhibits early stages of the human immunodeficiency virus type 1 replication cycle. J Virol. 1992;66:1321–1328. doi: 10.1128/jvi.66.3.1321-1328.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Colonna M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol Rev. 2010;234:142–162. doi: 10.1111/j.0105-2896.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Bowie AG. The interplay between viruses and innate immune signaling: recent insights and therapeutic opportunities. Biochem Pharmacol. 2008;75:589–602. doi: 10.1016/j.bcp.2007.07.043. [DOI] [PubMed] [Google Scholar]
- Wang Y, Abel K, Lantz K, Krieg AM, McChesney MB, Miller CJ. The Toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J Virol. 2005;79:14355–14370. doi: 10.1128/JVI.79.22.14355-14370.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler LA, Trifonova R, Vrbanac V, Basar E, McKernan S, Xu Z, Seung E, Deruaz M, Dudek T, Einarsson JI, et al. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. J Clin Invest. 2011;121:2401–2412. doi: 10.1172/JCI45876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler LA, Vrbanac V, Trifonova R, Brehm MA, Gilboa-Geffen A, Tanno S, Greiner DL, Luster AD, Tager AM, Lieberman J. Durable Knockdown and Protection From HIV Transmission in Humanized Mice Treated With Gel-formulated CD4 Aptamer-siRNA Chimeras. Mol Ther. 2013;21:1378–1389. doi: 10.1038/mt.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, KewalRamani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol. 2006;6:859–868. doi: 10.1038/nri1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Cherepanov P, Daigle JE, Engelman A, Lieberman J. The SET Complex Acts as a Barrier to Autointegration of HIV-1. PLoS Pathog. 2009;5:e1000327. doi: 10.1371/journal.ppat.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Bloch N, Nguyen LA, Kim B, Landau NR. SAMHD1 restricts HIV-1 replication and regulates interferon production in mouse myeloid cells. PLoS One. 2014;9:e89558. doi: 10.1371/journal.pone.0089558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, Reinhart TA, Rogan M, Cavert W, Miller CJ, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 1999;286:1353–1357. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.