

Abstract
Objective:
We sought to determine the underlying cortical gene expression changes associated with Parkinson dementia using a next-generation RNA sequencing approach.
Methods:
In this study, we used RNA sequencing to evaluate differential gene expression and alternative splicing in the posterior cingulate cortex from neurologically normal control patients, patients with Parkinson disease, and patients with Parkinson disease with dementia.
Results:
Genes overexpressed in both disease states were involved with an immune response, whereas shared underexpressed genes functioned in signal transduction or as components of the cytoskeleton. Alternative splicing analysis produced a pattern of immune and RNA-processing disturbances.
Conclusions:
Genes with the greatest degree of differential expression did not overlap with genes exhibiting significant alternative splicing activity. Such variation indicates the importance of broadening expression studies to include exon-level changes because there can be significant differential splicing activity with potential structural consequences, a subtlety that is not detected when examining differential gene expression alone, or is underrepresented with probe-limited array technology.
Parkinson disease (PD) is the second most common neurologic disorder, characterized by degeneration of midbrain dopaminergic neurons and a high prevalence of dementia. Neuroinflammation, oxidative stress, mitochondrial dysfunction, protein processing, and aberrant alternative splicing are among the commonly considered pathways of dysfunction in PD.1
Gene expression studies of PD and other neurologic disorders, via microarray and next-generation, high-throughput RNA sequencing (RNA-seq), have facilitated the expansion of gene expression knowledge. We previously found that axonal transport, cell adhesion, and mRNA splicing are the most prevalent dysregulated processes in PD cortical neurons, all occurring before the onset of dementia.2 Because previous expression profiling provided evidence of significant spliceosome alterations, in the present study, we leveraged mRNA-seq to incorporate splice variant analysis.
We report here that differential alternative splicing in the cortex is associated with PD and Parkinson disease with dementia (PD-D), possibly contributing to the etiology of these diseases. We profiled posterior cingulate cortex, because the spread of α-synuclein pathology to this cortical region is associated with dementia in PD.3 We found that genes displaying the highest degree of alternative splicing are somewhat disconnected from the greatest expression changes, although overlapping cellular processes are evident. Compared with gene expression profiling alone, alternative splice analysis of mRNA-seq data affords precision profiling in addition to an overview of global expression changes.
METHODS
Tissue collection.
Posterior cingulate cortex (Brodmann Area 23) samples were obtained from the Banner Sun Health Research Institute Brain Bank. All patients signed informed consent and were prospectively followed until death and autopsied according to previously published protocols.4 PD clinical diagnoses were made using UK Brain Bank criteria and DSM-IV criteria for cognitive diagnoses. Neuropathologic diagnoses were made using the Unified Staging System for Lewy body disorders.5 Cohorts studied were PD (N = 13; mean age 79.3 years ± 6.8, 23% female), PD-D (N = 10; mean age 75.5 years ± 7.0, 30% female), or healthy controls (N = 11; mean age 77.9 years ± 7.9, 9% female). Samples were selected with a postmortem interval less than 3 hours. Posterior cingulate cortex was sectioned at 8-µm thickness and placed into standard 1.5-mL microcentrifuge tubes and stored at −80°C until RNA extraction.
RNA extraction.
TRIzol reagent was used for initial RNA isolation per the manufacturer's protocol. The RNA solution was then processed through the Qiagen RNeasy Mini kit, with DNase treatment, using the manufacturer's protocol.
mRNA-seq library preparation.
One microgram of total RNA was used to generate mRNA-seq libraries for sequencing using Illumina's RNA Sample Prep Kit (catalog #FC-122-1001) using the manufacturer's protocol. Poly-A selection was performed using oligo(dT) magnetic beads, and double-stranded cDNA was generated and fragmented to a target size of 400 bp using sonication on the Covaris. Fragmented samples were end repaired and adenylated at the 3′ end, and TruSeq Indexed adapters were ligated on. Libraries were enriched using the TruSeq PCR Master Mix and primer cocktail. Final libraries were cleaned and quantified using the Agilent Bioanalyzer and Invitrogen Qubit. Libraries were equimolarly pooled for sequencing.
Paired-end sequencing.
Denatured and diluted libraries with a 1% phiX spike-in were used to generate clusters on Illumina's HiSeq Paired End v3 flowcell on the Illumina cBot using Illumina's TruSeq PE Cluster Kit v3 (catalog #PE-401-3001). The clustered flowcell was sequenced by synthesis on the Illumina HiSeq 2000 for paired 100-bp reads using Illumina's TruSeq SBS Kit V3 (catalog #FC-401-3001).
Sequencing and differential expression analysis.
An average of 109 million paired-end, passing-filter reads (90 mers) were generated for each sample. Reads were trimmed of adapter sequences and aligned to Homo sapiens GRCh37.62 with TopHat (version 2.0.8, bowtie version 0.12.7) using default parameters. A table of read counts was assembled in R with the easyRNASeq package. Pairwise differential expression analyses between the groups was conducted with the DESeq6 package.
Gene ontology.
GeneMANIA7 is a functional association tool used for data visualization and statistical analysis. It builds gene ontologies (GO)8 from gene input lists and is freely available as a web application.
Alternative splicing analysis.
We used SpliceSeq9 (version 2.1), developed for alternative splicing detection in RNA-seq data, to investigate significant differential splicing events in disease groups (PD or PD-D) relative to controls (CON). SpliceSeq returns gene RPKM and exon OPKM (observations per kilobase of exon/splice per million aligned reads) normalized read values, defined, respectively, as “reads” and “observations” per kilobase of transcript length per million aligned reads. OPKM provides a measure of exon expression; a read that contains at least 4 bases of an exon is “observed.”
Reads “spliced in,” i.e., reads that map to an exon, are calculated against the total number of reads that span the flanking exons, but skip it, plus the spliced in reads. This is the “percent spliced in” (PSI) value assigned to each exon. The difference between the average exon PSI of “group 2” and “group 1” (e.g., PD minus CON) is termed delta PSI (dPSI). dPSI sample calculation:
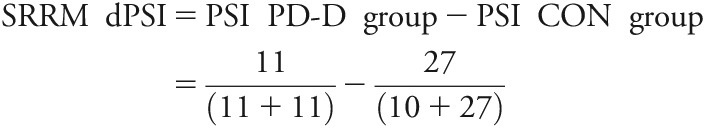 |
Quantitative real-time PCR validation.
RNA extracted (described above) from PD, PD-D, and control tissues was processed with the SuperScript III First-Strand Synthesis kit (Life Technologies, Carlsbad, CA) for cDNA synthesis, per the manufacturer's protocol. Quantitative real-time (qRT) PCR was completed using the Roche LightCycler 480 with SYBR Green detection.
RESULTS
We performed mRNA-seq on RNA samples isolated from the posterior cingulate cortex of individuals with either PD or PD-D or from healthy controls. Data analysis for this study focused on differential gene expression (DE) using DESeq, and alternative splicing (AS) events using SpliceSeq. DE and AS changes in PD and PD-D were quantified relative to CON. Genes from DE and AS analyses were compared for overlapping results, the DE data were run through GeneMANIA, and certain AS genes were validated by qRT-PCR.
Gene ontology and differential expression.
We first compared gene-level expression profiles across disease groups. DE lists were restricted to the top 20 genes differentially expressed by fold change (fc) ≥ |0.2| and p ≤ 0.001 (multiple test-corrected with the Benjamini-Hochberg procedure), as determined by DESeq analysis (table 1 and table e-1 at Neurology.org/ng). The top 20 DE genes from PD and PD-D vs CON were compared with gene networks for GO analysis. Functions (table e-2) associated with the top 20 overexpressed PD genes included cell chemotaxis, cytokine receptor binding, cAMP-mediated signaling, and ion homeostasis, whereas overexpressed PD-D genes were involved in unfolded or incorrectly folded protein responses. Underexpressed PD and PD-D genes shared a common involvement in hormone signaling.
Table 1.
Top 20 differentially expressed genes by disease group
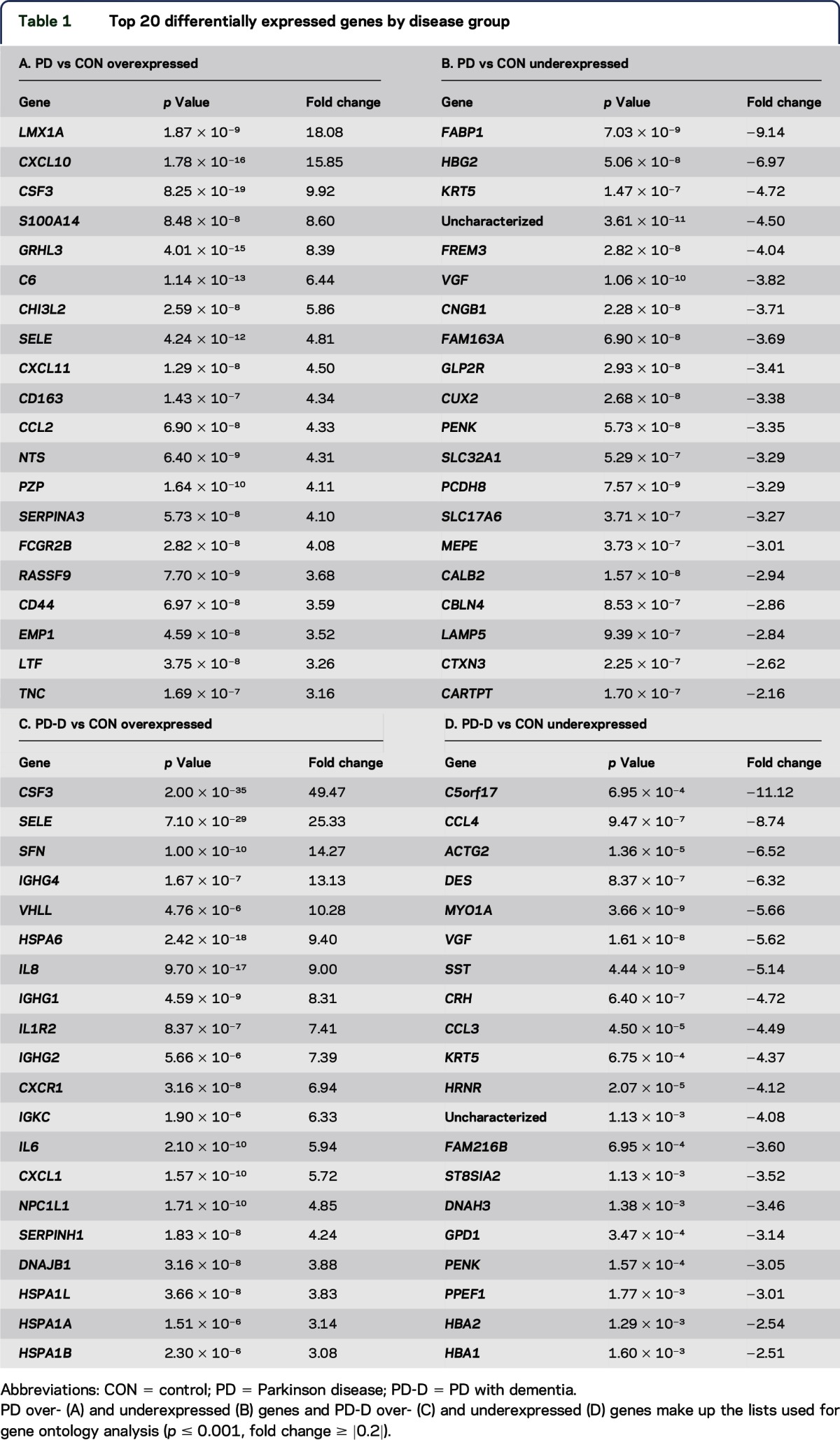
Comparison of PD with CON revealed changes in immune function and neuropeptide hormone function, including upregulation of chemokine (C-X-C motif) ligand 10 (CXCL10) and downregulation of proenkephalin (PENK). In the comparison of PD-D with CON, genes involved in the unfolded protein response were significantly upregulated, reflected by increased expression of dnaJ (Hsp40) homolog (DNAJB1) and several heat shock protein genes, and an overall downregulation of hormone activity and ion transport, indicated by decreased expression of corticotropin-releasing hormone (CRH), PENK, somatostatin (SST), chemokine (C-C motif) ligand 3, and chemokine (C-C motif) ligand 4.
Predominant pathways shared by both PD and PD-D included upregulated inflammatory responses, with increased expression of granulocyte colony-stimulating factor (CSF3), selectin-E (SELE), and additional cytokines. Shared downregulated genes for signaling and cytoskeletal structure included decreased expression of PENK and keratin 5 (KRT5); Vgf nerve growth factor (VGF) was also downregulated across disease states.
Alternative splicing.
SpliceSeq identified significant disease-associated differences in transcript variants. SpliceSeq-predicted events were initially filtered for significance (p < 0.01) and dPSI > |0.2|, where dPSI is the change in exon expression of disease (PD or PD-D) vs CON. Additional filtering parameters considered the occurrence of the splicing event across all transcripts in the group (magnitude) and the number of samples within the group experiencing the event (percent observed) – both set to > |0.8| (1.0 = 100%). Finally, genes with the highest RPKM within the filtered lists (15–100 RPKM) were preferentially picked for validation (table e-3).
Five genes (total = 40) from the PD-D/CON comparison (ATXN2, DST, HSPH1, RELA, and SRRM1) and 2 genes (total = 10) from the PD/CON comparison (leucine-rich repeat flightless-interacting protein 1 [LRRFIP1] and tripartite motif 9 [TRIM9]) were chosen for qRT-PCR validation of the specific AS events (figure 1). Heat shock protein 105 kDa (HSPH1), rel-like domain-containing (RELA), LRRFIP1, and TRIM9 may be grouped by processes of cellular maintenance or stress response (proliferation, apoptosis, etc.), and immune responses. RELA, LRRFIP1, and TRIM9 also code for transcription factors. Ataxin-2 (ATXN2) and serine/arginine repetitive matrix 1 (SRRM1) perform RNA-processing functions. Dystonin (DST) is a cytoskeletal linking protein.
Figure 1. SpliceSeq “splice graphs” for the 7 qRT-PCR tested genes.

RNA-seq transcript reads are aligned to composite gene graphs built from all possible isoforms existing in the Ensembl genome database. The graphs in this figure are partial splice graphs showing alternatively spliced regions. Values on graphs are splice observations per kilobase of transcript length per million aligned reads. Exon highlighting (determined by a log2 exon expression ratio) indicates up- or downregulated exon expression or splicing as follows: green exons and splice arcs represent increased activity in the CON group and red exons and splice arcs represent either the PD or PD-D disease group. Gene splice graphs shown are specific for PD-D vs CON (ATXN2, DST, HSPH1, RELA, and SRRM1) or PD vs CON (LRRFIP1 and TRIM9). CON = control; PD = Parkinson disease; PD-D = PD with dementia; qRT = quantitative real-time.
We performed qRT-PCR on exons predicted by SpliceSeq to experience an event, either exclusion from, or inclusion in the final transcript (table 2 for exon sites and primer sequences). SpliceSeq-predicted exon-level expression changes (figure 2) were validated by comparison with qRT-PCR fc (2−ΔΔCt). Significant fc values <1 for primers covering exon skip regions and >1 for the exon inclusion event were considered validated.
Table 2.
Splicing events and qRT-PCR primer sequences

Figure 2. dPSI values for SpliceSeq-predicted alternatively spliced exons within the labeled genes.
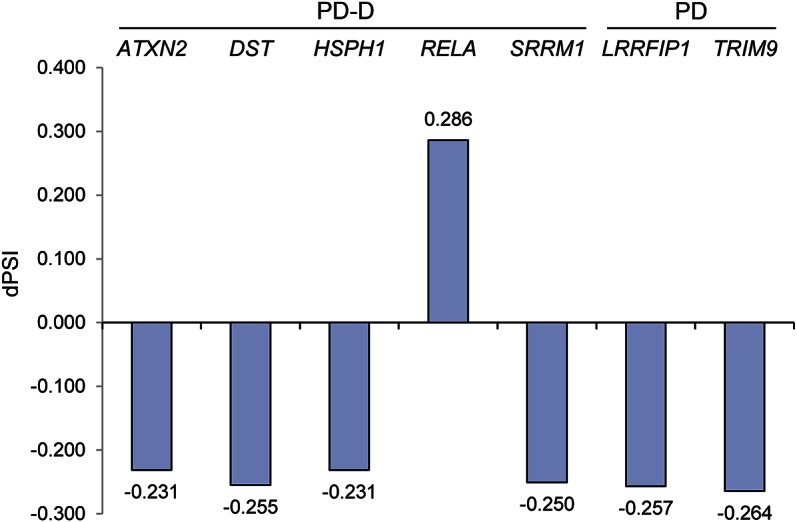
Exon dPSI represents the difference in percent spliced in, disease vs CON (see table 2 for exon details). CON = control; dPSI = delta percent spliced in; PD = Parkinson disease; PD-D = PD with dementia.
In PD-D, skips were predicted by SpliceSeq at ATXN2 exon 21, DST exon 104, HSPH1 exon 13, and SRRM1 exon 16, which disrupts a region reported as necessary for speckles and matrix localization. Exon 10.2 of RELA was predicted to be spliced in. This is a 687-bp inclusion containing a premature stop site that would result in a 377-amino acid truncated isoform and consequently eliminate an activation domain.
In PD, skips were predicted in LRRFIP1 exons 18–19 within a coiled-coil structural region and TRIM9 exons 8–10 within a fibronectin type III and B30.2/SPRY domain.
Quantitative real-time PCR.
The fc of the predicted alternative splice sites were compared with fc of adjacent sites lacking any predicted splicing activity, referred to as “no-splice” (table e-4). Six of the 7 genes were predicted to have exon skips and 1 was predicted to have an exon inclusion (RELA).
At splice-predicted sites of ATXN2, HSPH1, and SRRM1, fc were <1 relative to CON, in addition to having reduced expression compared with sites without predicted splice events. This is consistent with exon exclusion events in these transcripts. RELA, at the predicted inclusion site, was greater than 1 and overexpressed relative to the no-splice site. This indicates specific inclusion of exon 10.2. A change in DST was indiscernible from CON and its no-splice site.
LRRFIP1 and TRIM9 fc values (<1) likewise indicate reduced exon expression of the predicted exon for each gene. Figure 3 summarizes these results by comparing the fc of alternatively spliced transcripts to the main transcript isoforms (no-splice) as expression ratios, represented by percent exon inclusion of the alternative splice site. Exon skips are less than 100%, whereas the inclusion is greater than 100%. Results demonstrate that the vast majority of predicted AS events selected for validation were confirmed by secondary qRT-PCR measures.
Figure 3. qRT-PCR fold change comparison, disease vs CON, of splice vs no-splice primer sites.

Exon regions with expected alternative splicing activity are compared with flanking exon regions without predicted alternative splicing, “no-splice.” The dotted line indicates 100% inclusion (of a queried exon). CON = control; PD = Parkinson disease; PD-D = PD with dementia; qRT = quantitative real-time.
DISCUSSION
We present an mRNA-seq reference data set to further characterize the cortical molecular etiology of PD and PD-D. In addition to significant gene-level differential mRNA expression, RNA-seq clearly reveals the underlying differential alternative splicing in the posterior cingulate cortex during the course of PD and PD-D. Alternative splicing of ATXN2, HSPH1, SRRM1, RELA, LRRFIP1, and TRIM9 suggests dysregulation of genes within immune and inflammation responses and transcription and RNA processing. Compared with this alternative splicing profile, genes among those that were the most significantly differentially expressed (CSF3, SELE, PENK, VGF, KRT5, CRH, and SST) illustrate the breadth of PD gene dysregulation, beyond one level of expression analysis. In the overall expression changes, immune activity is prominent, but there is also evidence of disrupted neuronal signaling pathways. Immune responses and inflammation are consistently implicated in the course of many neurodegenerative disorders, including PD and PD-D, where microglia are commonly activated and inflammatory cytokines are overexpressed.10–12
CSF3 and SELE were overexpressed in both PD and PD-D relative to CON. Upregulation of both CSF3 (or G-CSF) and SELE in patients with PD, before considerable pathology in the posterior cingulate cortex, is consistent with an early role of immune induction in the progression of PD.
CSF3 is a major cytokine regulator of neutrophils and promotes their increased production in bone marrow.13,14 It can induce neuronal differentiation from adult progenitor cells and also protect neurons from apoptosis at the same time.15 SELE is a cell adhesion molecule (CAM) that aids leukocytes in attaching to the endothelium; monocyte CAMs are activated in neurodegeneration.16 Elevation of serum SELE has been detected in Guillain-Barré syndrome and MS, but when investigated in patients with Alzheimer disease, levels did not differ.17 We are not aware of other instances of induction in the cortex of patients with PD.
PENK, VGF, and KRT5 were underexpressed in both PD and PD-D relative to CON, which suggests potential deficits in neurotransmitter signaling and altered cytoskeletal function.
PENK is a precursor protein of signal peptides that function as neurotransmitters, autocrine and paracrine factors, and hormones.18 Consistent with our expression results, evidence of significant downregulation was reported, relative to young CON (mean age 31), in both aging CON (mean age 77) and in patients with PD.19 PENK is also linked to bipolar disorder.20 VGF is a neurosecretory protein most often expressed in neuroendocrine cells and neurons.21 VGF expression in parietal cortex is reportedly reduced in PD,22 and there is support for VGF as a biomarker of neurodegenerative and other diseases for its differential CF expression and cell type–specific profiles within neuroendocrine organs.21 KRT5 encodes a major keratin of the basal cell layer as part of the intermediate filament cytoskeleton in basal keratinocytes.23,24
CRH and SST were uniquely underexpressed in PD-D. Downregulation of certain hormone proteins suggests a potential hormone imbalance associated with dementia onset. CRH, SST, and PENK are underexpressed, all of which are indicated by GO to be involved with an altered hormone-signaling state.
The genes that emerged from alternative splicing analyses did not exhibit a high degree of differential expression when assessed at the gene level. However, within the AS group, there are a variety of overlapping functions or instances of close networking.
Tripartite motif (TRIM) proteins belong to a subgroup of RING finger proteins, a structural feature that functions as a ubiquitin ligase (E3) to conjugate ubiquitin to target proteins.25,26 TRIM9 is thought to be a targeting signal for proteasomal degradation, may be involved in neuron development and synaptic vesicle exocytosis, and is a negative regulator of transcription factor nuclear factor κB (NF-κB) activation.25–27 It has been found in Lewy bodies of both dementia with Lewy bodies (DLB) and PD; expression is decreased in DLB and rabies virus–infected brains, another case in which degeneration occurs.25 Skipping of exons 8–10 approaching the C terminus (protein position 550–636), shown in our data, affects a fibronectin type III and B30.2/SPRY domain. This domain is associated with microtubule binding among the RBCC (N-terminal RING finger/B-box/coiled coil) protein family, to which TRIM9 and many other TRIM proteins belong. Mutations in the C-terminal B30.2-like domain of another RBCC protein, MID1, disrupt localization and ubiquitin targeting.27
The p65 protein is encoded by RELA and is a subunit of the NF-κB transcription factor complex. They are part of a family of proteins involved in immune and inflammatory responses, apoptosis, and cell proliferation and differentiation.28 NF-κB nuclear localization, from the cytosol, is inhibited by IκB association with the p50-p65 heterodimer, which is liberated upon phosphorylation of IκB.29 Of the splicing activity predicted and validated here, the exon inclusion (exon 10.2, protein position 347–381) within RELA causes a protein truncation and the deletion of an activation domain, which would be predicted to cause overproduction of a truncated, inactive form of the protein.
HSPH1, a 105-kDa heat shock protein (HSP105), directs caspase-3–mediated apoptosis after ER stress and interacts with α-tubulin to suppress disorganization during heat shock.30,31 HSP105β is a truncated isoform (92 kDa) induced specifically at 42°C.32 Our data show a skip (exon 13, protein position 529–572), elevated in PD-D compared with CON, consistent with this β-isoform. Normal physiologic temperature ranges from peripheral 33°C to fever-induced 39°C and does not reach the extreme temperature (42°C) associated with a heat shock response.33 If the high-heat–responding β-isoform is overproduced instead of the constitutively active form, signaling for apoptosis could be dampened as a result of reduced functional activity within the normal physiologic temperature range.
SRRM1 (SR-related matrix protein of 160 kDa), is a pre-mRNA splicing coactivator that associates with the nuclear matrix along with its binding partner SRm300, aiding in recruitment of splicing factors to speckled regions and assembly of complexes.34 Disruption to a sequence portion that is necessary for speckle and matrix localization could hinder recruitment of pre-mRNA to these areas and therefore disrupt proper splicing and processing to mature mRNA. In the patients with PD-D, exon 16 (protein position 558–570), which is within the region encoding speckles and matrix localization specificity, is preferentially excluded in the mature mRNA.
ATXN2 is an RNA-binding protein involved in RNA processing and endocytosis, affecting membrane receptor presence. In vivo knockout of the mouse homolog, SCA2, leads to increased insulin production with a concurrent reduction in insulin receptors, resulting in obesity. Exclusion of exon 21 (protein position 1127–1145) was also previously identified by a broad sequencing and characterization study, although the effect of this particular event is unknown. Although a polyQ expansion within ATXN2 is a notorious culprit of genetic malfunction, the exon 21 skip is far downstream (>2 kbp) from the well-studied region in exon 1. Considering the aforementioned mouse study, alternatively spliced ATXN2 might interfere with cellular responses by reducing receptor production or membrane incorporation. However, the specific functional effect of this alternative splicing event is unclear.
LRRFIP1 is a promoter-binding transcriptional repressor that regulates EGFR and may also regulate the proinflammatory cytokine TNF.35 These functions suggest that it is tied to cell damage and injury response. Exon 18 and 19 (protein position 398–459) skipping was increased in our PD group. Together, they partially encode coiled-coil domains and a prefoldin superfamily domain, protein features that promote protein-protein interactions and molecular chaperoning, respectively.36,37 Exon skipping in this region has the potential to decrease binding affinity with LRRFIP1 targets and to interrupt any chaperone activity this protein may provide.
Although alternative splicing is a normal mechanism of gene regulation, the pathways in which it is relatively increased suggest altered responses in the disease state. A number of scenarios are possible. An aberrantly spliced transcript may drive a pathway to become overactive, contributing to disease onset or progression. Another possibility is that cells in distress during the progression of PD and PD-D may experience altered splicing as a consequence of widespread dysfunction. Distinguishing between these possibilities is an important goal of future work.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona, for the provision of human biological materials.
GLOSSARY
- ATXN2
ataxin-2
- CAM
cell adhesion molecule
- CON
controls
- CRH
corticotropin-releasing hormone
- CSF3
granulocyte colony-stimulating factor
- DE
differential gene expression
- DLB
dementia with Lewy bodies
- dPSI
delta PSI
- DSM-IV
Diagnostic and Statistical Manual of Mental Disorders, 4th edition
- DST
dystonin
- fc
fold change
- GO
gene ontology
- HSPH1
heat shock protein 105 kDa
- KRT5
keratin 5
- LRRFIP1
leucine-rich repeat flightless-interacting protein 1
- NF-κB
nuclear factor κB
- OPKM
observations per kilobase of transcript length per million aligned reads
- PD
Parkinson disease
- PD-D
Parkinson disease with dementia
- PENK
proenkephalin
- PSI
percent spliced in
- qRT
quantitative real-time
- RBCC
N-terminal RING finger/B-box/coiled coil
- RELA
rel-like domain-containing
- RNA-seq
RNA sequencing
- RPKM
reads per kilobase of transcript length per million aligned reads
- SELE
selectin-E
- SRRM1
serine/arginine repetitive matrix 1
- SST
somatostatin
- TRIM
tripartite motif
- TRIM9
tripartite motif 9
- VGF
Vgf nerve growth factor
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Adrienne Henderson-Smith contributed to laboratory work, experimental design, interpretation of data, and manuscript drafting and revision. Jason J. Corneveaux contributed to data analysis and interpretation. Matthew De Both contributed to data analysis. Lori Cuyugan contributed to experimental design. Winnie S. Liang and Matthew Huentelman contributed to manuscript revision for intellectual content. Charles Adler and Erika Driver-Dunckley contributed to study design. Thomas G. Beach contributed to study design and manuscript revision for intellectual content. Travis L. Dunckley contributed to study conceptualization and design, interpretation of data, and manuscript drafting and revision.
STUDY FUNDING
The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson's Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer's Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson's Disease Consortium), and the Michael J. Fox Foundation for Parkinson's Research.
DISCLOSURE
Dr. Henderson-Smith reports no disclosures. Dr. Corneveaux—deceased. Dr. De Both and Dr. Cuyugan report no disclosures. Dr. Liang holds patents for Long insert-based whole genome sequencing for identification of structural variants and Characterization of somatic events and viral integration in cancer. Dr. Huentelman has received research support from NIH/National Institute of Neurological Disorders and Stroke. Dr. Adler has received travel/speaker honoraria from the Movement Disorder Society; has received publishing royalties from Humana Press; has been a consultant for Abbvie, Acadia, Allergan, Cynapsus, Impax, Ipsen, Lilly, Lundbeck. Merz, and Teva; and has received research support from Avid Radiopharmaceuticals, NIH/National Institute of Neurological Disorders and Stroke, and the Michael J. Fox Foundation for Parkinson's Disease Research. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ. Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol 2012;8:518–530. [DOI] [PubMed] [Google Scholar]
- 2.Stamper C, Siegel A, Liang WS, et al. Neuronal gene expression correlates of Parkinson's disease with dementia. Mov Disord 2008;23:1588–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kraybill ML, Larson EB, Tsuang DW, et al. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 2005;64:2069–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beach TG, Adler CH, Sue LI, et al. Arizona study of aging and neurodegenerative disorders and brain and body donation program. Neuropathol 2015;35:354–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beach TG, Adler CH, Lue L, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 2009;117:613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montojo J, Zuberi K, Rodriguez H, et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 2010;26:2927–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan MC, Cleland J, Kim R, Wong WC, Weinstein JN. SpliceSeq: a resource for analysis and visualization of RNA-Seq data on alternative splicing and its functional impacts. Bioinformatics 2012;28:2385–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Botta-Orfila T, Sanchez-Pla A, Fernandez M, Carmona F, Ezquerra M, Tolosa E. Brain transcriptomic profiling in idiopathic and LRRK2-associated Parkinson's disease. Brain Res 2012;1466:152–157. [DOI] [PubMed] [Google Scholar]
- 11.De Rosa P, Marini ES, Gelmetti V, Valente EM. Candidate genes for Parkinson disease: lessons from pathogenesis. Clin Chim Acta 2015;449:68–76. [DOI] [PubMed] [Google Scholar]
- 12.Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol 2014;14:463–477. [DOI] [PubMed] [Google Scholar]
- 13.Fife M, Gibson MS, Kaiser P. Identification of chicken granulocyte colony-stimulating factor (G-CSF/CSF3): the previously described myelomonocytic growth factor is actually CSF3. J Interferon Cytokine Res 2009;29:339. [DOI] [PubMed] [Google Scholar]
- 14.Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and “emergency” hematopoiesis. Cytokine 2008;42:277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider A, Kuhn HG, Schäbitz WR. A role for G-CSF (granulocyte-colony stimulating factor) in the central nervous system. Cell Cycle 2005;4:1753–1757. [DOI] [PubMed] [Google Scholar]
- 16.Hochstrasser T, Weiss E, Marksteiner J, Humpel C. Soluble cell adhesion molecules in monocytes of Alzheimer's disease and mild cognitive impairment. Exp Gerontol 2010;45:70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rentzos M, Michalopoulou M, Nikolaou C, et al. The role of soluble intercellular adhesion molecules in neurodegenerative disorders. J Neurol Sci 2005;228:129–135. [DOI] [PubMed] [Google Scholar]
- 18.Denning GM, Ackermann LW, Barna TJ, et al. Proenkephalin expression and enkephalin release are widely observed in non-neuronal tissues. Peptides 2008;29:83–92. [DOI] [PubMed] [Google Scholar]
- 19.Backman CM, Shan L, Zhang Y, Hoffer BJ, Tomac AC. Alterations in prodynorphin, proenkephalin, and GAD67 mRNA levels in the aged human putamen: correlation with Parkinson's disease. J Neurosci Res 2007;85:798–804. [DOI] [PubMed] [Google Scholar]
- 20.Altar CA, Vawter MP, Ginsberg SD. Target identification for CNS diseases by transcriptional profiling. Neuropsychopharmacology 2009;34:18–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferri GL, Noli B, Brancia C, D'Amato F, Cocco C. VGF: an inducible gene product, precursor of a diverse array of neuro-endocrine peptides and tissue-specific disease biomarkers. J Chem Neuroanat 2011;42:249–261. [DOI] [PubMed] [Google Scholar]
- 22.Cocco C, D'Amato F, Noli B, et al. Distribution of VGF peptides in the human cortex and their selective changes in Parkinson's and Alzheimer's diseases. J Anat 2010;217:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chamcheu JC, Navsaria H, Pihl-Lundin I, Liovic M, Vahlquist A, Torma H. Chemical chaperones protect epidermolysis bullosa simplex keratinocytes from heat stress-induced keratin aggregation: involvement of heat shock proteins and MAP kinases. J Invest Dermatol 2011;131:1684–1691. [DOI] [PubMed] [Google Scholar]
- 24.Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet 2006;78:510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanji K, Kamitani T, Mori F, Kakita A, Takahashi H, Wakabayashi K. TRIM9, a novel brain-specific E3 ubiquitin ligase, is repressed in the brain of Parkinson's disease and dementia with Lewy bodies. Neurobiol Dis 2010;38:210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi M, Cho H, Inn KS, et al. Negative regulation of NF-kappaB activity by brain-specific TRIpartite Motif protein 9. Nat Commun 2014;5:4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Short KM, Cox TC. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J Biol Chem 2006;281:8970–8980. [DOI] [PubMed] [Google Scholar]
- 28.Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol 2009;29:1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharif-Askari E, Vassen L, Kosan C, et al. Zinc finger protein Gfi1 controls the endotoxin-mediated Toll-like receptor inflammatory response by antagonizing NF-kappaB p65. Mol Cell Biol 2010;30:3929–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meares GP, Zmijewska AA, Jope RS. HSP105 interacts with GRP78 and GSK3 and promotes ER stress-induced caspase-3 activation. Cell Signal 2008;20:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saito Y, Yamagishi N, Ishihara K, Hatayama T. Identification of α-tubulin as an hsp105α-binding protein by the yeast two-hybrid system. Exp Cell Res 2003;286:233–240. [DOI] [PubMed] [Google Scholar]
- 32.Ishihara K, Yasuda K, Hatayama T. Molecular cloning, expression and localization of human 105 kDa heat shock protein, hsp105. Biochim Biophys Acta 1999;1444:138–142. [DOI] [PubMed] [Google Scholar]
- 33.Katkere B, Rosa S, Caballero A, Repasky EA, Drake JR. Physiological-range temperature changes modulate cognate antigen processing and presentation mediated by lipid raft-restricted ubiquitinated B cell receptor molecules. J Immunol 2010;185:5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagner S, Chiosea S, Nickerson JA. The spatial targeting and nuclear matrix binding domains of SRm160. Proc Natl Acad Sci USA 2003;100:3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodall AH, Burns P, Salles I, et al. Transcription profiling in human platelets reveals LRRFIP1 as a novel protein regulating platelet function. Blood 2010;116:4646–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rikiyama T, Curtis J, Oikawa M, et al. GCF2: expression and molecular analysis of repression. Biochim Biophys Acta 2003;1629:15–25. [DOI] [PubMed] [Google Scholar]
- 37.Siegert R, Leroux MR, Scheufler C, Hartl FU, Moarefi I. Structure of the molecular chaperone prefoldin: unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 2000;103:621–632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.