

Abstract
RES-529 (previously named Palomid 529, P529) is a phosphoinositide 3-kinase (PI3K)/AKT/mechanistic target of rapamycin (mTOR) pathway inhibitor that interferes with the pathway through both mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) dissociation. This compound is currently being developed in oncology and ophthalmology. The oncology focus is for the treatment of glioblastoma, where it has received orphan designation by the US Food and Drug Administration, and prostate cancer. We present a review of the PI3K/AKT/mTOR pathway, its role in tumorigenesis, and the potential of RES-529 in cancer treatment. RES-529 inhibits mTORC1/mTORC2 activity in various cancer cell lines, as noted by decreased phosphorylation of substrates including ribosomal protein S6, 4E-BP1, and AKT, leading to cell growth inhibition and death, with activity generally in the range of 5–15 μmol/l. In animal tumor models where the PI3K/AKT/mTOR pathway is abnormally activated (i.e. glioblastoma, prostate cancer, and breast cancer), RES-529 reduces tumor growth by as much as 78%. RES-529 treatment is synergistic with radiation therapy, chemotherapy, and hormonal therapy in reducing tumor growth, potentially by preventing PI3K/AKT/mTOR pathway activation associated with these treatments. Furthermore, this compound has shown antiangiogenic activity in several animal models. mTORC1 and mTORC2 have redundant and distinct activities that contribute toward oncogenesis. Current inhibitors of this pathway have primarily targeted mTORC1, but have shown limited clinical efficacy. Inhibitors of mTORC1 and mTORC2 such as RES-529 may therefore have the potential to overcome the deficiencies found in targeting only mTORC1.
Keywords: AKT, glioblastoma, mTOR, mTORC1, mTORC2, P529, Palomid 529, PI3K, RES-529
Introduction
The phosphoinositide 3-kinase (PI3K)/AKT/mechanistic target of rapamycin (mTOR) pathway plays an essential role in the regulation of cell growth, survival, and proliferation in both physiological and pathological conditions 1–9. Inhibitors of this pathway have the potential to treat diseases such as cancer, which is associated with pathway dysregulation. This review summarizes the activity and potential of one such inhibitor, RES-529, which targets both mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) through complex dissociation, in the treatment of cancer. RES-529 was developed by RestorGenex Corporation. As of January 2016, RestorGenex Corporation merged with Diffusion Pharmaceuticals LLC to form Diffusion Pharmaceuticals Inc.
PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway involves the coordinated activation of multiple molecules leading to the stimulation of various cellular processes, including transcription, translation, and cell survival (Fig. 1). This pathway is activated through various receptors and agents, including receptor tyrosine kinases, G-protein-coupled receptors, and amino acid stimulation (Fig. 1) 1–4,6,7,10,11.
Fig. 1.
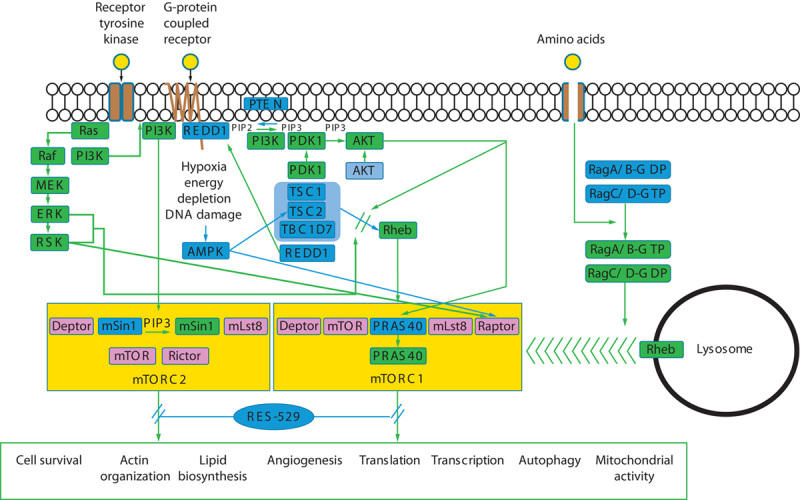
PI3K/AKT/mTOR pathway. Ligand (yellow circle) stimulation of receptor tyrosine kinases or G-protein-coupled receptors or influx of amino acids (yellow circle) leads to the activation of proteins or pathways (indicated by green boxes or lines) or the release of the inhibitory activity of proteins or pathways (indicated by blue boxes or lines, with release indicated by green lines and or green boxes) associated with PI3K/AKT/mTOR pathway activation 1–4,6,7,10,11. RES-529 inhibits this pathway and subsequent downstream biological effects by promoting mTORC1 and mTORC2 complex dissociation. AMPK, adenosine monophosphate-activated protein kinase; Deptor, DEP domain-containing mTOR-interacting protein; ERK, extracellular-signal-regulated kinase; MEK, mitogen-activated protein kinase (MAPK)/ERK kinase; mLst8, mammalian lethal with Sec13 protein 8; mSin1, mammalian stress-activated protein kinase interacting protein 1; mTOR, mechanistic target of rapamycin; mTORC1/2, mTOR complex 1/2; PDK1, 3-phosphoinositide-dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-triphosphate; PRAS40, protein-rich AKT/PKB substrate 40 kDa; PTEN, phosphatase and TENsin homolog deleted on chromosome 10; REDD1, regulated in development and DNA damage responses; Raptor, regulatory-associated protein of mTOR; Rictor, rapamycin-insensitive companion of mTOR; Rheb, Ras homolog enriched in brain; RSK, ribosomal protein S6 kinase; TBC1D7, TBC1, domain family member 7; TSC1/2, tuberous sclerosis complex 1/2.
Stimulation of receptor tyrosine kinases and G-protein-coupled receptors leads to the activation of class IA PI3Ks. Class I PI3Ks are heterodimeric proteins that consist of a catalytic subunit and a regulatory subunit 1–5,7. Before activation, PI3K resides in the cytoplasm. Upon cell stimulation, the regulatory subunit interacts with phosphotyrosine residues on the activated receptor, resulting in a relief of its inhibition of the catalytic subunit and the positioning of the catalytic subunit at the plasma membrane, where its substrate, phosphatidylinositol-4,5-bisphosphate (PIP2), resides 1–5,12. PI3K phosphorylates PIP2 to produce phosphatidylinositol-3,4,5-triphosphate (PIP3) 1–5. This activity can be antagonized by the lipid phosphatase PTEN (phosphatase and TENsin homolog deleted on chromosome 10) through the hydrolysis of PIP3 to PIP2 1–4,13–15.
PIP3 recruits proteins that contain a pleckstrin homology domain to cellular membranes, including the serine–threonine kinase AKT and its activating kinase, 3-phosphoinositide-dependent protein kinase 1 (PDK1), leading to AKT phosphorylation and activation 1,2,5. AKT has more than 100 substrates, including tuberous sclerosis complex 2 (TSC2) and protein-rich AKT/PKB substrate 40 kDa (PRAS40), two proteins that are associated with the regulation of mTORC1 1,3,16,17.
mTORC1 is one of two mTOR complexes, the other being mTORC2 1,3,18,19. mTOR is a serine–threonine kinase that is a member of the PI3K-related kinase family 18. mTORC1 is inhibited by the anticancer drug rapamycin, but mTORC2 is not sensitive to rapamycin, except when treated long term in a cell-type-dependent manner 18,20. mTORC1 is composed of five proteins: mTOR, DEP domain-containing mTOR-interacting protein (Deptor), mammalian lethal with Sec13 protein 8 (mLst8), PRAS40, and regulatory-associated protein of mTOR (Raptor) 1,18. The composition of mTORC2 is mTOR, Deptor, mLst8, rapamycin-insensitive companion of mTOR (Rictor), mammalian stress-activated protein kinase interacting protein 1 (mSin1), and protein observed with Rictor (Proctor) 18.
TSC2 forms a complex with TSC1 and TBC1 domain family member 7 (TSC complex) and acts as a negative regulator of mTORC1 signaling 1,3,18. The TSC complex is a GTPase-activating protein (GAP) for the small GTPase Ras homolog enriched in the brain (Rheb). Phosphorylation of TSC2 by AKT results in the inhibition of the GAP activity of the TSC complex, leading to Rheb activation 1,18. The activated Rheb directly binds to mTORC1 and enhances its kinase activity, along with acting as a scaffold to promote the interaction of mTOR with its substrates 1,18,21. In addition, mTORC1 is activated by AKT through the phosphorylation of PRAS40, an inhibitory component of mTORC1, leading to dissociation of PRAS40 from the complex 1,18.
Another pathway stimulated by growth factors that leads to mTORC1 activation is the Ras-extracellular-signal-regulated kinase (ERK) pathway 1,22. The Ras–ERK pathway activates mTORC1 through at least two mechanisms: the ERK and ribosomal protein S6 kinase (RSK)-mediated phosphorylation and inactivation of TSC2 and RSK phosphorylation of Raptor.
Amino acids activate mTORC1 through Rag GTPases. Upon amino acid stimulation, a GTP–GDP exchange occurs between the Rag GTPase subunits (RagA/RagB and RagC/RagD), leading to its activation 1,18,23. The activated Rag GTPase complex then recruits mTORC1 to the surface of lysosomes through binding to Raptor, whereupon mTORC1 is activated through its interaction with Rheb on the lysosome. Additional components of this pathway include Ragulator, which interacts with Rag GTPase and promotes its GTP–GDP exchange and lysosomal association; SLC38A9, a putative lysosomal arginine sensor for the mTORC1 pathway; and vacuolar H+-ATP 23–27.
Another mechanism that G-protein-coupled receptors use to activate mTORC1 activity, besides stimulating PI3K, is by promoting the sequestering to the plasma membrane of the mTORC1 inhibitory protein, regulated in development and DNA damage responses 1 (REDD1) 28, and preventing its association with mTORC1. REDD1 negatively regulates mTORC1 activity in a TSC complex-dependent and 14-3-3 protein-dependent manner and is induced under hypoxic conditions 18,29.
mTORC1 function is also regulated under stress conditions (such as hypoxia, energy depletion, and DNA damage) by adenosine monophosphate-activated protein kinase (AMPK) 1,18,30. Under stress conditions, AMPK is activated, leading to the phosphorylation of TSC2 and Raptor, which promotes Raptor-14-3-3 protein association, and subsequent mTORC1 inactivation.
mTORC1 plays an important role in regulating mRNA translation through its phosphorylation of 4E-BP1 and p70 ribosomal protein S6 kinase 1 (S6K1) 18,22,31. 4E-BP1 is an endogenous inhibitor of eukaryotic translation initiation factor 4E (eIF4E), a protein that promotes translation initiation by binding to the 5’ cap structure of mRNA. Phosphorylation of 4E-BP1 by mTORC1 prevents its ability to bind to eIF4E and therefore enables cap-dependent translation initiation. Phosphorylation of S6K1 by mTORC1 stimulates S6K1 kinase activity, leading to the phosphorylation and activation of proteins associated with mRNA translation and splicing, including ribosomal protein S6, eukaryotic translation initiation factor 4B (eIF4B), programmed cell death-4 (PDCD4), eukaryotic translation elongation factor 2 kinase (eEF2K), and S6K1 Aly/REF-like target (SKAR).
Additional functions associated with mTORC1 include regulation of transcription, lipid biosynthesis, autophagy induction, and mitochondrial activity 18,31–33. mTORC1 promotes ribosome biogenesis and RNA polymerase I-mediated, II-mediated, and III-mediated transcription, in part through its activation of S6K1 18. Lipid biosynthesis is regulated positively by mTORC1, mainly through its effect on sterol regulatory element-binding protein-1c (SREBP-1c), a major transcription factor associated with lipid biosynthesis 18,31,32. mTORC1 prevents autophagy by phosphorylating proteins, such as ULK1 and autophagy-related protein 13 (Atg13), which are involved in the initial steps of autophagy induction 18,31,33. Autophagy is a pathway for cell survival by which, under conditions of nutrient deprivation, lysosomal degradation of cellular components occurs to supply nutrients. Mitochondrial function is regulated by mTORC1 through the modulation of various mitochondrial and nuclear-encoded mitochondrial genes, including mitochondrial ribosomal proteins 31.
Unlike mTORC1, the mechanism by which mTORC2 is activated following cellular stimulation is not as well defined. Like mTORC1, it is activated through growth factor stimulation of cells 18,34. Recently, Liu et al. 35 identified a link between growth factor stimulation of PI3K and mTORC2 activation. They found that the PH domain of mSin1, a component of mTORC2, interacts with the kinase domain of mTOR to suppress its activity. PIP3, which is produced by activated PI3K, interacts with the PH domain of mSin1 to repress its inhibitory activity to mTOR, leading to mTORC2 activation.
mTORC2 plays an important role in regulating cell survival through the phosphorylation and activation of several AGC family kinases, including AKT, serum and glucocorticoid-induced kinase 1 (SGK1), and protein kinase C (PKC) 18,34,36. In addition, mTORC2 regulates actin organization through the phosphorylation of PKCα and paxillin and the activation of Ras homolog gene family, member A (RhoA), and Ras-related C3 botulinum toxin substrate-1 (Rac1) 18,34. Lipid biosynthesis also appears to be positively regulated by mTORC2, in part through the AKT-mediated activation of SREBP-1c 18,31,37. mTORC2 also regulates mitochondrial function following its growth factor-stimulated recruitment to the mitochondrial-associated endoplasmic reticulum membrane 31,38.
The PI3K/AKT/mTOR pathway is relevant in promoting angiogenesis. Vascular endothelial growth factor (VEGF) receptor activity requires the stimulation of the PI3K/AKT/mTOR pathway 39,40. In addition, VEGF expression is induced by the PI3K/AKT/mTOR pathway through hypoxia-inducible factor 1α (HIF-1α)-dependent and HIF-1α-independent mechanisms, leading to increases in VEGF protein levels 41–44.
Role of PI3K/AKT/mTOR pathway in cancer
Genetic changes in the PI3K/AKT/mTOR pathway leading to its constitutive activation are highly prevalent in a many tumor types, including glioblastoma and prostate, breast, ovary, colon, and lung cancer 2,4,45–51. Mutations in this pathway exist in 86% of glioblastomas, and 42% of primary and 100% of metastatic prostate cancers 45,46.
The PI3K/AKT/mTOR pathway is genetically activated through various elements in the pathway and by different mechanisms. Activating mutations of the PI3K catalytic subunit p110α gene PIK3CA occur in various cancers, including colon, brain, gastric, breast, and lung 2,4,51–53, and amplification of this subunit has also been found 4,7,54,55. Mutations of the regulatory subunit of PI3K resulting in constitutive activity exist in brain, colon, and ovarian cancer 2,4,45,49,51,56,57. Furthermore, alterations in the PI3K antagonist PTEN, including loss-of-function mutations, deletions, and epigenetic silencing of the gene, have been identified in various cancers 51,58–61.
Activating mutations and amplification of the AKT genes have also been found in different types of cancers 62–65, and PDK1 kinase domain mutations have been identified in colon cancer 64. Mutations that increase mTORC signaling, including mTOR, TSC1, and TSC2, and Rheb mutations have been found in various cancers 66–68.
Besides mutations in the pathway itself, the overexpression and mutational alteration of upstream receptors and molecules that promote the PI3K/AKT/mTOR pathway activation, such as receptor tyrosine kinases, occur in cancer 45,50. Thus, in summary, given the significant types and number of mutations in this pathway associated with cancer, identification of compounds that target this pathway is highly relevant.
Rationale for mTORC1 and mTORC2 complex formation inhibitors
mTOR inhibitors are currently approved for the treatment of renal cell carcinoma and pancreatic neuroendocrine tumors [Afinitor (everolimus; Novartis, East Hanover, New Jersey, USA); Torisel (temsirolimus; Pfizer, Philadelphia, Pennsylvania, USA)] 69,70. However, these agents are rapamycin analogs (rapalogs) that target mTORC1, but not mTORC2, and have shown limited clinical efficacy in other tumor types, including prostate cancer and glioblastoma 71–75.
One reason for the lack of clinical efficacy of rapalogs is that they can upregulate the PI3K pathway through the induction of insulin receptor substrate-1 (IRS-1) expression, resulting in AKT activation through Ser473 phosphorylation, an mTORC2 substrate, and subsequent downstream signaling including mitogen-activated protein kinase (MAPK)/ERK activation 76–80. Paradoxically, in various cancer cells types, mTORC1 inhibition also promotes eIF4E phosphorylation, potentially through the MAP/ERK pathway and the activation of Mnk1 (MAPK-interacting serine–threonine kinase 1) 78,81. eIF4E plays an important role in translation initiation and phosphorylation enhances this activity.
Targeted silencing of either mTORC1 or mTORC2 by small interfering RNA (siRNA) has also shown a potential utility of targeting both kinases for cancer 82. In a recent report by Gravina et al. 82, the silencing of mTORC2 through siRNA knockdown of Rictor led to relevant growth inhibition of human prostate 22rv1 cells, whereas siRNA knockdown of Raptor (mTORC1 silencing) had no effect. Furthermore, mTORC1 silencing led to increased expression of the androgen receptor and phosphorylation of Ser473 of AKT, whereas mTORC2 silencing decreased their levels. Overall, these results suggest that a combined mTORC1 and mTORC2 inhibitor may be more efficacious than an mTORC1 inhibitor.
Pharmacology of RES-529
RES-529 (previously named Palomid 529, P529) is a PI3K/AKT/mTOR pathway inhibitor that targets both mTORC1 and mTORC2 through mTOR complex dissociation. It is a modification of a dibenzo[c]chromen-6-one-anti-estrogen derivative (Fig. 2) 83. The concept behind the initial development of RES-529 was the observation that antiestrogens have antiangiogenic and antiproliferative activities that are not because of their antagonism of estrogen receptor function 84,85. Although initially developed using an antiestrogen scaffold, RES-529 has no antiestrogenic activity. At 10 μmol/l concentrations, RES-529 reduces the binding of 0.5 nmol/l [3H]estradiol to estrogen receptor (ER)α and ERβ by 3% or less 83.
Fig. 2.
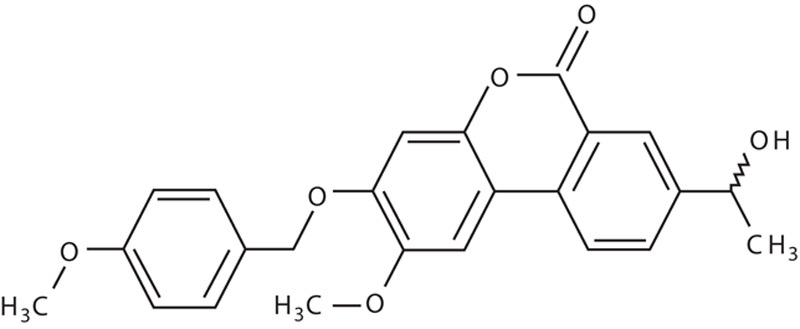
Structure of RES-529.
RES-529 is an orally administered compound that has good blood–brain penetration and lacks affinity to ATP-binding cassette, subfamily B, member 1 (ABCB1) and ATP-binding cassette, subfamily G, member 2 (ABCG2) drug efflux transporters 86. RES-529 has been evaluated in two phase I open-label trials in patients with neovascular age-related macular degeneration (NCT01271270 and NCT01033721) 87–89. In these clinical studies, RES-529 was administered as an ocular injection. The drug was shown to be generally well tolerated and there were no drug-related systemic adverse events 87. Development for age-related macular degeneration through subconjunctival administration is ongoing. The oral formulation of RES-529 is currently being developed for the treatment of glioblastoma, for which it has received orphan designation by the US Food and Drug Administration, and prostate cancer 90.
RES-529 was identified as a PI3K/AKT/mTOR pathway inhibitor in studies determining its mechanism of action in the antiproliferative and apoptotic activity on glioblastoma, endothelial, and prostate tumor cells along with keloid dermal fibroblasts, as described in more detail in the following section 83,91,92. Treatment of human prostate PC-3 cells with RES-529 led to the time-dependent inhibition of ribosomal protein S6 (Ser235/236), 4E-BP1 (Thr37/46), glycogen synthase kinase-3β (GSK-3β, Ser9,) forkhead box protein O1a (Foxo1a, Ser256), mouse double minute 2 homolog (MDM-2, Ser166), and p70S6K (Thr389) phosphorylation (Fig. 3a) 91. RES-529 did not inhibit the phosphorylation of AKT (Thr308) or PDK1 (Ser241), indicating that it does not affect PDK1. Besides inhibiting the phosphorylation of mTORC1 targets, such as ribosomal protein S6 and 4E-BP1 18,22,91, RES-529 treatment has also been shown in various studies to inhibit AKT (Ser473) phosphorylation, an mTORC2-specific substrate (Fig. 3b) 77,83,91,92.
Fig. 3.
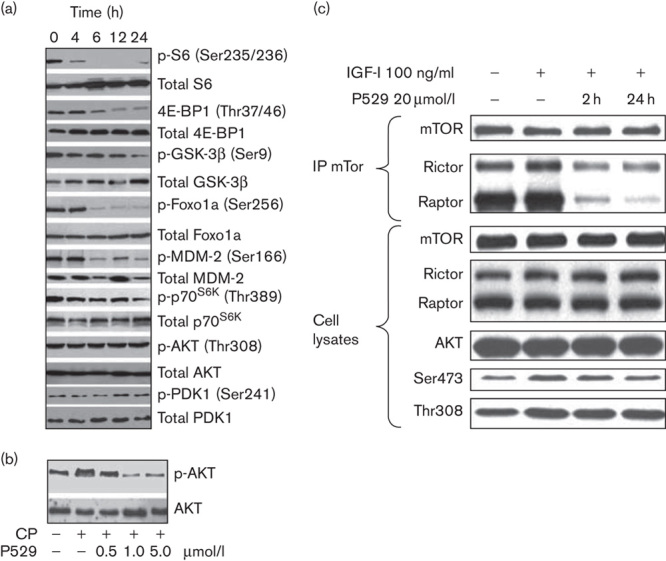
Inhibition of PI3K/AKT/mTOR pathway activity by RES-529 83,91. (a) Western blot analysis of PI3K/AKT/mTOR pathway signaling in PC-3 human prostate cells treated with 1 μmol/l RES-529 for the indicated time points. Adapted from Gravina et al. 91. Copyright © 2011, Society for Endocrinology. (b) Western blot analysis of AKT and p-AKT (Ser473) from extracts of PC-3 human prostate cells treated with 10 μg/ml CP and indicated concentrations of RES-529 (P529). Reproduced with permission from Gravina et al. 91. Copyright © 2011, Society for Endocrinology. (c) Immunoprecipitation of mTOR, followed by Rictor or Raptor western blot analysis to determine mTORC1 and mTORC2 complex formation following stimulation of C6V10 glioblastoma cells with IGF-1 and RES-529 (P529) treatment. Reproduced from Xue et al. with permission from AACR 83. CP, cisplatin; Foxo1a, forkhead box protein O1a; GSK-3β, glycogen synthase kinase-3β; IGF-1; insulin-like growth factor-1; MDM-2, mouse double minute 2 homolog; mTOR, mechanistic target of rapamycin; PDK1, 3-phosphoinositide-dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; Raptor, regulatory-associated protein of mTOR; Rictor, rapamycin-insensitive companion of mTOR.
Because the phosphorylation of GSK-3β on Ser9 is associated with GSK-3β inactivation 93, the effects of RES-529 treatment on GSK-3β activity and modulation of downstream markers were evaluated by Gravina et al. 94 in PC-3 and 22rv1 cells. RES-529 treatment significantly increased GSK-3β activity in cells (P<0.05). Active GSK-3β is known to promote apoptosis by the reduced expression and increased nuclear translocation of survivin 94,95. Furthermore, Gravina et al. 94 showed that radiation treatment acted synergistically with RES-529 to further increase GSK-3β activity and decrease survivin levels. Treatment of cells with the GSK-3β inhibitor SB216761 prevented the decrease in survivin levels with RES-529 and radiation, indicating that the activity of RES-529 on survivin levels was through a GSK-3β-dependent process.
RES-529 was also shown to regulate levels of components of the cell cycle, MAPK, and apoptotic pathways through PI3K/AKT/mTOR pathway inhibition 91. RES-529 treatment generally resulted in dramatically reduced levels of the cell cycle-associated proteins cyclin B1, cyclin D1, cyclin-dependent kinase 4 (cdk4), and cdk6 in PC-3 and 22rv1 cells along with a concomitant increase in p21 and p27 levels, on the basis of western blot analysis. An increase in ERK and p38 MAPK activity and reduced c-Jun N-terminal kinase (p-JNK) activity was also observed in these cells. Finally, changes in protein levels consistent with apoptosis were observed, with increases in Bcl-Xs and Bax [BCL-2 (B-cell lymphoma-2)-associated] protein levels and reduced amounts of Bcl-2, B-cell lymphoma extra long (Bcl-XL), and phosphorylated BCL-2-associated death promoter (BAD).
Insight into the mechanism by which RES-529 acts as a PI3K/AKT/mTOR pathway inhibitor emerged from the work of Xue et al. 83, who showed that treatment of C6V10 rat neuroblastoma cells with 20 μmol/l RES-529 promoted the dissociation of the mTORC1 and mTORC2 on the basis of immunoprecipitation experiments from lysates of treated cells that were stimulated with insulin-like growth factor-1 (Fig. 3c). Further studies are ongoing in various laboratories to further evaluate the mechanism of RES-529 action.
Antiangiogenic activity of RES-529
RES-529 has shown antiangiogenic potential in both cellular and animal models. RES-529 inhibited both VEGF-stimulated and β fibroblast growth factor-stimulated human umbilical vein endothelial (HUVEC) cell proliferation with half-maximal inhibitory concentrations (IC50) of ∼10 and 30 nmol/l, respectively 83. Treatment of HUVEC cells with RES-529 also resulted in a four-fold induction of apoptosis on the basis of DNA fragmentation 83.
The antiangiogenic activity of RES-529 was shown in two different mouse models 83. In a neonatal P7 mouse model in which pathologic retinal angiogenesis was induced by placing the mice into hyperoxia conditions, followed by normal conditions, RES-529 (RES-529 150 mg/kg/day, 5 days, intraperitoneal) inhibited pathologic angiogenesis, as indicated by a decrease in the number of glomeruloid tufts by ∼50% 83. In a model where angiogenesis was induced by VEGF-A produced by an injection of an adenovirus vector containing the VEGF-A gene into the mouse ear, treatment of mice with RES-529 (50−200 mg/kg/2 days, intraperitoneal) before the adenovirus injection resulted in a dose-dependent reduced number and size of blood vessels as well as decreased vessel permeability. At the higher dose of RES-529, angiogenesis and vascular permeability were reduced by ∼50% compared with the control on the basis of Evans blue staining. Furthermore, PI3K/AKT/mTOR pathway signaling was shown to be reduced in this model 83.
Antitumor activity of RES-529: cellular models
Growth inhibition was observed with RES-529 treatment in various cancer cell lines from the National Cancer Institute-60 (NCI-60) tumor panel, with IC50 ranges of 5–15 μmol/l for central nervous system cancer cells and 5–30 μmol/l for prostate cancer cells (Fig. 4) 96. Growth-inhibitory activity of RES-529 treatment was also observed in both PTEN-positive and PTEN-negative prostate cancer cell lines, although PTEN-negative cells appeared to be more sensitive 91.
Fig. 4.
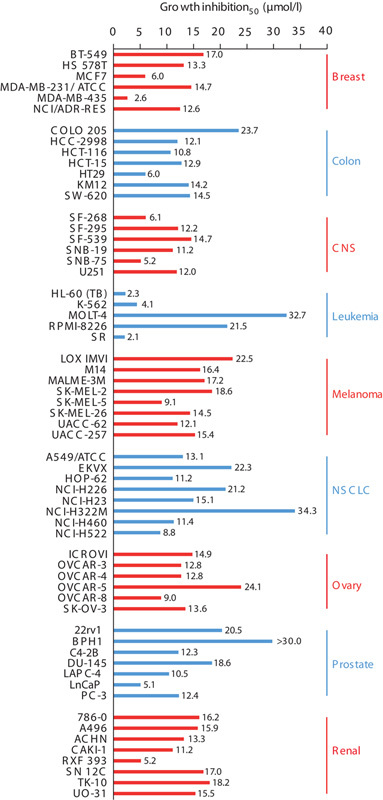
Cell growth inhibition of NCI-60 tumor cell line panel by RES-529 91,96. CNS, central nervous system; NSCLC, non-small-cell lung cancer. *Gravina et al. 91.
Growth inhibition was also accompanied by cell death. In the studies by Gravina et al. 91,94, RES-529 treatment of PC-3 and 22rv1 cells resulted in a notable induction of apoptosis, as determined by an increase in caspase-3 activity and annexin V staining.
Antitumor activity of RES-529: animal models
RES-529 has shown antitumor activity in a variety of mouse models, including those for glioblastoma 83, and prostate 91 and breast cancer 97. In a C6V10 glioblastoma subcutaneous xenograft model, mice pretreated with RES-529 (200 mg/kg/2 days, intraperitoneal) 1 week before and for 3 weeks after a tumor cell injection showed an ∼70% decrease in tumor volume compared with the control (Fig. 5) 83. In another glioblastoma tumor model using human U87 cells, mice treated with micronized RES-529 3 days after a tumor cell injection showed a reduction in tumor growth by ∼78 and 29% with 50 and 25 mg/kg/2 days, intraperitoneal, RES-529, respectively, after 24 days compared with the control 83. Furthermore, no noticeable toxicity was observed with this treatment 83. The antitumor activity in the U87 glioblastoma mouse xenograft model was confirmed in a recent report by Lin et al. 86, in which a significant decrease in tumor volume was observed in mice treated with 54 mg/kg, intraperitoneal, micronized RES-529 compared with the control (P<0.05) starting at 11 days of treatment to the end of the study (18 days), with tumor volume in the RES-529-treated mice being ∼52% lower than the control.
Fig. 5.
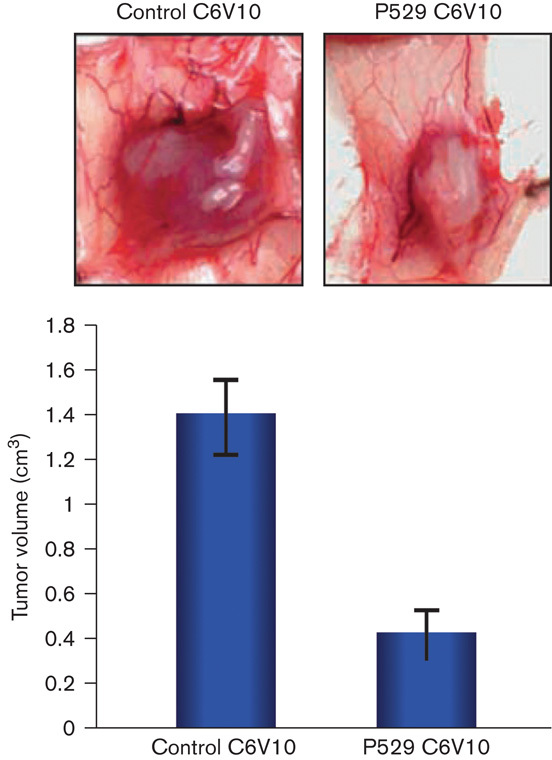
RES-529 activity in mouse glioblastoma xenograft models 83. Growth of C6V10 glioblastoma tumor in mice treated with RES-529 (P529, 200 mg/kg/2 days, intraperitoneal). Reproduced from Xue et al. with permission from AACR 83.
In PC-3 and 22rv1 mouse prostate xenograft models, a significant reduction in tumor mass was observed with RES-529 treatment (P<0.001; Fig. 6) 91. In the PC-3 xenograft model, a 10, 47.6, and 59.3% tumor volume reduction was observed with RES-529 50, 100, and 200 mg/kg, oral, treatment, respectively, with similar results found in the 22rv1 xenograft model. These reductions in tumor mass were accompanied by tumor cell apoptosis in both models. Furthermore, a potent effect on angiogenic neovascularization was observed in the PC-3 xenograft model, as shown by a marked decrease in the number, size, and stability of the blood vessel bed (as indicated by decreased staining for fibrin aggregates and CD31-positive microvessels) and a decrease in VEGF-positive tumor cells (Fig. 6).
Fig. 6.
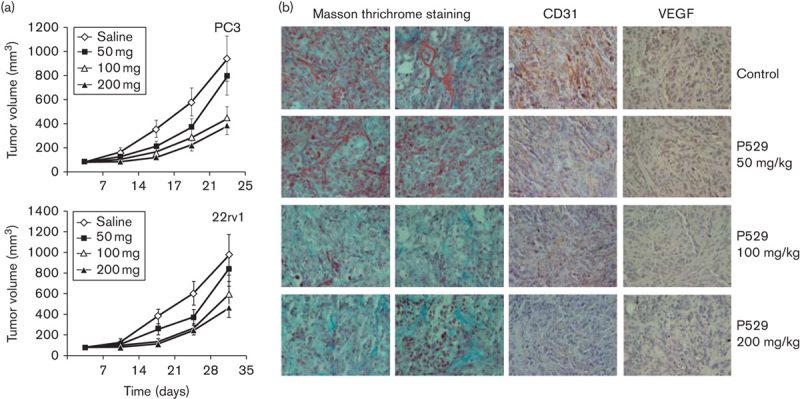
RES-529 activity in mouse prostate xenograft models 91. (a) Treatment of PC-3 or 22rv1 prostate mouse xenografts with RES-529 (P529) 50, 100, or 200 mg/kg/5 days/week with tumor volumes measured weekly. (b) Staining of PC-3 tumor tissue for fibrin deposits (Masson trichrome; orange/red stain), CD31-positive endothelial cells, and VEGF-positive tumor cells. Reproduced with permission from Gravina et al. 91. Copyright © 2011, Society for Endocrinology. VEGF, vascular endothelial growth factor.
RES-529 was also evaluated in breast cancer using two mouse xenograft models 97. One model utilized a human MCF-7 breast cancer cell line and the second used embryonic fibroblast cells from mice expressing a truncated Brca1 allele lacking the BRCT repeats (Brca1tr/tr) and with higher levels of p-AKT than wild-type mouse embryonic fibroblasts 97. RES-529 was shown to significantly inhibit tumor growth in both models (P<0.001) as well as decrease AKT and ribosomal S6 phosphorylation.
Synergistic activity of RES-529: cellular and animal models
Various anticancer therapies, including radiation therapy, chemotherapy, and hormonal therapy, have been shown to activate the PI3K/AKT/mTOR pathway 91,96. Therefore, studies have been performed in cell and animal models to determine whether inhibition of this pathway through treatment with RES-529 can have synergistic activity with these treatments.
The synergistic action of RES-529 with radiation treatment has been shown in a number of prostate cell and tumor models 94,96. In PC-3 cells, 2 μmol/l RES-529 along with 2 Gy radiation reduced cell survival by 70% compared with 15% for radiation alone (P<0.001) 96. A reduction in the clonogenic capacity of PC-3 cells was also shown to be greater with RES-529 when used in combination with 2 or 4 Gy radiation compared with radiation alone (P<0.05 and <0.01, respectively). This effect was at least partially mediated by the complete inhibition by RES-529 of the more than 10-fold radiation-induced phosphorylation of AKT. In addition, RES-529 treatment reduced the radiation-induced expression of inhibitor of differentiation-1 (id1), a molecule associated with radioresistance 98; VEGF; matrix metalloproteinase (MMP)9; and MMP2.
The synergistic activity of RES-529 with radiation in prostate cancer was further expanded in a recent paper by Gravina et al. 94. In this study, the decrease in the clonogenic capacity of prostate cancer cell lines LAPC-4, LnCaP, 22rv1, C4-2B, PC-3, and DU145 by radiation was further enhanced with 1 μmol/l RES-529. This was also accompanied by a significant enhancement of tumor autophagy compared with individual treatments (P<0.05), as measured by higher Beclin-1 protein expression, and increased apoptosis, on the basis of increased cleaved caspase-3 activity. In addition, there was an increase in tumor cell senescence, which was related to tumor autophagy, and a significant increase in the percentage of DNA damage (P<0.05) when RES-529 was combined with radiation treatment compared with radiation treatment alone. This increase in DNA damage was believed to be associated with negative effects on the homologous repair and nonhomologous end-joining DNA repair pathways through the reduced expression of Rad51, Ku70, and p-DNA–PKCs by RES-529 treatment. The increased efficacy in cell growth inhibition with this combination was considered to be associated with a combined inhibitory effect on c-Myc levels as well as the ability of RES-529 to inhibit the expression of radiation-induced cyclin D1.
The synergistic effects observed in prostate cell culture models with RES-529 and radiation therapy were also observed in animal models 94,96. RES-529 (20 mg/kg, q3d) and radiation (single 6 Gy dose 1 week after injection) treatment in a mouse PC-3 tumor model reduced tumor volume by 77% compared with the control, and treatment with the individual agents reduced growth by 43–53% after 4 weeks 96. In histological examination, tumors from mice treated with RES-529 and radiation showed more extensive tumor tissue damage compared with single therapy, including tumor cell loss, cells with pyknotic nuclei, and extensive fibrosis. Treatment with RES-529 and radiation also resulted in a significant reduction in proliferating cell nuclear antigen-positive cells, indicative of apoptosis, compared with the control (17.1±12.2 vs. 40.9±5.5%, P<0.01). This significant increase in apoptosis with proliferating cell nuclear antigen staining was correlated with caspase activity changes, with 8.7% caspase-3 positive cells present with the combination treatment compared with the ∼5.7 and 3.3% positive cells for the individual and control treatments (P<0.01 and <0.001, respectively).
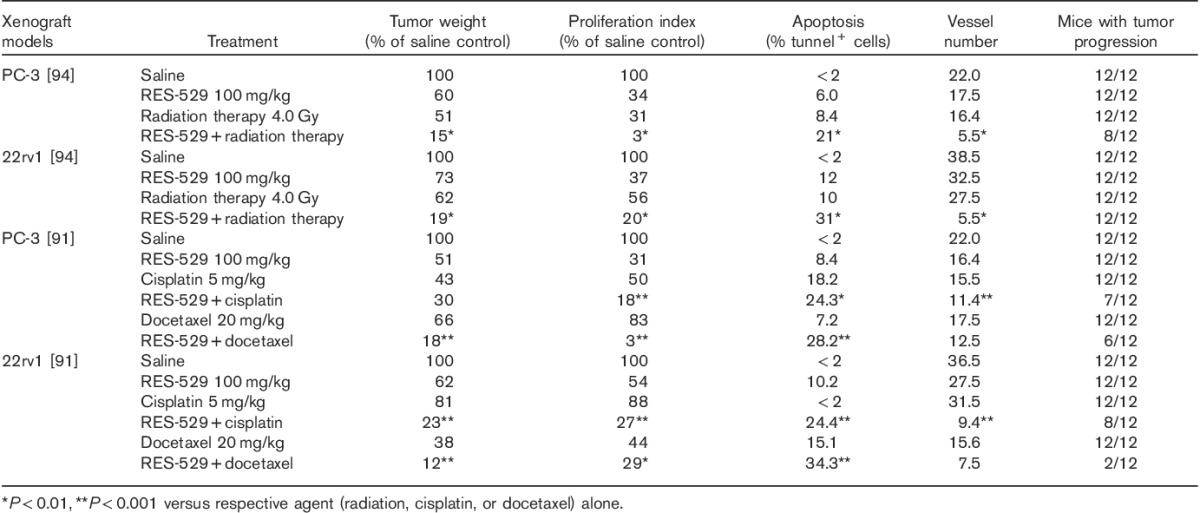
Similar results were observed in the study by Gravina et al. 94, where RES-529 enhanced the antitumor activity of radiation in mouse PC-3 and 22rv1 human prostate xenograft models (Table 1). A significant reduction in tumor volume was observed when RES-529 100 mg/kg, oral, 5 days/week, was combined with radiation (4 Gy) compared with the individual treatments alone (P<0.05). In addition, the number of mice with tumor progression was significantly fewer with combination therapy (P<0.05). A significant delay in the median time to progression was observed with RES-529 and radiation treatment compared with either treatment alone (P<0.001). This delay correlated with a significant decrease in the proliferation index and an increase in the number of apoptotic cells (P<0.01 for monotherapies vs. combination). In addition, in the PC-3 model, the mean absolute number of vessels in the tumor decreased significantly with the combination therapy (P<0.01 for monotherapies vs. combination).
Table 1.
Synergistic activity of RES-529 with radiation, cisplatin, or docetaxel in mouse prostate and glioblastoma xenograft models 91,94
Synergy of RES-529 treatment with radiation therapy was also observed in glioblastoma xenograft and intracranial orthotopic mouse models using the human cell line U251 (Fig. 7) 99. RES-529 plus radiation delayed the growth of xenografted U251 tumors compared with radiation alone (4 Gy) by 2.2 and 4 days with 25 and 50 mg/kg treatment, respectively. In addition, there was a significant increase in the survival of mice implanted intracranially with U251 cells with the combination of RES-529 50 mg/kg and radiation (4 Gy) treatment compared with RES-529 (P=0.016) or radiation (P=0.021) alone.
Fig. 7.
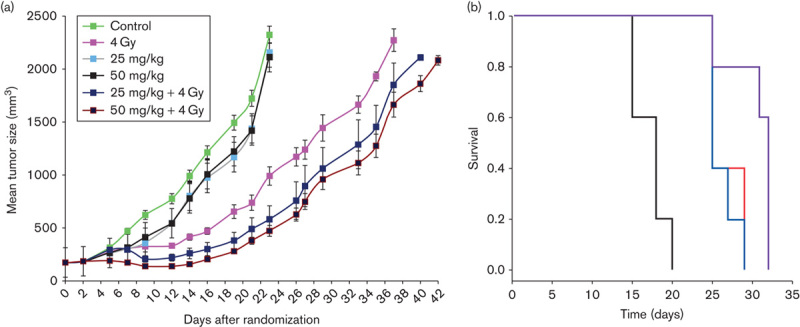
Synergistic activity of RES-529 with radiation in mouse glioblastoma xenograft model 99. (a) Effect of RES-529 treatment and radiation therapy on U251 tumor growth in a mouse xenograft model. (b) Kaplan–Meier curve of effect of radiation (4 Gy, blue), RES-529 (50 mg/kg, red), and RES-529, and radiation (purple) on survival versus control (black). Reproduced with permission from Cerna et al. 99.
In addition to radiation treatment, RES-529 was also shown to have synergistic activity with cisplatin and docetaxel in 22rv1 and PC-3 cellular and mouse xenograft models 91 and hormonal therapy in 22rv1 mouse xenograft models 82. Treatment of cells with RES-529 and either cisplatin or docetaxel, administered either in combination or sequentially, enhanced apoptosis compared with the individual administration of these agents. In mouse xenograft model studies where synergy was evaluated by calculating the combination index (CI) 100, RES-529 (100 mg/kg, oral) in combination with cisplatin (5 mg/kg, intraperitoneal) was synergistic in 22rv1 xenografts (CI=0.69) and additive for PC-3 xenografts (CI=1.13). RES-529 (100 mg/kg, oral) in combination with docetaxel (20 mg/kg, intraperitoneal) was synergistic in both 22rv1 and PC-3 xenograft models (CI=0.50 and 0.34, respectively). This synergistic/additive effect was also observed when evaluated by the log cell kill. Furthermore, combination therapy significantly increased the time to progression compared with individual therapies (P<0.0001). The combination of RES-529 with either cisplatin or docetaxel decreased the number of mice with tumors in progression from 100% with the individual treatments to 67 and 17%, respectively, in the 22rv1 xenograph model and 58 and 50%, respectively, in the PC-3 tumor xenograft model (Table 1). Recently, Gravina et al. 82 reported synergy of RES-529 with the 5α-reductase inhibitor dutasteride, the androgen synthesis inhibitor abiraterone, and the androgen receptor inhibitor bicalutamide in mouse 22rv1 xenograft models.
Basis for clinical evaluation of RES-529 in glioblastoma
For the clinical development of RES-529 in oncology, the initial focus is to target relevant tumors for which there is a high unmet medical need, such as glioblastoma. The current median survival of patients with glioblastoma is 9.7 months, with the existing treatment options limited to surgery, radiotherapy, and chemotherapy, such as temozolomide 101,102.
The potential of treating glioblastoma with inhibitors of the PI3K/AKT/mTOR pathway has been shown through the identification of pathway-activating mutations in patients with glioblastoma and activity of pathway inhibitors in preclinical glioblastoma models, such as those presented for RES-529. Activating mutations in the PI3K/AKT/mTOR pathway are found in a majority of patients with glioblastoma 45,51,103. In an analysis of 206 glioblastomas, 86% of the samples had at least one genetic event in the receptor tyrosine kinase/PI3K pathway 45. In addition, mutations and deletions of PTEN or mutations/amplification of epidermal growth factor receptor, both of which are frequent in glioblastoma, lead to the dysregulation of the PI3K pathway 104–106.
In addition to the studies described above for RES-529, other mTOR inhibitors that target mTORC1 and mTORC2 have shown efficacy in mouse glioblastoma xenograft models 107,108. In mouse orthotopic xenograft models using the glioblastoma cell line CD133+ GBMJ1, treatment with AZD2014, an ATP-competitive mTOR inhibitor that targets mTORC1 and mTORC2, along with radiation resulted in a significant increase in survival compared with either control or radiation alone (P=0.014 and 0.03, respectively) 107. AZD8055, another mTOR inhibitor targeting mTORC1 and mTORC2, inhibited tumor growth in subcutaneous human brain tumor-initiating cell mouse xenografts and mTORC1 and mTORC2 signaling 108. Although no detailed safety results have been reported for these mouse glioblastoma xenograft studies, Xue et al. 83 observed no toxicity with RES-529.
Currently, at least 17 PI3K/AKT/mTOR pathway inhibitors are being evaluated in clinical trials for glioblastoma. However, other than RES-529, none are believed to work through the dissociation of both mTOR complexes (mTORC1 and mTORC2). Furthermore, inhibitors that target only mTORC1 have shown poor efficacy in clinical trials 73–75.
Conclusion
PI3K/AKT/mTOR inhibitors have the potential to treat various tumor types, including glioblastoma and prostate and breast cancer. On the basis of past experience with mTORC1 inhibitors, there is a need for a dual mTORC1 and mTORC2 inhibitor using various approaches, including compounds that promote complex dissociation or ATP-competitive inhibition. As a dual mTORC1 and mTORC2 inhibitor, RES-529 potentially has a number of advantages over mTORC1-specific inhibitors. mTORC1-specific inhibitors have shown efficacy that is essentially limited to renal cell carcinoma and pancreatic neuroendocrine tumors, whereas RES-529 has shown efficacy in animal tumor models for glioblastoma and prostate cancer, two cancers resistant to mTORC1 inhibitors 71–75. Anticancer therapies, including radiation therapy, chemotherapy, and hormonal therapy, activate the PI3K/AKT/mTOR pathway and, in particular, mTORC2, as noted by increased AKT (Ser473) phosphorylation 91,96. RES-529 inhibits anticancer therapy-induced AKT activation and acts synergistically with these agents in animal tumor models. One mechanism of resistance to mTORC1 inhibitors occurs through the upregulation of receptor tyrosine kinase signaling adapter proteins, leading to AKT activation through Ser473 phosphorylation 76, which could be circumvented by inhibiting mTORC2. However, another mechanism of resistance to mTORC1 inhibitors through MAPK pathway activation is not prevented by genetic inactivation of mTORC2 79. Thus, there is the potential of resistance to RES-529 through MAPK pathway activation. MAPK pathway activation has been observed previously in PC-3 and 22rv1 cells treated with RES-529 91. A potential disadvantage of RES-529 compared with mTORC1-specific inhibitors is the potential for increased adverse events because of inhibition of both mTORC1 and mTORC2 activity. Therefore, it will be important to determine the clinical efficacy of RES-529 in relevant cancers such as glioblastoma and prostate cancer, and how it compares with other mTOR inhibitors, both mTORC1-specific and ATP-competitive mTORC1–mTORC2 inhibitors, with respect to safety and potency.
Acknowledgements
Medical writing assistance was provided by Alan Saltzman, PhD (Fishawack Communications, Horsham, Pennsylvania, USA), and funded by RestorGenex. Acknowledgement is given to David Sherris, PhD, former chief scientific officer of RestorGenex and former chief executive officer of Paloma Pharmaceuticals, for his activity in facilitating many of the experiments conducted with RES-529. RES-529 was developed by RestorGenex Corporation. As of January 2016, RestorGenex Corporation merged with Diffusion Pharmaceuticals LLC to form Diffusion Pharmaceuticals Inc.
Conflicts of interest
M.W. was an employee of RestorGenex Corporation during the development of the manuscript.
Footnotes
As of January 2016, RestorGenex Corporation merged with Diffusion Pharmaceuticals LLC to form Diffusion Pharmaceuticals Inc.
References
- 1.Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 2015; 25:545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci 2015; 40:88–100. [DOI] [PubMed] [Google Scholar]
- 3.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014; 13:140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015; 15:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene 2008; 27:5486–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 2009; 27:2278–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010; 28:1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duzgun Z, Eroglu Z, Biray Avci C. Role of mTOR in glioblastoma. Gene 2016; 575 (Pt 1):187–190. [DOI] [PubMed] [Google Scholar]
- 9.Xia P, Xu XY. PI3K/Akt/mTOR signaling pathway in cancer stem cells: from basic research to clinical application. Am J Cancer Res 2015; 5:1602–1609. [PMC free article] [PubMed] [Google Scholar]
- 10.Skolnik EY, Margolis B, Mohammadi M, Lowenstein E, Fischer R, Drepps A, et al. Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell 1991; 65:83–90. [DOI] [PubMed] [Google Scholar]
- 11.Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT. A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein beta gamma subunits. Cell 1994; 77:83–93. [DOI] [PubMed] [Google Scholar]
- 12.Rordorf-Nikolic T, Van Horn DJ, Chen D, White MF, Backer JM. Regulation of phosphatidylinositol 3′-kinase by tyrosyl phosphoproteins. Full activation requires occupancy of both SH2 domains in the 85-kDa regulatory subunit. J Biol Chem 1995; 270:3662–3666. [DOI] [PubMed] [Google Scholar]
- 13.Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci USA 1998; 95:13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 1998; 273:13375–13378. [DOI] [PubMed] [Google Scholar]
- 15.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998; 95:29–39. [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Renuse S, Sahasrabuddhe NA, Zahari MS, Chaerkady R, Kim MS, et al. Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nat Commun 2014; 5:4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahara T, Maeda T. Evolutionarily conserved regulation of TOR signalling. J Biochem 2013; 154:1–10. [DOI] [PubMed] [Google Scholar]
- 19.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 2002; 10:457–468. [DOI] [PubMed] [Google Scholar]
- 20.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22:159–168. [DOI] [PubMed] [Google Scholar]
- 21.Sato T, Nakashima A, Guo L, Tamanoi F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J Biol Chem 2009; 284:12783–12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009; 10:307–318. [DOI] [PubMed] [Google Scholar]
- 23.Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 2012; 18:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012; 150:1196–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015; 347:188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rebsamen M, Pochini L, Stasyk T, de Araújo ME, Galluccio M, Kandasamy RK, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015; 519:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011; 334:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michel G, Matthes HW, Hachet-Haas M, El Baghdadi K, de Mey J, Pepperkok R, et al. Plasma membrane translocation of REDD1 governed by GPCRs contributes to mTORC1 activation. J Cell Sci 2014; 127 (Pt 4):773–787. [DOI] [PubMed] [Google Scholar]
- 29.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 2008; 22:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shackelford DB, Shaw RJ. The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9:563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J, Topisirovic I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015; 14:473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol 2009; 19:R1046–R1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle 2011; 10:2305–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, et al. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov 2015; 5:1194–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006; 127:125–137. [DOI] [PubMed] [Google Scholar]
- 37.Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 2012; 15:725–738. [DOI] [PubMed] [Google Scholar]
- 38.Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 2013; 110:12526–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008; 453:662–666. [DOI] [PubMed] [Google Scholar]
- 40.Trinh XB, Tjalma WA, Vermeulen PB, Van den Eynden G, Van der Auwera I, Van Laere SJ, et al. The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br J Cancer 2009; 100:971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 2000; 60:1541–1545. [PubMed] [Google Scholar]
- 42.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 2001; 21:3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pore N, Jiang Z, Shu HK, Bernhard E, Kao GD, Maity A. Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol Cancer Res 2006; 4:471–479. [DOI] [PubMed] [Google Scholar]
- 44.Maity A, Pore N, Lee J, Solomon D, O’Rourke DM. Epidermal growth factor receptor transcriptionally up-regulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3’-kinase and distinct from that induced by hypoxia. Cancer Res 2000; 60:5879–5886. [PubMed] [Google Scholar]
- 45.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010; 18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 2008; 68:6084–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell 2013; 155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304:554. [DOI] [PubMed] [Google Scholar]
- 53.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci USA 2005; 102:18443–18448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun X, Huang J, Homma T, Kita D, Klocker H, Schafer G, et al. Genetic alterations in the PI3K pathway in prostate cancer. Anticancer Res 2009; 29:1739–1743. [PubMed] [Google Scholar]
- 55.Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 2004; 64:7678–7681. [DOI] [PubMed] [Google Scholar]
- 56.Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res 2001; 61:7426–7429. [PubMed] [Google Scholar]
- 57.Mizoguchi M, Nutt CL, Mohapatra G, Louis DN. Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol 2004; 14:372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275:1943–1947. [DOI] [PubMed] [Google Scholar]
- 59.Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res 2002; 8:1178–1184. [PubMed] [Google Scholar]
- 60.Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res 2004; 64:3014–3021. [DOI] [PubMed] [Google Scholar]
- 61.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012; 13:283–296. [DOI] [PubMed] [Google Scholar]
- 62.Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer 2008; 99:1265–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim MS, Jeong EG, Yoo NJ, Lee SH. Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br J Cancer 2008; 98:1533–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, et al. Colorectal cancer: mutations in a signalling pathway. Nature 2005; 436:792. [DOI] [PubMed] [Google Scholar]
- 65.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, et al. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA 1992; 89:9267–9271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 2014; 4:554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knowles MA, Habuchi T, Kennedy W, Cuthbert-Heavens D. Mutation spectrum of the 9q34 tuberous sclerosis gene TSC1 in transitional cell carcinoma of the bladder. Cancer Res 2003; 63:7652–7656. [PubMed] [Google Scholar]
- 68.Martignoni G, Pea M, Reghellin D, Zamboni G, Bonetti F. PEComas: the past, the present and the future. Virchows Arch 2008; 452:119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Novartis Pharmaceuticals Corporation. Afinitor® prescribing information. Available at: https://www.pharma.us.novartis.com/product/pi/pdf/afinitor.pdf. [Accessed 13 October 2015].
- 70.Wyeth Pharmaceuticals Inc. Torisel® prescribing information. Available at: http://www.pfizerpro.com/content/showlabeling.asp?id=490. [Accessed 13 October 2015].
- 71.Armstrong AJ, Shen T, Halabi S, Kemeny G, Bitting RL, Kartcheske P, et al. A phase II trial of temsirolimus in men with castration-resistant metastatic prostate cancer. Clin Genitourin Cancer 2013; 11:397–406. [DOI] [PubMed] [Google Scholar]
- 72.Nakabayashi M, Werner L, Courtney KD, Buckle G, Oh WK, Bubley GJ, et al. Phase II trial of RAD001 and bicalutamide for castration-resistant prostate cancer. BJU Int 2012; 110:1729–1735. [DOI] [PubMed] [Google Scholar]
- 73.Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, II, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol 2010; 96:219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma DJ, Galanis E, Anderson SK, Schiff D, Kaufmann TJ, Peller PJ, et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol 2015; 17:1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol 2014; 16:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science 2005; 307:1098–1101. [DOI] [PubMed] [Google Scholar]
- 78.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65:7052–7058. [DOI] [PubMed] [Google Scholar]
- 79.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118:3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang X, Hawk N, Yue P, Kauh J, Ramalingam SS, Fu H, et al. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors’ anticancer efficacy. Cancer Biol Ther 2008; 7:1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X, Sun SY. Enhancing mTOR-targeted cancer therapy. Expert Opin Ther Targets 2009; 13:1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gravina GL, Marampon F, Colapietro A, Scarsella L, Jitariuc A, Sanità P, et al. RES-529, a dual TORC1/TORC2 inhibitor, potentiates the in vitro and in vivo efficacy of hormone manipulations in the 22rv1 prostate cancer cell model. In Proceedings of the 25th Annual Meeting of the Italian Society of Uro-oncology (SIUrO); 2015 June 21–23; Rome, Italy [abstract #3644]. Anticancer Res 2015; 35:3638–3639. [Google Scholar]
- 83.Xue Q, Hopkins B, Perruzzi C, Udayakumar D, Sherris D, Benjamin LE. Palomid 529, a novel small-molecule drug, is a TORC1/TORC2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer Res 2008; 68:9551–9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001; 6:469–477. [DOI] [PubMed] [Google Scholar]
- 85.Bogush T, Dudko E, Bogush E, Polotsky B, Tjulandin S, Davydov M. Tamoxifen non-estrogen receptor mediated molecular targets. Oncol Rev 2012; 6:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin F, Buil L, Sherris D, Beijnen JH, van Tellingen O. Dual mTORC1 and mTORC2 inhibitor Palomid 529 penetrates the blood-brain barrier without restriction by ABCB1 and ABCG2. Int J Cancer 2013; 133:1222–1233. [DOI] [PubMed] [Google Scholar]
- 87.Dalal M, Jacobs-El N, Nicholson B, Tuo J, Chew E, Chan CC, et al. Subconjunctival Palomid 529 in the treatment of neovascular age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol 2013; 251:2705–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Clinical trials.gov. Phase I study of Palomid 529, a dual Torc 1/2 inhibitor of the PI3K/Akt/mTOR pathway for advanced age-related macular degeneration (P52901). Available at: https://www.clinicaltrials.gov/ct2/show/NCT01033721?term=palomid&rank=2. [Accessed 13 October 2015].
- 89.Clinical trials.gov. Palomid 529 in patients with neovascular age-related macular degeneration. Available at: https://www.clinicaltrials.gov/ct2/show/NCT01271270?term=palomid&rank=1. [Accessed 13 October 2015].
- 90.RestorGenex Press Release 28 January 2015. RestorGenex granted orphan drug designation for RES-529 for treatment of glioblastoma multiforme. Available at: http://globenewswire.com/news-release/2015/01/28/700603/10117109/en/RestorGenex-Granted-Orphan-Drug-Designation-for-RES-529-for-Treatment-of-Glioblastoma-Multiforme.html. [Accessed 13 October 2015].
- 91.Gravina GL, Marampon F, Petini F, Biordi L, Sherris D, Jannini EA, et al. The TORC1/TORC2 inhibitor, Palomid 529, reduces tumor growth and sensitizes to docetaxel and cisplatin in aggressive and hormone-refractory prostate cancer cells. Endocr Relat Cancer 2011; 18:385–400. [DOI] [PubMed] [Google Scholar]
- 92.Syed F, Sherris D, Paus R, Varmeh S, Singh S, Pandolfi PP, Bayat A. Keloid disease can be inhibited by antagonizing excessive mTOR signaling with a novel dual TORC1/2 inhibitor. Am J Pathol 2012; 181:1642–1658. [DOI] [PubMed] [Google Scholar]
- 93.Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev 2001; 101:2527–2540. [DOI] [PubMed] [Google Scholar]
- 94.Gravina GL, Marampon F, Sherris D, Vittorini F, Di Cesare E, Tombolini V, et al. Torc1/Torc2 inhibitor, Palomid 529, enhances radiation response modulating CRM1-mediated survivin function and delaying DNA repair in prostate cancer models. Prostate 2014; 74:852–868. [DOI] [PubMed] [Google Scholar]
- 95.Li J, Xing M, Zhu M, Wang X, Wang M, Zhou S, et al. Glycogen synthase kinase 3beta induces apoptosis in cancer cells through increase of survivin nuclear localization. Cancer Lett 2008; 272:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Diaz R, Nguewa PA, Diaz-Gonzalez JA, Hamel E, Gonzalez-Moreno O, Catena R, et al. The novel Akt inhibitor Palomid 529 (P529) enhances the effect of radiotherapy in prostate cancer. Br J Cancer 2009; 100:932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xiang T, Jia Y, Sherris D, Li S, Wang H, Lu D, Yang Q. Targeting the Akt/mTOR pathway in Brca1-deficient cancers. Oncogene 2011; 30:2443–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang H, Rosdahl I. Ultraviolet A and B differently induce intracellular protein expression in human skin melanocytes – a speculation of separate pathways in initiation of melanoma. Carcinogenesis 2003; 24:1929–1934. [DOI] [PubMed] [Google Scholar]
- 99.Cerna D, Carter D, Flaherty S, Cao L, Sherris D, Yoo S. Palomid 529, a PI3K/Akt/mTOR dual TORC1/2 inhibitor, is a radiation enhancer with effect in both subcutaneous and orthotopic U251 glioblastoma tumor xenograph models. In Proceedings of the 101st Annual Meeting of the American Association for Cancer Research; 2010 April 17–21; Washington DC. Philadelphia (PA): AACR [abstract]. Cancer Res 2010; 70:2506. [Google Scholar]
- 100.Bruzzese F, Di Gennaro E, Avallone A, Pepe S, Arra C, Caraglia M, et al. Synergistic antitumor activity of epidermal growth factor receptor tyrosine kinase inhibitor gefitinib and IFN-alpha in head and neck cancer cells in vitro and in vivo. Clin Cancer Res 2006; 12:617–625. [DOI] [PubMed] [Google Scholar]
- 101.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol 2012; 107:359–364. [DOI] [PubMed] [Google Scholar]
- 102.National Comprehensive Cancer Network (NCCN). Clinical practice guidelines in oncology (NCCN Guidelines®) central nervous system cancers, version 1; 2015. Available at: http://www.nccn.org/professionals/physician_gls/pdf/cns.pdf. [Accessed 13 October 2015].
- 103.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, et al. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res 2003; 63:2742–2746. [PubMed] [Google Scholar]
- 105.Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, Parsons R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res 1997; 57:4183–4186. [PubMed] [Google Scholar]
- 106.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 2000; 60:1383–1387. [PubMed] [Google Scholar]
- 107.Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K, Tofilon PJ. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol 2014; 16:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luchman HA, Stechishin OD, Nguyen SA, Lun XQ, Cairncross JG, Weiss S. Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival. Clin Cancer Res 2014; 20:5756–5767. [DOI] [PubMed] [Google Scholar]