

Abstract
Knowledge of the transfer free energy of amino acids from aqueous solution to a lipid bilayer is essential for understanding membrane protein folding and for predicting membrane protein structure. Here we report a computational approach that can calculate the folding free energy of the transmembrane region of outer membrane β-barrel proteins (OMPs) by combining an empirical energy function with a reduced discrete state space model. We quantitatively analyzed the transfer free energies of 20 amino acid residues at the center of the lipid bilayer of OmpLA. Our results are in excellent agreement with the experimentally derived hydrophobicity scales. We further exhaustively calculated the transfer free energies of 20 amino acids at all positions in the TM region of OmpLA. We found that the asymmetry of the gram-negative bacterial outer membrane as well as the TM residues of an OMP determine its functional fold in vivo. Our results suggest that the folding process of an OMP is driven by the lipid-facing residues in its hydrophobic core, and its NC-IN topology is determined by the differential stabilities of OMPs in the asymmetrical outer membrane. The folding free energy is further reduced by lipid A and assisted by general depth-dependent cooperativities that exist between polar and ionizable residues. Moreover, context-dependency of transfer free energies at specific positions in OmpLA predict regions important for protein function as well as structural anomalies. Our computational approach is fast, effcient and applicable to any OMP.
Graphical TOC Entry

Introduction
Membrane proteins account for about 30% of all proteins in a typical genome1 and serve a multitude of essential cellular functions such as immune response, metabolite transport and energy transduction.2 Among the two major classes of membrane proteins, α-helical membrane proteins are predominantly located in the plasma membrane of eukaryotic cells, the inner membranes of eukaryotic organelles and prokaryotes. In contrast, β-barrel membrane proteins, or outer membrane proteins (OMPs), are found in the outer membranes of gram-negative bacteria, mitochondria, and chloroplasts. Both types of membrane proteins are involved in several life-threatening diseases either through altered function or dysfunction.3 Understanding the principles governing membrane protein folding and stability, as well as identifying their functional form is therefore of fundamental importance.
Despite significant differences in their biogenesis,4 the insertion of α-helical and β-strand TM segments is dictated by its partition free energy from the aqueous environment into the membrane.5,6 A central question in membrane protein biology is therefore the assessment of the transfer free energy of amino acids from solution into a lipid bilayer. The transfer free energies of 20 amino acid residues, often called hydrophobicity scales, have been determined in several experimental systems5–8 and have generate considerable insight. However, experimental measurement of transfer free energy is technically challenging, as identifying conditions of reversible folding can be time-consuming.7,8 Conditions at which reversible folding were observed have been reported only for three OMPs.9–13 It is therefore important to develop methods that allow fast generation of effective transfer free energies applicable to membrane proteins in general.
Computational studies can complement experimental methods and expand our knowledge of the governing principles of membrane protein folding.14,15 Knowledge-based hydrophobicity scales have been derived in several studies.16–18 The EZα and EZβ empirical potentials can position and orient TM segments, discriminate side-chain decoys, and identify protein-lipid interfaces.17,18 However, these scales do not consider physical interactions between residues either from neighboring helices/strands or from the same helix/strand. Such interactions are important for membrane protein insertion and folding.19,20 Detailed molecular interactions in membrane proteins can be investigated using molecular dynamics (MD) simulations.21–23 However, the choice of the reference unfolded state remains a challenging problem as reproducing transfer free energies of different amino acids require different reference states.22 In this study, we describe a computational method that incorporates energies of depth-dependent membrane burial, intra- and inter-strand interactions, native as well as exhaustively enumerated non-native conformations with different strand registrations, which allows calculation of the transfer free energy of the TM section of OMPs. Inspired by the Moon-Fleming “whole-protein-scale”,12 we used OmpLA as a client OMP to derive the transfer free energy of 20 amino acid residues at the center of the bilayer. Our results are in excellent agreement with the experimentally derived scales. We have further expanded our studies to include residues at other depths in the bilayer to address key questions in OMP folding. Our findings suggest that the asymmetric nature of the gram-negative bacterial outer membrane as well as the specific amino acid composition in the TM region of an OMP are critical for adopting a functional form in the cell. Our results indicate that lipid-facing residues in the core region provide the driving force for OMP folding. Our method represents a new approach to evaluate the effects of single or multiple residue substitutions on the stability, structure and function of OMPs.
Results
Our goal was to develop a method for high-throughput computation of the transfer free energies of amino acid residues, accounting for key physical interactions in the TM region, positions of residues in the bilayer, and the type of OMP. Our approach is based on the TMSIP empirical energies of burial and inter-strand interactions developed from detailed combinatorial analysis of OMP structures,24 with further improvement incorporating intra-strand side-chain interactions. The configuration space of OMPs is represented by a discrete state model,25 in which each strand move up or down several positions from its native position. We enumerate all possible configurations in this reduced discrete state space for the TM β-strands of an OMP, with the probability of a specific configuration following the Boltzmann distribution. We calculated transfer free energies in the context of OMPs from the ensemble of enumerated conformations of the TM β-strands.
A general computational transfer free energy scale of OmpLA
We first examined if our computational method can reproduce the experimentally derived transfer free energy scales. Inspired by the work of Moon and Fleming,12 we determined the transfer free energies of 20 amino acid residues by substituting the host residue A210 in OmpLA with the remaining 19 amino acid residues. Folding free energy of OmpLA with specific amino acid substitution was calculated and the difference between the wild type Ala and the substitution was assigned as the transfer free energy at the host residue A210. Importantly, key physical interactions in the TM region of OmpLA were taken into account in the calculations (Fig S1). While OmpLA has been reported to form a dimer for enzymatic activity,26,27 the monomeric form of OmpLA in the outer membrane under normal condition26 is used in deriving the transfer free energy scale. This computational transfer free energy scale, termed ΔΔGA210, correlates well with the experimentally determined transfer free energy scales (R2 = 0.81 for all 20 amino acids) with the Moon-Fleming scale12 (Fig 1a). Compared to Ala, Pro is more hydrophilic in our scale but more hydrophobic in the Moon-Fleming scale. Once Pro is excluded, the correlation between the two scales increases to 0.91. Furthermore, our scale also agrees well with Hessa et al’s biological scale (R2 = 0.86 for all amino acids, R2 = 0.88 after excluding Pro), and with the Wimley-White octanol scale (R2 = 0.67 for all amino acids, R2 = 0.68 after excluding Pro). This general agreement indicates that our reduced state model and the empirical energy function can capture key determinants of contribution of the amino acids to the stability of OMPs.
Fig 1.

Transfer free energy ΔΔGA210 and general position specific transfer free energy scales ΔΔG(i) were derived using host residues in OmpLA. (a) ΔΔGA210 (blue bar) calculated at host residue A210 of OmpLA was compared with the experimentally measured Moon-Fleming whole-protein scale12 (red bar). ΔΔGA210 correlates well with the whole protein scale 12 ( R2 = 0.91 excluding Pro), the biological scale of Hessa et al28 (R2 = 0.88 excluding Pro), and the Wimley-White octanol scale ΔΔGOctanol5 (R2 = 0.68 excluding Pro). (b)ΔΔG(i) are derived for all 9 positions in the TM region. For position i, ΔΔG derived from multiple host residues at this same depth-position are averaged to obtain ΔΔG(i). Host residues with strong context dependency in their transfer free energy scales were excluded from calculation.
We then derived exhaustively transfer free energy scales using every lipid-facing TM residue as the host residue in OmpLA (52 hosts in addition to A210) (Fig S2 and Table S1). Overall, the transfer free energy values are very similar for residues at the same depth positions in the bilayer of the bacterial outer membrane, and a general transfer free energy scale emerges that is applicable to 41 out of the 53 TM residues. This general transfer free energy scale ΔΔG(i) is then calculated by averaging the scales derived from host residues among the 41 residues that are at the same bilayer depth i (Fig 1b).
Strong context dependency of transfer free energies allows identification of residues important for enzymatic function of OmpLA
Host residues whose transfer free energy scales deviate from the general scale were detected by analyzing the correlation coeffcient between the transfer free energy scales derived from host residues at the same depth positions (Table S2). If the average correlation coeffcient increases significantly (> 10%) upon excluding the scale of a host residue, this scale strongly deviates from the rest. Overall, we found that scales from 12 host residues, D36, N38, L40, Y42, Q94, H142, N156, P175, V235, L237, V241, and L265, strongly deviated from the remaining 41 host residues (Fig 2a-b). Six of them (D36, N38, L40, P175, L237, and L265) are located in the hydrophobic core region, while the other six are located in the headgroup region.
Fig 2.
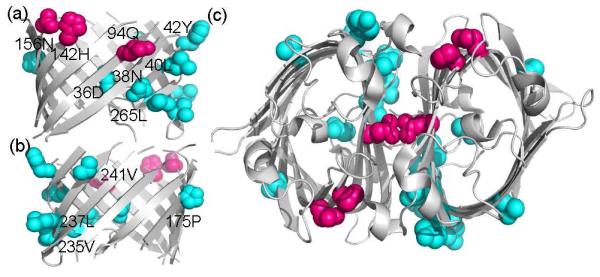
Strong context dependency of transfer free energies detects amino acid residues involved in OmpLA function. Side (a and b) view on and top (c) view on OmpLA dimer. Residues with strong context dependency are either located in a structurally deformed environment (cyan) or are functionally important residues (pink). These residues, except P175, V237, and V241, are located in the dimerization interface of OmpLA.
Except Q94, H142, and N156, all are located in a structurally deformed environment. Residues Q94, H142, and N156 are functionally important residues. Active-site residues H142 and N156 are part of the catalytic triad of OmpLA.29 Q94, V235, and L265 provide intermolecular interactions for OmpLA dimerization (Fig S4), which is required for enzymatic function.26 Overall, most of these residues are found in the dimerization interface of OmpLA (Fig 2c). The significant deviation from the general transfer free energy scale implies that our computational hydrophobicity scale can be context dependent for certain host residues. We further suggest that such context dependency can be utilized to detect either possible structural anomaly or assign functional residues in OMPs of unknown tertiary structure.
The inner leaflet of the bacterial outer membrane imposes an energetic barrier to insertion of polar and ionizable amino acid residues
Experimental studies have shown that the energetic cost of transferring amino acids into a lipid bilayer is affected by the composition of the local membrane environment to which they are transferred as well as their depth in the bilayer.6 As the gram-negative outer membrane is strongly heterogeneous in its composition, with the outer leaflet consisting solely of lipopolysaccharide (LPS) and the inner leaflet comprised of phospholipids,30 an important question is how this asymmetric environment affects OMP folding.
We first examined the depth-dependent profiles of Leu and Arg and compared them to the experiment values according to ref.12 Here we assigned an index i to every host residue, with i = 0 corresponding to the center of the bilayer. Hosts located in the outer leaflet have i > 0, and hosts located in the inner leaflet have i < 0. Fig 3a demonstrates that the energetic cost to transfer Leu into any depth of the asymmetric bacterial OM is comparable to the symmetric DLPC bilayer used by Moon and Fleming.12 In contrast, transfer of Arg into the outer leaflet (i.e., the LPS containing leaflet) is much more favorable than insertion into the inner leaflet (i.e., the phospholipid containing leaflet), as well as a DLPC leaflet. For example, ΔΔG of Arg at position i = 2 is 2.38 kcal/mol, which correlates well with the experimentally measured value of 2.35 kcal/mol. However, Arg at position i = 3 or i = 4 is much more favorable in an LPS than DLPC environment (0.19 kcal/mol in LPS vs 2.07 kcal/mol in DLPC at i = 3; 0.08 kcal/mol in LPS vs 0.61 kcal/mol in DLPC at i = 4). Our results therefore capture the effect of the asymmetric composition of the bacterial OM on OMP folding.
Fig 3.

Depth-dependent profiles of amino acid transfer free energies exhibit asymmetric distribution in the bacterial OM. (a) ΔΔGs of Leu (red closed circle) calculated using six host residues (L120, L164, A210, G212, Y214, and A223) of OmpLA are similar to measured values (open circle).12 However, ΔΔGs of Arg (blue closed circle) are lower than measured values at outer leaflet (i > 0) of the bacterial OM, suggesting LPS favors insertion of Arg. (b) ΔΔGaa(i) is plotted against the position index i for each individual residue type (cyan dots). Ionizable residues Arg and Lys, polar residues Gin, Asn, Ser, and Thr, aromatic residues Tyr and Trp have lower ΔΔGaa(i) in the outer leaflet (i > 0) compared to that in the inner leaflet (i < 0). Blue lines are fitted single or double asymmetric Gaussian curves. i is set to 0 at the bilayer center. It increases towards outer leaflet headgroup, and decreases towards the inner leaflet headgroup. Details of ΔΔGaa(i) can be found in Table S3.
We then evaluated the effect of residue depth on the transfer free energy of all 20 amino acid residues. From the position specific general transfer free energy scale ΔΔG(i), we can directly obtain the depth-dependent profiles of all 20 residues ΔΔGaa(i) using an asymmetric Gaussian function (Fig 3b). As expected, aliphatic residues L, I, V, M, and aromatic residues F, Y, W exhibit favorable transfer free energies at all bilayer depths, whereas ionizable and polar residues R, K, H, D, E, N, Q, S, T, as well as G and P show unfavorable transfer free energies throughout the membrane. In general, hydrophobic residues are most favorable at the center of the bilayer, whereas hydrophilic residues are most unfavorable at the center of the membrane, consistent with previous experimental study.6 However, we found that ionizable residues, R and K, and polar residues, N, Q, S and T, have higher free energy cost in the inner leaflet than the outer leaflet (Fig 3b), thus, exhibiting asymmetric depth-dependency. These findings suggest that the inner leaflet of the bacterial OM imposes an energetic barrier to the insertion of polar and ionizable residues, whereas the outer leaflet favors transfer of these residues.
Presence of Lipid A lowers the folding free energy of OMPs
The asymmetry in the depth-dependent profiles of ionizable and polar amino acid residues demonstrated a clear correlation between the transfer free energies of these residues and the heterogeneity of the bacterial OM. To assess the effect of this phenomenon on the folding of bacterial OMPs, we built a model of a symmetric OM by implicitly substituting lipid A, the membrane part of LPS, in the outer leaflet with the phospholipids of the inner leaflet (Fig 4). Symmetric membranes are biologically relevant, as bacterial OMPs are capable of folding into the symmetric lipid bilayer of the mitochondrial OM.31 We approximated the folding free energy of OMPs with the summation of the transfer free energy of lipid-facing TM residues of the OMPs and evaluated the effect of the asymmetric composition of the OM on the stability of OMPs. For the asymmetric bacterial OM, we used the depth-dependent profiles ΔΔGaa(i) to calculate the total transfer free energy (ΔΔGasym) of a given client OMP. For the symmetric OM model, we first modified the asymmetric depth-dependent profiles to a symmetric profile , in which the values in the outer leaflet (i > 0) were replaced by those in the inner leaflet (i < 0), and the respective total transfer free energy of a client OMP (ΔΔGsym) were then calculated.
Fig 4.
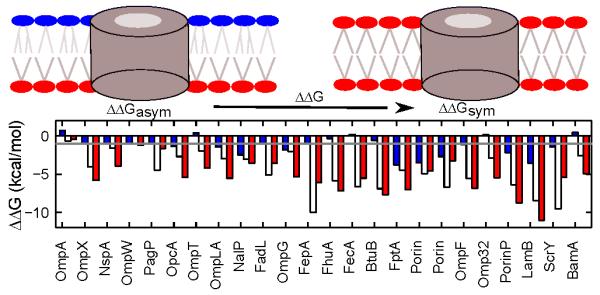
Lipid A contributes to the thermodynamic stability of bacterial OMPs. The stability differences ΔΔGs of 24 bacterial OMPs in the asymmetric bacterial outer membrane (OM) and a symmetric model OM were determined. With ∣ ΔΔG ∣ > lkcal/mol as the cutoff (grey line), all except OmpA have higher stability in the asymmetric membrane than in the symmetric membrane using either the total transfer free energy of all lipid-facing TM residues (white bar), or the folding free energy of the TM region (red bar). Only 15 OMPs show higher stability in the asymmetric OM if the total single burial energy of all lipid-facing TM residues is used (blue bar).
With the exception of OmpA, all OMPs are more stable in the asymmetric membrane by an ∣ΔΔG ∣ > 1kcal/mol (Fig 4). The same results are reached when folding free energies of the TM regions instead of transfer free energies are used. To rule out the possibility that our observations arise from the asymmetry of the empirical potential function, itself derived from OMP structures in asymmetric outer membranes, we evaluated protein stabilities using the only asymmetric term in our energy function, namely, the single body burial term.24 We found that only 15 out of the 24 OMPs are more stable in the asymmetric membrane with an ∣ ΔΔG ∣ > 1kcal/mol (Fig 4). Therefore, we conclude that the asymmetry in the potential function is insuffcient to determine the asymmetric stabilities of OMPs. Rather, physical interactions of TM residues in the ensemble of native and non-native configurations collectively give rise to the asymmetric thermodynamic stabilities. We further conclude that the presence of LPS in the outer leaflet not only serves the Gram-negative cell as a defense mechanism against solubilization by hydrophobic molecules, but further plays an important role in OMP biogenesis as it contributes to their thermodynamic stability This conclusion is consistent with a number of experimental findings. For example, PhoE was found to refold more efficiently in the phospholipid/LPS bilayer than in phospholipid/phospholipid bilayer,32 and LPS accelerates OmpA folding and insertion into lipid vesicles.33
Differential stabilities of TM regions in determining native topology of OMPs
The observation that LPS contributes to the thermodynamic stability of an OMP leads us to an interesting question about the native topology of OMPs. All bacterial OMPs adopt a membrane topology where the N- and C- terminus are located in the periplasmic space, termed here as the “NC-IN topology”. In contrast, the orientation of mitochondrial OMPs in the symmetric mitochondrial OM is highly debated and likely of dual-topology.34 We examined this issue by first approximating the folding free energy in the TM region following an additive model based on the computed transfer free energies of lipid-facing residues. In this additive model, the stability of an OMP is calculated as the summation of the transfer free energy of lipid-facing residues in the native configuration. Fig 5 shows that out of 24 representative bacterial OMPs examined, 17 of them exhibit higher stability in the native NC-IN topology.
Fig 5.

Native bacterial OMP topology is driven by the asymmetric nature of the OM as well as the lipid-facing residues in the head-group region, (a) Precursor sequence of an unfolded OMP inserts into the bacterial OM to either the native NC-IN topology or the non-native NC-OUT topology. OmpLA is used here as an example at different discrete depth position, (b) Difference of folding free energy of OmpLA in two topologies is calculated as the difference of total transfer free energy of all lipid-facing TM residues, (c) Stability differences of OMPs adopting native NC-IN topology and non-native NC-OUT topology are calculated. 17 OMPs have higher stability with NC-IN topology using the total transfer free energy of all lipid-facing TM residues (white bar) under the additive model. All 24 OMPs have higher stability in native NC-IN topology (red bar) using the non-additive model, which directly calculates the folding free energy of the TM region. 20 OMPs show higher stability in native topology if the total single burial energy of all lipid-facing residues is used (grey bar).
As this additive model failed to account for the native topology of 7 bacterial OMPs, we next adopt an alternative approach and directly estimate the thermodynamic stability of OMPs by computing the folding free energy of the TM region. In this case, non-additive effects that depend on local inter- and intra-strand interactions in native and non-native configurations are taken into account. Our results determine that all 24 bacterial OMPs in our analysis have lower folding free energy, thus higher thermodynamic stability, with NC-IN topology than that of NC-OUT topology (Fig 5c). Our findings suggest that the native NC-IN topology of bacterial OMPs is driven by the interplay of membrane asymmetry and interactions between amino acids in the TM region. Nevertheless, a few OMPs show very small differences in folding free energy between the two topologies. This suggests that in addition to the stability of the TM-region, there are other factors influencing OMP topology. For example, OmpW has long extended β-strands in the extracellular domain to form a hydrophobic channel.35 PagP has a periplasmic α-helix that out-clamps the barrel.25,36 These special structural features not incorporated in our model may also contribute to the native topologies of OMPs in the membrane.
Cooperativity among ionizable and polar residues
It is well documented that charged residues in OMPs are preferred in the extracellular side.24,30 This is different from helical membrane proteins, where charged residues are preferred in the cytoplasmic side.37 Non-additive effects may play an important role in transferring these amino acid residues across or into the lipid bilayer.38,39 For example, the cooperative nature of inserting multiple Arg residues has long been of interests in the studies of membrane protein biophysics.12,40,41 Cooperativities between aromatic residues have also been recently examined.39 However, the mechanism of inserting or translocating multiple ionizable or polar residues into or across highly hydrophobic membrane bilayer is not clear, and the role of cooperativities between ionizable and polar residues beyond Arg is not known in general. To investigate non-additive effects between residues on insertion into the TM region, we derived the cooperativity between any two ionizable or polar residues at different position of the bilayer. The sum of the energy costs of the single variants at a host pair is compared to the energy cost of the double variant, and the difference quantifies the cooperativity. Basic residues Arg, Lys, and His, acidic residues Asp and Glu along with two highly polar residues Asn and Gln were included in this analysis. We chose two adjacent lipid-facing residues in strands β4, β8, and β9 as the host residue pairs, and derived the cooperativity at the respective positions (Fig 6a). These strands are chosen as their lipid facing residues do not show strong context dependency. We further averaged cooperativity values of ionizable or polar residue at host pairs of the same position from all three strands.
Fig 6.
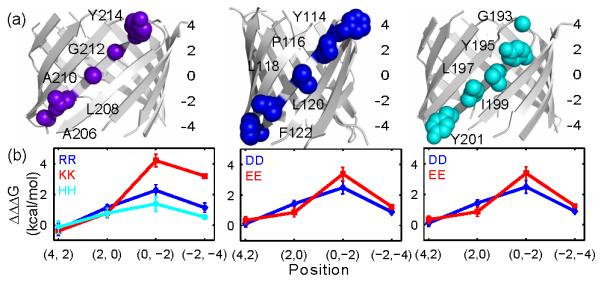
Positive cooperativity is general, asymmetric, and depth-dependent for ionizable or polar residue pairs’, (a) Four host pairs are selected from β9, β4, and β8 to derive the cooperativity. Here (i + 2, i) are the locations indices of the host pairs, i.e., Y214-G212 in strand β9 has location index (4, 2). (b) Positive cooperativity was observed for most residue pairs (Table S5). The average cooperativity at a specific position is plotted with its standard error. The strongest cooperativity is observed in the inner leaflet core region at (0, −2).
We found cooperativity between two ionizable or highly polar residues exist at most of the positions (Fig 6b). The strongest cooperativity was observed in pairs located in the inner leaflet core region (position (0, −2)), followed by pairs in the inner leaflet headgroup region (position (−2, −4)). Overall, the cooperativity decreases as the host pair moves away from the core region. Lys-Lys has the highest cooperativity (ΔΔΔG = 4.20 kcal/mol) at position (0, −2), while cooperativity in the outer leaflet headgroup region (positions 4, 2) is negligible (Fig 6b). Our results suggest that positive and depth-dependent cooperativity is a general property for ionizable or highly polar residue pairs. Located in the TM or the extracellular loop region, these residues encounter an energetic barrier during insertion into or translocate the hydrophobic lipid bilayer. Due to this cooperativity, clustering of these residues may reduce the energetic cost during their insertion, providing an additional driving force for the folding and stability of OMPs. Coopeartivity may play a similar role in the translocation of Arg-rich cell penetrating peptides.
Spontaneous insertion of OMPs is driven by the lipid-facing residues in the hydrophobic core
It is well known that bacterial OMPs can fold spontaneously without an external energy source to their native state.42 However, it is unclear what occurs thermodynamically during insertion as the inner leaflet imposes an energetic barrier to insertion of ionizable and polar residues. To address this question, we used a simplified folding model with 9 discrete steps (Fig 7a), based on the concerted folding mechanism proposed by Kleinschmidt et al.43 We calculated the total transfer free energy of lipid-facing residues inserted into the bilayer in each step. While pore-facing residues and their interactions also contribute to the overall protein stabilities, our analysis shows that lipid-facing residues contribute approximately 5 times more that the pore-facing residues in OmpLA. Therefore, we focus on lipid-facing residues that are directly involved in insertion and folding in the membrane environment. To identify the origin of the favorable free energy, we decomposed the overall free energy into contributions from the hydrophobic core residues and from the headgroup residues (Fig 7b).
Fig 7.

Lipid-facing residues in the hydrophobic core region drive spontaneous OMP insertion, (a) OMP insertion is described by 9 sequential discrete steps. The depth position (−4 to 4) up to where the TM region inserts into is regarded as the reaction coordinate, (b) Folding free energies of bacterial OMPs are approximated as the total transfer free energy of lipid-facing residues inserted into the bilayer (red line). The energetic barrier caused by the lipid-facing residues in the headgroup region (blue diamond) is compensated by the favorable folding free energy of the lipid-facing residues in the hydrophobic core (green circles).
Indeed, we found that the folding free energy of all investigated OMPs during the insertion of the TM region is overall favorable with ΔΔG < 0 (kcal/mol) (red line in Fig 7b), in agreement with many studies indicating spontaneous OMP insertion and folding. If the head-group region residues face an energetic barrier during OMP insertion, the hydrophobic core residues compensate their costs by reducing the overall folding free energy, thus enabling spontaneous OMP insertion (Fig 7b). Therefore, we conclude that lipid-facing residues in the hydrophobic core region of OMPs provide the main driving forces for OMP folding into the lipid membrane. Our results suggest that assembly machinery proteins such as BAM complex,44 or periplasmic chaperons such as skp45 may accelerate the folding process but are unlikely to affect the overall mechanism of the process. These results are in agreement with the experimental folding study on OmpA by Kleinschmidt et al.43
Discussion
Our results demonstrated that the computational model presented here is capable of capturing key factors determining the stability of the TM region of bacterial OMPs and therefore enabled us to reproduce experimentally measured transfer free energy scales for studying thermodynamic properties of bacterial OMPs. While several computational hydrophobicity scales specific for OMPs have been obtained by converting observed abundance of a TM residue at a particular depth position into empirical free energy,17,24,30 these empirical scales agree poorly with the whole protein Moon-Fleming 20 amino acid transfer free energy scale (Fig S4a), as the protein-specific details of strong interactions between residues on neighboring strands are not considered in these scales. In contrast, our computational scale has excellent agreement with the Moon-Fleming scale. This is due to the fact that in addition to the single-body burial energy term,24 we also consider intra- and inter-strand interactions. Furthermore, we examine all energies of configurations of different strand registrations through enumeration, with Boltzmann contributions from both native and non-native configurations treated alike in calculating ensemble properties. Detailed configuration enumeration has been successfully applied to predict and engineer oligomerization states of OMPs,25,46 to predict protein-protein interactions interface in the TM region,25 and to predict three-dimensional structures of the TM region of OMPs.47 By computationally substituting specific residues in the TM region of an OMP, we can determine desired hydrophobicity scales at any position in the lipid bilayer. We used this approach to determine systematically the transfer free energies of 20 amino acids at all possible TM lipid-facing positions of the OMP OmpLA. We noted both the Moon-Fleming scale as well as our computational scale derived in the context of an OMP correlate reasonably well with the biological scale derived in the context of a TM helix (R2 = 0.73 and R2 = 0.86 after excluding Pro, respectively). This suggests that important aspects of the overall thermodynamic properties of residue side-chains are preserved in both types of membrane proteins.
Our results show that there exists a general depth-dependent transfer free energy scale that is applicable to most lipid-facing positions of OmpLA. We also found that deviations from the general transfer free energy values correlated with lipid-facing amino acid residues important for enzymatic function of OmpLA (i.e., dimerization as well as formation of the active site), or for structural anomalies. Deformed structures are known to be relevant for the function of OMPs. For example, BamA, a conserved OMP responsible for the assembly of OMPs in bacterial outer membrane, induces the thinning of the bilayer around the short β-strands 1 and 16, thus creating a local membrane defect. This defect is hypothesized to facilitate OMP folding.44 Our computational method can therefore be used to predict the locations of residues that are important for OMP function.
Success in detecting such a residue requires examination of the profile of the 19 transfer free energy values at the position of this residue. A naive approach such as examining the only depth-dependent single-body burial energy term Eb in the potential function does not work, as this term only depends on the depth position and does not contribute to the observed context dependency. While evaluating the full empirical energy of the native structure incorporating additional terms can successfully identify unstable strands implicated in protein-protein interactions,25 it is inadequate as this approach detects only 2 (D36 and N38 on the weakly stable β1 strand) of the 12 residues (see Fig S5). Neither D36 in the lumen of the barrel nor N38 before the beginning of strand β1 participates in forming β-strands, and are not at lipid-facing positions as would be expected from a canonical model of a TM strand (Fig S1). While our single-body burial energy term Eb is suffcient to detect the structurally deformed local environments of these two residues, overall it is the collective effects of the burial, the intra-strand and inter-strand interaction terms, the ensemble of 7N = 712 native and non-native configurations involving the host strand, as well as the effects of mutating the host position to all 19 other amino acid residues that distinguish these deformed or functional residues. Furthermore, our method can be applied to OMPs with no known structures to identify residues in deformed environment and residues involved in function, as the calculation of transfer free energies requires only knowledge of the TM sequences.
We also demonstrated that the depth-dependent profiles of amino acids can be used to determine the correct topology of OMPs in the cell outer membrane, and further to evaluate the folding free energy contributed by the TM segments during the insertion of OMPs. While asymmetric distributions of residues in different regions of the membrane bilayer are apparent from statistical analysis,24 profiles of empirical energy values of Arg and Leu at different depth positions using either potential converted from their asymmetric distributions or the full energy values evaluated on the native structure25 were unable to accurately reproduce the experimentally measured energy costs of transferring these residues to different positions of the bilayer (Fig S4b). In contrast, profiles of computed transfer free energies of Arg and Leu have excellent agreement with those obtained from experimental measurements. In addition, our results show that the energy cost of ionizable and polar residues in the inner leaflet is higher than that in the outer leaflet, suggesting that the inner leaflet creates an energy barrier for OMP folding. Further analysis showed that lipid-facing TM residues located in the headgroup region need to overcome an energy barrier to translocate through the hydrophobic core region during the insertion process. Experimental folding studies have indeed demonstrated that folding OMPs in the native lipids of E. Coli is very ine cient.44 In vivo, OMPs folding and insertion is accomplished with the existence of BAM complex. BamA may reduce the energy barrier created by the outer membrane through destabilizing the local membrane.48
Our analysis also suggests that Lipid A of the LPS in the outer leaflet of bacterial outer membrane contributes to the thermodynamic stability of OMPs, as the folding free energy of the TM region of OMPs is lower in the asymmetric membrane formed by phospholipids inner leaflet and LPS outer leaflet than the folding free energy in the symmetric membrane whose leaflets are both formed by phospholipids. Although the difference in thickness of inner and outer leaflet is not incorporated in this study, LPS outer leaflet with thinner hydrophobic core49 may further increase the stability of the TM region. In addition, the O-antigen polysaccharides chain of LPS provides a densely confined space in the extracellular domain of the cell membrane, further increasing the stability of the OMPs.50 The stabilization effect may be further enhanced by the favorable interaction between the LPS and the extracellular loops.49,51 Therefore, asymmetric bacterial outer membrane plays an important role in the OMP folding through stabilizing both TM and non-TM region of OMPs.
Approximation of the stability of OMPs in the membrane bilayer using the total transfer free energy of lipid-facing residues was suffcient to capture the preference of OMPs in the asymmetric OM. While asymmetric Gaussian functions were used to represent the depth-dependent transfer free energy profiles of lipid-facing residues, alternative parametric models based on 2nd- and 3rd-degree polynomials also showed higher OMP stabilities in the asymmetric outer membrane (Fig S6). The importance of lipid-facing residues in maintaining the stability of OMPs is also reflected in their substitution patterns. A detailed evolutionary analysis of the substitution rates showed that lipid-facing residues have a conserved pattern of allowed and forbidden substitutions across different OMP families.52 While pore-facing residues are also under strong selection pressure as they perform important biological functions such as channel conductance, substrate binding, and substrate transport, the pattern of conservation is more specific to individual protein families whose members are of similar functions.
As the asymmetric bacterial outer membrane provides a stabilizing environment to bacterial OMPs, one important question is why such asymmetry is not observed in eukaryote outer membrane. This can be explained by the difference in the biogenesis of bacterial OMPs and eukaryote OMPs. In bacteria, OMP is translocated to the periplasmic space after its synthesis in the cytoplasm.4 Energy sink in the bacterial outer membrane is necessary to sort the OMPs precursors to the outer membrane.13 This indicates that OMP folding in bacteria is controlled by the physical nature of the OM. This is critical, as a porin would destroy the proton motive force if spontaneously fold into the inner membrane (IM), resulting in ions and small molecules diffusing through its nonspecific channel, which will lead to cell death. In mitochondria, on the other hand, OMP folding is already tightly controlled throughout it biogenesis, as folding is directly coupled to OMP synthesis in the cytosol and translocation across the OM by the TOM-SAM supercomplex.53 This well-controlled process of folding would ensure that OM is the only environment a mitochondria OMP would encounter. Unlike bacterial OMP, energy sink may not be necessary for sorting eukaryotic OMP to the mitochondria OM.
In our study, cooperativity is broadly observed in pairs of ionizable and polar residues when exposed to the lipids. The depth-dependent cooperativity is stronger when the residue pairs are closer to the hydrocarbon core of the bilayer. Studies of the non-additive effect of the translocation of Arg-Arg pair through molecular dynamic simulations by MacCallum et al40 suggest that water-filled defect appears when an isolated Arginine residue is inserted into the center of the hydrocarbon core, which drastically reduces the energetic cost of inserting the second Arg residue.21,40 Similar observations on water-filled defect were found for TM helix and strand.22,54,55 Using a continuum mechanical model of membrane, a study on the insertion of a single TM helix suggests that the cooperativity between Arg residues arise from the fact that once membrane bends to accommodate the first charged residue, no further bending is needed for the second charged residue.56 However, MD simulation studies showed that Arg residue is special, as it is the only ionizable residue that can maintain water-filled defects when placed in the center of the bilayer, whereas such defects dissipate when other ionizable residues are placed close to the bilayer center.21 It is unclear whether non-additivity of other ionizable residues beyond Arg in the TM segment of β-barrel membrane protein observed in this study is governed by the same mechanism as found in ref.40 Our study suggests that cooperativity between ionizable residues may partly arise from favorable neighboring interactions in an overall depth-dependent membrane environment. It is possible that such favorable interactions are related mechanistically to water-filled defects and membrane deformation, and contribute to the cooperativities of ionizable residue pairs in β-barrel outer membrane proteins.
We also studied the OMP membrane-insertion and folding as a discrete process based on the reported synchronized translocation of β-hairpins and the concerted folding model of OmpA proposed in reference.43 We calculated the energetic cost of inserting the OMPs at each step of the insertion process. This model is a highly simplified model and important details such as how membrane would reorganize during each insertion step are not accounted for. Nevertheless, our results enabled a number of useful findings. We found that folding of the TM-region of an OMP into the bacterial outer membrane is energetically favorable, and there is no significantly energy barrier observed during the insertion. This agrees well with the fact that OMPs can fold spontaneously both in vitro and in vivo without any external energy source. The ability of spontaneous folding is also preserved in the OMPs forming oligomers in the outer membrane. In fact, similar folding effciency of the trimeric OmpF and the engineered monomeric OmpF was observed in in vitro study,46 which indicates that folding an individual subunit and oligomerization are two independent processes. All these observations support that OMPs behave as autonomous folding domains, in agreement with the viewpoint of Popot and Engelman.57 Interestingly, spontaneous insertion into bacterial outer membrane is also predicted for mitochondrial outer membrane protein VDAC (Fig S7). This is consistent with experimental in vivo refolding study of VDAC into bacterial outer membrane58 and in vitro refolding into PC bilayer.59
To summarize, we first validated our method by computationally reproducing the measured transfer free energy scale of Ala210 (Fig 1a) and the depth-dependent profiles of Arg and Leu (Fig 3a).12 We then applied our method and predicted a new set of transfer free energy scales for all other 52 lipid-facing residues in OmpLA as hosts, which currently have no experimental measurements (Fig 1b). These scales lead to the depth-dependent profiles of the 20 amino acids (Fig 3b), which complement the two known profiles of Arg and Leu.12 Our method can also predict TM residues either in a structural deformed environment or are related to functions of OmpLA (Fig 2). As only TM sequences are required, our method can be applied to other OMPs without requiring knowledge of their structures. Another prediction is the general cooperativity between all charged TM residue pairs and their depth-dependency, complementing current knowledge of cooperativity between Arg pairs.12 Our method also predicts that the asymmetric nature of the bacterial OM stabilizes OMPs (Fig 4), which can be experimentally tested once techniques for generating asymmetric lipid bilayer become more practical. Our method also explains several known facts, including the basis of the correct NC-IN topology of OMPs, as well as the mechanism of the spontaneous folding of OMPs in the bacterial OM. In addition, our method can be used to study the behavior of eukaryotic outer membrane proteins, such as the spontaneous insertion of mitochondrial protein VDAC in bacterial OM.58,59
In conclusion, this work introduced a new computational approach to derive transfer free energy scale in the context of an OMP and to evaluate the thermodynamic stability of OMPs, which can be used to reveal important biological insight. Future directions include deriving transfer free energy scales for other OMPs and relate to their properties. As OMPs are found in gram-negative bacteria, mitochondria, and chloroplasts, our method can aid in understanding the general biophysical principles of structure, stability, and function of this important class of membrane proteins. Because of its computational effciency, our approach can also be useful in de novo design of outer membrane proteins as nanodevices for biotechnological applications such as DNA and RNA sequencing60,61 and single molecule sensing.62–65 Our current method is designed for β-barrel membrane proteins, as the empirical potential function was developed specifically for outer membrane proteins in their native membrane environments. As the lipid bilayer is not modeled explicitly, the effectiveness of evaluating the thermodynamic properties of OMPs in non-natural membranes with lipid compositions different from that of their native host membranes is unknown. It is possible to generalize our approach to α-helical membrane proteins,14,15,66,67 which would require the development of an appropriate discrete state model and an enumeration method or an effective sampling method, with empirical potential function specifically constructed for the state model upon removal of confounding effects.66–68
Methods
The state space Ω of the native and non-native configurations of the TM region of an OMP is defined following reference,25 where each strand has 16 residues and can slide up to 3 residues away from its center position. Each TM strand residue interacts with neighboring strand residues through strong H-bond, non H-bond, or weak H-bond following previous studies.24,69,70 In addition, neighboring TM residues on the same strand facing the same side of the OMPs (lipid-facing or pore-facing) interacts. For a specific configuration d, an empirical strand energy function incorporating single residue burial energy EB, strong H-bond interaction energy ESH, non H-bond interaction energy ENH, weak H-bond interaction energy EWH, and intra-strand interaction energy EIntra with proper weights is used to calculate the energy of the TM region E(d). The folding free energy Glipid of the TM segment of an OMP was then calculated from the partition function Zlipid, which is the summation of the the Boltzmann factors of residues in the TM region over all discrete configurations in the state space Glipid = −kBT ln . Given a lipid-facing host residue of OmpLA, the differences in the TM folding free energy of the Ala substitution compared to the other 19 amino acid substitutions were used to construct the transfer free energy scale of that host residue. Following an additive model, the total transfer free energy of lipid-facing residues was used to approximate the folding free energy and the thermodynamic stability of an OMP. The calculated stability of an OMP in the native asymmetric membrane is then compared to the calculated stability in a symmetric membrane. The total transfer free energy was also used to test whether it is suffcient to determine the native NC-IN topology of OMPs. Furthermore, membrane protein insertion was analyzed using the total transfer free energy of lipid-facing residues upon insertion. More details of our method can be found in SI Method and Material.
Supplementary Material
Acknowledgement
This work was supported by NIH grant GM079804, NSF grants DMS-0800257 and DBI-1062328, and Chicago Community Trust grant Catalyst-2013-9.
Footnotes
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Wallin E, von Heijne G. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fairman J, Noinaj N, Buchanan S. Curr Opin Struct Biol. 2011;21:523–531. doi: 10.1016/j.sbi.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sanders C, Nagy J. Curr Opin Struct Biol. 2000;10:438–442. doi: 10.1016/s0959-440x(00)00112-3. [DOI] [PubMed] [Google Scholar]
- (4).Hagan C, Silhavy T, Kahne D. Annu Rev Biochem. 2011;80:189–210. doi: 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
- (5).Wimley WC, Creamer TP, White SH. Biochemistry. 1996;35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- (6).Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, Nilsson I, White SH, von Heijne G. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- (7).Moon C, Kwon S, Fleming K. J Mol Biol. 2011;413:484–494. doi: 10.1016/j.jmb.2011.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Otzen D, Andersen K. Arch Biochem Biophys. 2013;531:34–43. doi: 10.1016/j.abb.2012.10.008. [DOI] [PubMed] [Google Scholar]
- (9).Hong H, Tamm L. Proc Natl Acad Sci U S A. 2004;101:4065–4070. doi: 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hong H, Szabo G, Tamm L. Nat Chem Biol. 2006;2:627–635. doi: 10.1038/nchembio827. [DOI] [PubMed] [Google Scholar]
- (11).Huysmans G, Baldwin S, Brockwell D, Radford S. Proc Natl Acad Sci U S A. 2010;107:4099–4104. doi: 10.1073/pnas.0911904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Moon C, Fleming K. Proc Natl Acad Sci U S A. 2011;108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Moon C, Zaccai N, Fleming P, Gessmann D, Fleming K. Proc Natl Acad Sci U S A. 2013;110:4285–4290. doi: 10.1073/pnas.1212527110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liang J. Curr Opin Chem Biol. 2002;6:878–884. doi: 10.1016/s1367-5931(02)00393-9. [DOI] [PubMed] [Google Scholar]
- (15).Liang J, Naveed H, Jimenez-Morales D, Adamian L, Lin M. Biochim Biophys Acta. 2012;1818:927–941. doi: 10.1016/j.bbamem.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Koehler J, Woetzel N, Staritzbichler R, Sanders C, Meiler J. Proteins. 2009;76:13–29. doi: 10.1002/prot.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hsieh D, Davis A, Nanda V. Protein Sci. 2012;21:50–62. doi: 10.1002/pro.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schramm CA, Hannigan BT, Donald JE, Keasar C, Saven JG, Degrado WF, Samish I. Structure. 2012;20:924–935. doi: 10.1016/j.str.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Moore D, Berger B, DeGrado W. Structure. 2008;16:991–1001. doi: 10.1016/j.str.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hedin LE, Ojemalm K, Bernsel A, Hennerdal A, Illergard K, Enquist K, Kauko A, Cristobal S, von Heijne G, Lerch-Bader M, Nilsson I, Elofsson A. J. Mol. Biol. 2010;396:221–229. doi: 10.1016/j.jmb.2009.11.036. [DOI] [PubMed] [Google Scholar]
- (21).MacCallum J, Bennett W, Tieleman D. J Gen Physiol. 2007;129:371–377. doi: 10.1085/jgp.200709745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gumbart J, Roux B. Biophys. J. 2012;102:795–801. doi: 10.1016/j.bpj.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ulmschneider M, Ulmschneider J, Schiller N, Wallace B, von Heijne G, White S. Nat Commun. 2014;5:4863. doi: 10.1038/ncomms5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jackups R, Jr., Liang J. J Mol Biol. 2005;354:979–993. doi: 10.1016/j.jmb.2005.09.094. [DOI] [PubMed] [Google Scholar]
- (25).Naveed H, Jackups R, Jr., Liang J. Proc Natl Acad Sci U S A. 2009;106:12735–12740. doi: 10.1073/pnas.0902169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dekker N, Tommassen J, Lustig A, Rosenbusch J, Verheij H. J Biol Chem. 1997;272:3179–3184. doi: 10.1074/jbc.272.6.3179. [DOI] [PubMed] [Google Scholar]
- (27).Snijder H, Ubarretxena-Belandia I, Blaauw M, Kalk K, Verheij H, Egmond M, Dekker N, Dijkstra B. Nature. 1999;401:717–721. doi: 10.1038/44890. [DOI] [PubMed] [Google Scholar]
- (28).Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von Heijne G. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- (29).Kingma R, Fragiathaki M, Snijder H, Dijkstra B, Verheij H, Dekker N, Egmond M. Biochemistry. 2000;39:10017–10022. doi: 10.1021/bi000786d. [DOI] [PubMed] [Google Scholar]
- (30).Slusky J, Dunbrack R., Jr. Bioinformatics. 2013;29:2122–2128. doi: 10.1093/bioinformatics/btt355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Walther D, Papic D, Bos M, Tommassen J, Rapaport D. Proc Natl Acad Sci U S A. 2009;106:2531–2536. doi: 10.1073/pnas.0807830106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hagge S, de Cock H, Gutsmann T, Beckers F, Seydel U, Wiese A. J Biol Chem. 2002;277:34247–34253. doi: 10.1074/jbc.M201950200. [DOI] [PubMed] [Google Scholar]
- (33).Bulieris P, Behrens S, Holst O, Kleinschmidt J. J Biol Chem. 2003;278:9092–9099. doi: 10.1074/jbc.M211177200. [DOI] [PubMed] [Google Scholar]
- (34).Ujwal R, Cascio D, Chaptal V, Ping P, Abramson J. Channels (Austin) 2009;3:167–170. doi: 10.4161/chan.3.3.9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hong H, Patel D, Tamm L, van den Berg B. J Biol Chem. 2006;281:7568–7577. doi: 10.1074/jbc.M512365200. [DOI] [PubMed] [Google Scholar]
- (36).Ahn V, Lo E, Engel C, Chen L, Hwang P, Kay L, Bishop R, Prive G. EMBO J. 2004;23:2931–2941. doi: 10.1038/sj.emboj.7600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).von Heijne G. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- (38).Hristova K, Wimley W. J Membr Biol. 2011;239:49–56. doi: 10.1007/s00232-010-9323-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hong H, Park S, Jimenez R, Rinehart D, Tamm L. J Am Chem Soc. 2007;129:8320–8327. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- (40).MacCallum J, Bennett W, Tieleman D. Biophys J. 2011;101:110–117. doi: 10.1016/j.bpj.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Marks J, Placone J, Hristova K, Wimley W. J Am Chem Soc. 2011;133:8995–9004. doi: 10.1021/ja2017416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Burgess N, Dao T, Stanley A, Fleming K. J Biol Chem. 2008;283:26748–26758. doi: 10.1074/jbc.M802754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kleinschmidt J, den Blaauwen T, Driessen A, Tamm L. Biochemistry. 1999;38:5006–5016. doi: 10.1021/bi982465w. [DOI] [PubMed] [Google Scholar]
- (44).Gessmann D, Chung Y, Danoff E, Plummer A, Sandlin C, Zaccai N, Fleming K. Proc Natl Acad Sci U S A. 2014;111:5878–5883. doi: 10.1073/pnas.1322473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Denoncin K, Schwalm J, Vertommen D, Silhavy T, Collet J. Proteomics. 2012;12:1391–1401. doi: 10.1002/pmic.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Naveed H, Jimenez-Morales D, Tian J, Pasupuleti V, Kenney LJ, Liang J. J. Mol. Biol. 2012;419:89–101. doi: 10.1016/j.jmb.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Naveed H, Xu Y, Jackups R, Liang J. J. Am. Chem. Soc. 2012;134:1775–1781. doi: 10.1021/ja209895m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Noinaj N, Kuszak A, Gumbart J, Lukacik P, Chang H, Easley N, Lithgow T, Buchanan S. Nature. 2013;501:385–390. doi: 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wu E, Fleming P, Yeom M, Widmalm G, Klauda J, Fleming K, Im W. Biophys J. 2014;106:2493–2502. doi: 10.1016/j.bpj.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhou H, Dill K. Biochemistry. 2001;40:11289–11293. doi: 10.1021/bi0155504. [DOI] [PubMed] [Google Scholar]
- (51).Edrington T, Kintz E, Goldberg J, Tamm L. J Biol Chem. 2011;286:39211–39223. doi: 10.1074/jbc.M111.280933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jimenez-Morales D, Liang J. PLoS One. 2011;6:e26400. doi: 10.1371/journal.pone.0026400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Qiu J, Wenz L, Zerbes R, Oeljeklaus S, Bohnert M, Stroud D, Wirth C, Ellenrieder L, Thornton N, Kutik S, Wiese S, Schulze-Specking A, Zufall N, Chacinska A, Guiard B, Hunte C, Warscheid B, van der Laan M, Pfanner N, Wiedemann N, Becker T. Cell. 2013;154:596–608. doi: 10.1016/j.cell.2013.06.033. [DOI] [PubMed] [Google Scholar]
- (54).Dorairaj S, Allen TW. Proc. Natl. Acad. Sci. U.S.A. 2007;104:4943–4948. doi: 10.1073/pnas.0610470104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Fleming PJ, Freites JA, Moon CP, Tobias DJ, Fleming KG. Biochim. Biophys. Acta. 2012;1818:126–134. doi: 10.1016/j.bbamem.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Callenberg K, Latorraca N, Grabe M. J Gen Physiol. 2012;140:55–68. doi: 10.1085/jgp.201110766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Popot J, Engelman D. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- (58).Walther D, Bos M, Rapaport D, Tommassen J. Mol Biol Evol. 2010;27:887–895. doi: 10.1093/molbev/msp294. [DOI] [PubMed] [Google Scholar]
- (59).Shanmugavadivu B, Apell H, Meins T, Zeth K, Kleinschmidt J. J Mol Biol. 2007;368:66–78. doi: 10.1016/j.jmb.2007.01.066. [DOI] [PubMed] [Google Scholar]
- (60).Branton D, Deamer D, Marziali A, Bayley H, Benner S, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, Jovanovich S, Krstic P, Lindsay S, Ling X, Mastrangelo C, Meller A, Oliver J, Pershin Y, Ramsey J, Riehn R, Soni G, Tabard-Cossa V, Wanunu M, Wiggin M, Schloss J. Nat Biotechnol. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ayub M, Hardwick S, Luisi B, Bayley H. Nano Lett. 2013;13:6144–6150. doi: 10.1021/nl403469r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Gu L, Braha O, Conlan S, Cheley S, Bayley H. Nature. 1999;398:686–690. doi: 10.1038/19491. [DOI] [PubMed] [Google Scholar]
- (63).Braha O, Gu L, Zhou L, Lu X, Cheley S, Bayley H. Nat Biotechnol. 2000;18:1005–1007. doi: 10.1038/79275. [DOI] [PubMed] [Google Scholar]
- (64).Rotem D, Jayasinghe L, Salichou M, Bayley H. J Am Chem Soc. 2012;134:2781–2787. doi: 10.1021/ja2105653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Fahie M, Chen M. J Phys Chem B. 2015;119:10198–10206. doi: 10.1021/acs.jpcb.5b06435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Adamian L, Jackups R, Jr., Binkowski T, Liang J. J Mol Biol. 2003;327:251–272. doi: 10.1016/s0022-2836(03)00041-x. [DOI] [PubMed] [Google Scholar]
- (67).Adamian L, Nanda V, DeGrado W, Liang J. Proteins. 2005;59:496–509. doi: 10.1002/prot.20456. [DOI] [PubMed] [Google Scholar]
- (68).Jackups R, Jr., Liang J. IEEE/ACM Trans Comput Biol Bioinform. 2010;7:524–536. doi: 10.1109/TCBB.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Wouters MA, Curmi PM. Proteins. 1995;22:119–131. doi: 10.1002/prot.340220205. [DOI] [PubMed] [Google Scholar]
- (70).Ho B, Curmi P. J Mol Biol. 2002;317:291–308. doi: 10.1006/jmbi.2001.5385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.