

Abstract
Antisense oligonucleotide (AO)-mediated exon-skipping therapeutics shows great promise for Duchenne muscular dystrophy (DMD) patients. However, recent failure with drisapersen, an AO candidate drug in phase 3 trial, highlights the importance of exploring other effective AO chemistries for DMD. Previously, we demonstrated the appreciable biological activity of peptide nucleic acid (PNA) AOs in restoring dystrophin expression in dystrophin-deficient mdx mice intramuscularly. Here, we further explore the systemic potential and feasibility of PNA AOs in mediating exon skipping in mdx mice as a comprehensive systemic evaluation remains lacking. Systemic delivery of PNA AOs resulted in therapeutic level of dystrophin expression in body-wide peripheral muscles and improved dystrophic pathology in mdx mice without any detectable toxicity. Up to 40% of dystrophin restoration was achieved in gastrocnemius, to a less extent with other skeletal muscles, with no dystrophin in heart. Notably, comparable systemic activity was obtained between PNA AOs and phosphorodiamidate morpholino oligomer, a DMD AO chemistry in phase 3 clinical trial, under an identical dosing regimen. Overall, our data demonstrate that PNA is viable for DMD exon-skipping therapeutics with 20 mer showing the best combination of activity, solubility, and safety and further modifications to increase PNA aqueous solubility can enable longer, more effective therapeutics without the associated toxicity.
Keywords: antisense oligonucleotide, Duchenne muscular dystrophy, dystrophin, exon skipping, peptide nucleic acid
Introduction
Duchenne muscular dystrophy (DMD) is one of the most common and severe muscular dystrophies, caused by frame-disrupting mutations in the dystrophin gene, which result in loss of functional dystrophin protein.1 There is no effective treatment available for DMD patients clinically, though the average lifespan of DMD patients has been extended significantly with the improvement in medical care and the use of ventilator.2,3,4 A diversity of therapeutic interventions currently is under intensive scrutiny including virus-based gene replacement, stop codon read-through, uptrophin upregulation and antisense oligonucleotide (AO)–mediated exon-skipping therapeutics.5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20 Among them, the stop codon readthrough and exon-skipping therapeutics are the two most promising and well-developed approaches, with the former being conditionally approved in Europe and the latter currently in phase 3 clinical trials.21 However, the major limitation to the stop codon readthrough approach is that it is only amenable to nonsense mutations, which prevents its general use to most of DMD patients with other types of mutations. In contrast, AO-mediated exon-skipping therapeutics is applicable to a larger spectrum of mutations and has been estimated to be amenable to 72.5% of DMD patients.22
Currently, two AO drugs (eteplirsen and drisapersen) targeting at human DMD exon 51 are leading the way and showed encouraging clinical outcome from studies in man.23,24,25 However, recent report on the failure of drisapersen trial to meet the expected endpoints, which likely attributes to the insufficient systemic efficacy of the 2-O-methyl phosphorothioate (2'Ome RNA) chemistry as demonstrated in mdx mice,15 further underlines the importance of developing effective candidate chemistries for DMD. Previously we have demonstrated that peptide nucleic acid (PNA), a synthetic chemistry bearing appreciable biochemical properties and activity, in which the sugar backbone is replaced with the N-(2-aminoethyl)-glycine,20 triggered effective exon-skipping and dystrophin expression in mdx mice intramuscularly.26 Notably, PNA AOs showed superiority to 2'Ome RNA and comparable activity to phosphorodiamidate morpholino oligomer (PMO) from local intramuscular studies.27 However, the systemic potential of PNA in restoring dystrophin expression and rescuing the phenotypic pathology of mdx mice remains to be explored, particularly at doses comparable to those of 2'Ome RNA and PMO as reported.15,16
Here, we demonstrated for the first time that repeated administration of PNA AOs in mdx mice at the dose of 100 mg/kg intravenously induced therapeutic level of dystrophin expression and resulted in functional and phenotypic rescue of treated mdx mice. Furthermore, appreciable safety profiles were established with repeated injections of PNA AOs without any detectable drug-related toxicity and inflammatory response. Notably, comparable level of exon-skipping and dystrophin expression was obtained between PNA AOs and PMO in mdx mice systemically under an identical dosing regimen. Finally, we noted that longer PNA AOs such as 30 mer showed improved efficacy with significant improvement in the level of exon-skipping and dystrophin restoration compared to shorter ones in mdx mice intramuscularly. However, the increased acidity arose from long PNA purification, which is also likely required for solubility, resulted in toxicity and further improvement in PNA purification and solubility at neutral pH may enable safer and much better PNA-based therapeutics. Our study indicates that PNA is a promising chemistry for AO-mediated exon-skipping therapeutics for DMD and can be even more potent and competent alternatives with longer ones if the acidity can be overcome.
Results
PNA shows appreciable exon-skipping activity at relatively high doses in mdx mice
Previously, we have demonstrated that PNA AOs induced effective exon-skipping in mdx mice intramuscularly.26 However, the systemic potential of PNA AOs in triggering exon-skipping in mdx mice remains to be appropriately assessed. In order to ascertain the ability of PNA AOs to elicit exon-skipping and dystrophin expression in mdx mice systemically, we administered PNA AOs into adult mdx mice intravenously at the dose of 50 mg/kg for 3 weeks at weekly interval. An increase in the number of dystrophin-positive fibers was evident in quadriceps, gastrocnemius, and abdominal muscle with no dystrophin expression in heart (Figure 1a). Compared to PNA AOs, no difference was detected in the distribution and number of dystrophin-positive fibers in corresponding samples from mdx mice treated with PMO under an identical dosing regimen (Figure 1a). Consistent with the immunostaining results, detectable and comparable exon-skipping was observed in these corresponding muscles from mdx mice treated with either PNA AOs or PMO as revealed by RT-PCR (Figure 1b), though the exon-skipping efficiency varied between muscles as shown by quantification of exon 23 skipping efficiency (Figure 1c). Corroborating with the immunostaining and RT-PCR results, the level of dystrophin protein expression in the quadriceps and gastrocnemius from mdx mice treated with either PNA AOs or PMO was ~2% of normal control as shown in western blot (Figure 1d). Altogether, comparable activity was achieved between PNA AOs and PMO based on the level of exon-skipping and dystrophin expression under the identical dosing regimen (Figure 1), which is consistent with the previous intramuscular study.26
Figure 1.

Systemic evaluation of PNA AOs at the dose of 50 mg/kg with three weekly injections in mdx mice. (a) Immunostaining for dystrophin-positive fibers in body-wide periphearl muscles from mdx mice treated with PNA AOs and PMO at the dose of 50 mg/kg (bar = 100 μm). The top and second panels show for normal C57BL6 and untreated mdx mice. TA, tibialis anterior. (b) RT-PCR to detect dystrophin exon-skipping transcripts in treated tissues. –ve represents blank control; Δexon 23 is for the exon 23 skipped band. (c) Quantitative analysis of percentage of exon 23 skipping efficiency in different muscles from mdx mice treated with either PNA AOs or PMO. (d) Western blot to demonstrate the level of dystrophin restoration in treated samples with PNA AOs and PMO, respectively. 5% and 1% C57 represents 100% C57 protein extracted from TA muscle was diluted in 1 in 20 or 1 in 100, respectively; α-actinin was used as a loading control.
Repeated administration of PNA AOs results in therapeutic level of dystrophin expression in mdx mice
Given the low level of exon-skipping and dystrophin expression at 50 mg/kg dose, we next examined the systemic effect of PNA AOs at higher doses. A multiple dosing regimen of 100 mg/kg for five weekly injections was applied in mdx mice. Strikingly, a uniform distribution of dystrophin-positive fibers throughout the muscle sections of most peripheral muscle groups was achieved, with the highest expression observed in gastrocnemius and quadriceps (Figure 2a). Compared with single dose of PNA AOs, significant improvement was accomplished with repeated administration, indicating a cumulative effect of PNA AOs. Consistent with the immunostaining results, more pronounced exon-skipping was detected in muscles from mdx mice treated with repeated doses than counterparts treated with single injection of PNA AOs, with ~65% exon 23 skipping was obtained in gastrocnemius (Figure 2b,c). Up to 40% of normal level of dystrophin expression was yielded in gastrocnemius and quadriceps treated with repeated injections of PNA AOs as shown by western blot (Figure 2d), suggesting a therapeutic level of dystrophin protein can be achieved in body-wide skeletal muscles with multiple administration of PNA AOs. An increase in the level of exon-skipping and dystrophin expression was also found with PNA AOs at 100 mg/kg for three weekly injections compared to single dose (see Supplementary Figure S1 online), implying that repeated injections of PNA AOs will probably yield more beneficial effects.
Figure 2.

Restoration of dystrophin expression after repeated administration of PNA AOs at 100 mg/kg in mdx mice intravenously. (a) Immunohistochemistry for dystrophin protein expression in mdx mice treated with PNA AOs at 100 mg/kg for single or five weekly injections, respectively. (b) RT-PCR to detect exon-skipping transcripts in body-wide skeletal muscles. Δexon 23 is for the exon 23 skipped band. (c) Quantitative analysis of percentage of exon 23 skipping efficiency in different muscles from mdx mice treated with single or repeated doses of PNA AOs. (d) Western blot to detect dystrophin protein expression in skeletal muscles from treated mdx mice compared with C57BL6. 5% or 10% C57 represents 100% C57 protein extracted from TA muscle was diluted in 1 in 20 or in 10, respectively; α-actinin was used as a loading control.
PNA AOs promote functional rescue of mdx mice
Since a therapeutic level of dystrophin protein was restored with repeated administration of PNA AOs at the dose of 100 mg/kg, it is crucial to investigate whether the pathological progression of mdx mice can be halted with this treatment. The dystrophin-associated protein complex (DAPC) plays a key role in maintaining the integrity of sarcolemma and will diffuse into cytoplasm in the absence of dystrophin; therefore, assessment of DAPC localization is an important parameter for functional restoration in mdx mice.28 Key components of the DAPC complex including β-dystroglycan, α-sarcoglycan, β-sarcoglycan, and neuronal nitric oxide synthase were assayed with serial immunostaining. The results indicated that the DAPC complex relocalized correctly to the sarcolemmal membrane in the presence of dystrophin (Figure 3a). Grip strength measurements, used to examine the physical improvement as mdx mice lose strength with muscle deterioration, revealed a significant increase in mdx mice treated with PNA AOs compared to untreated mdx controls (Figure 3b). Creatine kinase (CK) is an important biochemical indicator for DMD and is elevated in mdx mice.29 A significant decline in the serum CK level was observed in mdx mice treated with repeated administration of PNA AOs compared to untreated mdx mice (Figure 3c), showing that repeated administration of PNA AOs can reverse the pathological process occurring in mdx mice.
Figure 3.

Functional and phenotypic correction in mdx mice following treatments with PNA AOs at 100 mg/kg for five weekly intravenous injections. (a) Serial immunostaining of dystrophin-associated protein complex (DAPC) in mdx mice. DAPC protein components β-dystroglycan, α-sarcoglycan, and β-sarcoglycan and neuronal nitric oxide synthase (nNOS) were detected in quadriceps from mdx mice treated with PNA AOs. Arrowheads point to the identical muscle fibers (bar = 200 μm). (b) Muscle function was assessed using grip strength test to determine the physical improvement of PNA AOs treated mdx mice. Significant force recovery was detected in treated mdx mice compared with untreated mdx controls (t-test, *P < 0.05; n = 6). (c) Serum CK levels were detected to indicate muscle membrane instability in treated mdx mice compared with the untreated control group (t-test, *P < 0.05; n = 6).
Repeated administration of PNA AOs does not elicit any overt toxicity
We monitored the animals closely during the experiment and there was no phenotypic and behavioral abnormality observed and the bodyweight of treated animals increased steadily as those of untreated mdx controls (Figure 4a). To examine whether repeated administration of PNA AOs could elicit any possible toxicity, we measured the level of serum indices of liver damage including serum aspartate aminotransferase and alanine aminotransferase. The results showed a significant drop in the serum level of aspartate aminotransferase and to a less extent with aspartate aminotransferase compared with untreated mdx mice (Figure 4b), suggesting that PNA AO treatment improves the phenotype of mdx mice without any detectable toxicity. In line with the serum biochemical parameters, further routine hematoxylin and eosin staining of liver and kidney tissue sections revealed no detectable morphological changes in the mdx mice treated with PNA AOs compared with untreated mdx controls (Figure 4c). To investigate whether the administration of PNA AOs trigger any inflammatory response, we examined the presence of CD3+ and CD68+ T lymphocytes in diaphragmatic sections of treated animals and the results showed only sporadic CD3+ and CD68+ T cells were observed in cross-sections from mdx mice treated with PNA AOs (Figure 4d). Overall, these results indicated that PNA AOs did not induce any overt toxicity or activation of the immune system at the systemic dose of 100 mg/kg for five weekly injections in mdx mice.
Figure 4.

Investigation of potential drug-associated toxicity and immune activation of PNA AOs at 100 mg/kg for five weekly injections in mdx mice. (a) Assessment of bodyweight from treated mdx mice and mdx controls. (b) Measurement of serum levels of aspartate aminotransferase and alanine aminotransferase enzymes in PNA-treated mdx mice compared with untreated mdx controls. (t-test, *P < 0.05; n = 6). (c) Hematoxylin and eosin staining staining of liver and kidney tissues sections from PNA AOs treated mdx mice, untreated mdx, and C57BL6 normal controls (bar = 200 μm). (d) Detection of CD3+ T lymphocytes and CD68+ macrophages in diaphragmatic sections from treated and untreated mdx mice (bar = 100 μm). Arrowheads indicate T lymphocytes or macrophages detected by CD3 or CD68 monoclonal antibodies, respectively.
Discussion
AOs-mediated exon-skipping therapeutics shows exciting prospects for DMD patients, particularly with encouraging clinical outcomes from phase 2 clinical trials reported previously.23,24,25 However, recent studies with drisapersen failed to meet primary endpoints30; eteplirsen, another promising DMD AO drug, await further extensional studies to obtain approval from the US Food and Drug Administration. Despite the striking results with another new DNA analog (tricyclo-DNA oligomer) in body-wide muscles and brain at extremely high doses in mdx mice, its toxicological profiles and potential off-target effects due to its high affinity remain to be established.31 Therefore, it leaves more space to develop other potential AO chemistries. Previously, we evaluated PNA in mdx mice intramuscularly and demonstrated its potential for exon-skipping therapeutics.32 Here we systemically investigated PNA AOs in mdx mice at relatively high dose. The results with repeated administration of PNA AOs at the dose of 100 mg/kg confirmed the beneficial effect of PNA AOs in eliciting exon-skipping and dystrophin restoration and functional improvement without any detectable toxicity in mdx mice.
PNA AOs induced effective exon-skipping and dystrophin expression in quadriceps and gastrocnemius throughout all the dosing regimens tested, suggesting the chemical property of PNA AOs somehow facilitates its uptake in these two tissues. However, there was no dystrophin expression in heart with five repeated injections of PNA AOs at 100 mg/kg, similar to the dosing regimens with PMO and 2'Ome RNA,15,16 indicating the same barrier might also exist for PNA AOs as for 2'Ome RNA and PMO. Of note, comparable activity was achieved for PNA AOs and PMO in our systemic study under the identical dosing regimen as demonstrated intramuscularly.32 Based on this finding, we speculate that it is likely dystrophin restoration can be achieved in heart either with targeting peptides or with much higher doses of PNA AOs since dystrophin only became detectable in heart with PMO when up to 300 mg/kg dose of PMO was repeatedly utilized.17 Thus, there may be merit in PNA AOs modified with heart-targeting peptides.
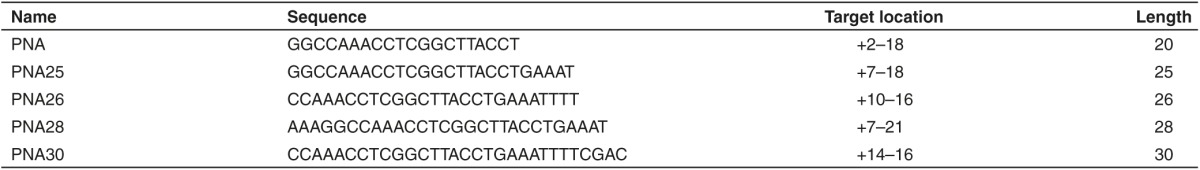
Given the length of PMO used in current clinical trials is 30-nucleotide long, we attempted to test the longest length of PNA AOs feasible with current PNA synthesis technology.24 Therefore, different lengths of PNA AOs were assessed in mdx mice intramuscularly in our current study including 26 (PNA26), 28 (PNA28), and 30 mer (PNA30) PNA AOs (Table 1). Although a dramatic increase in the number of dystrophin-positive fibers, the level of exon-skipping and dystrophin protein expression was achieved in tibialis anterior muscles from mdx mice treated with PNA30 compared to the other shorter versions (data not shown), suggesting a potentially length-dependent effect as shown previously,26 an acidity-related toxicity arose from subsequent systemic studies precluded further investigation on the systemic effect of longer PNA AOs in mdx mice. Also the possible influence of other parameters, including target locations, sequence composition and acidity-related issues, on the exon-skipping activity cannot be excluded and further detailed studies are warranted for a more definitive conclusion. Nevertheless, there is a potential with longer PNA AOs as an alternative DMD AO but its systemic evaluation and potential clinical use will depend on the improvement on the synthesis technology.
Table 1. Oligonucleotide nomenclature and sequences.
In summary, we assessed the systemic potential of PNA AOs at relatively high doses in mdx mice for the first time. Based on the findings, PNA is indeed a viable and promising chemistry for DMD exon-skipping therapeutics. Given the current purification approach for PNA AOs, 20 mer is the optimal length to use as reflected by the activity, solubility and safety in mdx mice systemically. Although longer PNA will be likely more potent, particularly PNA30, than shorter ones based on our observation from intramuscular studies, more improvement on the purification technology need to be undertaken prior to its systemic use in animals and clinical translation in man.
Materials and methods
Animals and antisense oligonucleotides. Mdx mice (6–8 weeks old) were used in all experiments (three mice in each of the test and control groups). The experiments were carried out in the animal unit (Tianjin Medical University, Tianjin, China), according to procedures authorized by the institutional ethical committee. Mice were killed by cervical dislocation at desired time-points, and muscles and other tissues were snap-frozen in liquid nitrogen-cooled isopentane and stored at −80 °C. Different lengths of PNA AOs were purchased from PNANAGENE Corporation (Daejeon, Korea) and the sequence, target location were noted in Table 1.
RNA extraction and nested RT-PCR. Sections were cut and collected into 1.5 ml Eppendorf tubes, snap-frozen in liquid nitrogen, and homogenized in Trizol reagent (Invitrogen, Carlsbad, CA). Total RNA was then extracted and 400ng of RNA template was used for a RT-PCR with OneStep RT-PCR kit (Qiagen, West Sussex, UK) as described previously.33 The primer sequences for the initial RT-PCR were Exon 20F0: 5′-CAGAATTCTGCCAATTGCTGAG-3′ and Exon 26R0: 5′-TTCTTCAGCTTTTGTGTCATCC-3′ for reverse transcription from mRNA and amplification of cDNA from exons 20–26. The cycling conditions were 95 °C for 1 minute, 55 °C for 1 minute, and 72 °C for 2 minutes for 25 cycles. The primer sequences for the second rounds were Exon 20F1: 5′-CCCAGTCTACCACCCTATCAGAGC-3′and Exon 24R1: 5′-CCTGCCTTTAAGGCTTCCTT-3′. The cycling conditions were 95 °C for 1 minute, 57 °C for 1 minute, and 72 °C for 1 minute for 25 cycles. Products were examined by electrophoresis on a 2% agarose gel. RNA extracted from tibialis anterior muscle of C57BL6 and mdx mice were used as controls.
Protein extraction and western blot. Protein extraction and western blot were carried out as previously described.33 Various amounts of protein from wild-type C57BL6 mice were used as positive controls and corresponding amounts of protein from muscles of treated or untreated mdx mice were loaded onto sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels (4% stacking, 6% resolving). The membrane was then washed and blocked with 5% skimmed milk and probed overnight with DYS1 (Abcam, Cambridge, UK) for the detection of dystrophin protein and α-actinin (Sigma, Shanghai, China) as a loading control. The bound primary antibody was detected by peroxidase-conjugated goat anti-mice IgG (Sigma, Shanghai, China) and the ECL western blot analysis system (Millipore, Billerica, MA). The intensity of the bands obtained from treated mdx muscles was measured by Image J software.
Immunohistochemistry and histology. Sections of 8 µm were cut from tibialis anterior, quadriceps, gastrocnemius, triceps, abdominal, diaphragm, and cardiac muscles. Sections were then examined for dystrophin expression with a polyclonal antibody 2166 against the dystrophin C-terminal region (the antibody was kindly provided by Professor Kay Davies, University of Oxford). Polyclonal antibodies were detected by goat antirabbit IgG Alexa Fluor 594 (Invitrogen, Carlsbad, CA). Routine hematoxylin and eosin staining was used to examine the overall liver and kidney morphology and assess the level of infiltrating mononuclear cells. The serial sections were also stained with a panel of polyclonal and monoclonal antibodies for the detection of DAPC protein components. Rabbit polyclonal antibody to neuronal nitric oxide synthase and mouse monoclonal antibodies to β-dystroglycan, α-sarcoglycan, and β-sarcoglycan were used according to manufacturer's instructions (Novocastra, Newcastle upon Tyne, UK). Polyclonal antibodies were detected by goat anti-rabbit IgGs Alexa 594 and the monoclonal antibodies by goat anti-mice IgGs Alexa 594 (Invitrogen, Carlsbad, CA). The M.O.M. blocking kit (Vector Laboratories, Burlingame, CA) was applied for the immunostaining of the DAPC.
Grip strength tests. Grip strength was assessed using grip strength meter consisting of horizontal forelimb mesh (BIOSEB, GT-31003004, Vitrolles, France). Each mouse was held 2 cm from the base of the tail, allowed to grip the metal mesh attached to the apparatus with their forepaws, and pulled gently until they released their grip. The force exerted was recorded, and five sequential tests were carried out for each mouse, averaged at 1 minute apart. Five successful forelimb strength measurements were recorded, and data were normalized to body weight and expressed as kilogram force.
Serum enzyme measurements. Mouse blood was taken immediately after cervical dislocation and centrifuged at 1,500 rpm for 10 minutes. Serum was separated and stored at −80 °C. Analysis of levels of serum creatinine kinase, aspartate aminotransferase, and alanine aminotransferase was performed by the clinical laboratory (Tianjin Huanhu Hospital, Tianjin, China).
Statistical analysis. All data are reported as mean values ± SEM. Statistical differences between treatment and control groups were evaluated by SigmaStat (Systat Software, London, UK) and the Student's t-test.
SUPPLEMENTARY MATERIAL Figure S1. Evaluation of dystrophin expression in mdx mice treated with PNA AOs at the dose of 100 mg/kg for 3 weekly intravenous injections.
Acknowledgments
The authors acknowledge Yiqi Seow (Biomedical Sciences Institutes, A*STAR, Singapore) for critical reading of the manuscript and Zhihong Shi (Tianjin Huanhu Hospital, Tianjin, China) for assistance with the clinical biochemistry assays. This work was supported by Chinese National Basic Research Program (973) (no. 2012CBA01305, 2012CB932503), National Natural Science Foundation of China (grant no. 81301526, 81361128013, and 81273420), and Action Duchenne UK.
The authors declare no conflict of interest.
Supplementary Material
Evaluation of dystrophin expression in mdx mice treated with PNA AOs at the dose of 100 mg/kg for 3 weekly intravenous injections.
References
- Hoffman, EP, Brown, RH Jr and Kunkel, LM (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928. [DOI] [PubMed] [Google Scholar]
- Bushby, K, Finkel, R, Birnkrant, DJ, Case, LE, Clemens, PR, Cripe, L et al.; DMD Care Considerations Working Group. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 9: 77–93. [DOI] [PubMed] [Google Scholar]
- Bushby, K, Finkel, R, Birnkrant, DJ, Case, LE, Clemens, PR, Cripe, L et al.; DMD Care Considerations Working Group. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 9: 177–189. [DOI] [PubMed] [Google Scholar]
- Hamada, S, Ishikawa, Y, Aoyagi, T, Ishikawa, Y, Minami, R and Bach, JR (2011). Indicators for ventilator use in Duchenne muscular dystrophy. Respir Med 105: 625–629. [DOI] [PubMed] [Google Scholar]
- Lostal, W, Kodippili, K, Yue, Y and Duan, D (2014). Full-length dystrophin reconstitution with adeno-associated viral vectors. Hum Gene Ther 25: 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, Y, Yue, Y, Liu, M, Ghosh, A, Engelhardt, JF, Chamberlain, JS et al. (2005). Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol 23: 1435–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabb, SA, Wells, DJ, Serpente, P and Dickson, G (2002). Adeno-associated virus vector gene transfer and sarcolemmal expression of a 144 kDa micro-dystrophin effectively restores the dystrophin-associated protein complex and inhibits myofibre degeneration in nude/mdx mice. Hum Mol Genet 11: 733–741. [DOI] [PubMed] [Google Scholar]
- Bostick, B, Shin, JH, Yue, Y, Wasala, NB, Lai, Y and Duan, D (2012). AAV micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in >21-m-old mdx mice, an end-stage model of Duchenne muscular dystrophy cardiomyopathy. J Mol Cell Cardiol 53: 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard, MT, Anderson, CB, Fass, U, Khatri, S, Gesteland, RF, Atkins, JF et al. (2004). Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol 55: 422–426. [DOI] [PubMed] [Google Scholar]
- Kayali, R, Ku, JM, Khitrov, G, Jung, ME, Prikhodko, O and Bertoni, C (2012). Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum Mol Genet 21: 4007–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley, J, Deconinck, N, Fisher, R, Kahn, D, Phelps, S, Gillis, JM et al. (1998). Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med 4: 1441–1444. [DOI] [PubMed] [Google Scholar]
- Perkins, KJ and Davies, KE (2002). The role of utrophin in the potential therapy of Duchenne muscular dystrophy. Neuromuscul Disord 12(suppl. 1): S78–S89. [DOI] [PubMed] [Google Scholar]
- Gillis, JM (2000). An attempt of gene therapy in Duchenne muscular dystrophy: overexpression of utrophin in transgenic mdx mice. Acta Neurol Belg 100: 146–150. [PubMed] [Google Scholar]
- Lu, QL, Mann, CJ, Lou, F, Bou-Gharios, G, Morris, GE, Xue, SA et al. (2003). Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med 9: 1009–1014. [DOI] [PubMed] [Google Scholar]
- Lu, QL, Rabinowitz, A, Chen, YC, Yokota, T, Yin, H, Alter, J et al. (2005). Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci USA 102: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter, J, Lou, F, Rabinowitz, A, Yin, H, Rosenfeld, J, Wilton, SD et al. (2006). Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med 12: 175–177. [DOI] [PubMed] [Google Scholar]
- Wu, B, Xiao, B, Cloer, C, Shaban, M, Sali, A, Lu, P et al. (2011). One-year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice. Mol Ther 19: 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus, A, Kaman, WE, Bremmer-Bout, M, Janson, AA, den Dunnen, JT, van Ommen, GJ et al. (2004). Comparative analysis of antisense oligonucleotide analogs for targeted DMD exon 46 skipping in muscle cells. Gene Ther 11: 1391–1398. [DOI] [PubMed] [Google Scholar]
- Orum, H and Wengel, J (2001). Locked nucleic acids: a promising molecular family for gene-function analysis and antisense drug development. Curr Opin Mol Ther 3: 239–243. [PubMed] [Google Scholar]
- Nielsen, PE, Egholm, M, Berg, RH and Buchardt, O (1991). Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 254: 1497–1500. [DOI] [PubMed] [Google Scholar]
- Bushby, K, Finkel, R, Wong, B, Barohn, R, Campbell, C, Comi, GP et al.; PTC124-GD-007-DMD STUDY GROUP. (2014). Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 50: 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus, A, Van Deutekom, JC, Fokkema, IF, Van Ommen, GJ and Den Dunnen, JT (2006). Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34: 135–144. [DOI] [PubMed] [Google Scholar]
- Mendell, JR, Rodino-Klapac, LR, Sahenk, Z, Roush, K, Bird, L, Lowes, LP et al.; Eteplirsen Study Group. (2013). Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 74: 637–647. [DOI] [PubMed] [Google Scholar]
- Cirak, S, Arechavala-Gomeza, V, Guglieri, M, Feng, L, Torelli, S, Anthony, K et al. (2011). Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans, NM, Tulinius, M, van den Akker, JT, Burm, BE, Ekhart, PF, Heuvelmans, N et al. (2011). Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med 364: 1513–1522. [DOI] [PubMed] [Google Scholar]
- Yin, H, Lu, Q and Wood, M (2008). Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol Ther 16: 38–45. [DOI] [PubMed] [Google Scholar]
- Cao, L, Han, G, Gu, B and Yin, H (2014). Wild-type mouse models to screen antisense oligonucleotides for exon-skipping efficacy in Duchenne muscular dystrophy. PLoS One 9: e111079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake, DJ, Weir, A, Newey, SE and Davies, KE (2002). Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 82: 291–329. [DOI] [PubMed] [Google Scholar]
- Thompson, MW, Murphy, EG and McAlpine, PJ (1967). An assessment of the creatine kinase test in the detection of carriers of Duchenne muscular dystrophy. J Pediatr 71: 82–93. [DOI] [PubMed] [Google Scholar]
- Lu, QL, Cirak, S and Partridge, T (2014). What can we learn from clinical trials of exon skipping for DMD? Mol Ther Nucleic Acids 3: e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyenvalle, A, Griffith, G, Babbs, A, El Andaloussi, S, Ezzat, K, Avril, A et al. (2015). Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med 21: 270–275. [DOI] [PubMed] [Google Scholar]
- Yin, H, Betts, C, Saleh, AF, Ivanova, GD, Lee, H, Seow, Y et al. (2010). Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol Ther 18: 819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X, Zhao, J, Han, G, Zhang, Y, Dong, X, Cao, L et al. (2014). Effective dystrophin restoration by a novel muscle-homing peptide-morpholino conjugate in dystrophin-deficient mdx mice. Mol Ther 22: 1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Evaluation of dystrophin expression in mdx mice treated with PNA AOs at the dose of 100 mg/kg for 3 weekly intravenous injections.