

Abstract
Hepatitis C virus (HCV) is the major cause of progressive liver disease such as chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Previously, we reported that a 29 nucleotide-long 2'-F pyrimidine modified RNA aptamer against the HCV nonstructural protein 5B efficiently inhibited HCV replication and suppressed HCV infectious virus particle formation in a cell culture system. In this study, we modified this aptamer through conjugation of cholesterol for in vivo availability. This cholesterol-conjugated aptamer (chol-aptamer) efficiently entered the cell and inhibited HCV RNA replication, without any alteration in gene expression profiling including innate immune response-related genes. Moreover, systemic administration of the chol-aptamer was well tolerated without any abnormalities in mice. To evaluate the pharmacokinetics of the chol-aptamer in vivo, dose proportionality, bioavailability, and pharmacokinetic parameters were evaluated by noncompartmental analyses in normal BALB/c mice. Population analysis was performed using nonlinear mixed effects modeling. Moreover, the pharmacokinetics of two different routes (intravenous, IV, versus intraperitoneal, IP) were compared. Cholesterol conjugation showed dose proportionality, extended the time that the aptamer was in the plasma, and enhanced aptamer exposure to the body. Noticeably, the IV route was more suitable than the IP route due to the chol-aptamer remaining in the plasma for a longer period of time.
Keywords: aptamer, cholesterol conjugation, HCV, NS5B, pharmacokinetics
Introduction
HCV infection affects more than 170 million people worldwide and is the source of 27% of liver cirrhosis cases and 25% of hepatocellular carcinoma cases.1,2 Until recently, HCV infection has been treated with a combination of pegylated interferon-α (PEG-IFNα) and ribavirin. However, this therapy is associated with many side effects, and also has numerous contraindications in the case of genotype 1, with only roughly 50% of patients being able to support a sustained viral response.3 In 2011, FDA approval of telaprevir (Incivek) and boceprevir (Victrelis), direct-acting antivirals targeting the HCV NS3 protease, gave confidence in conquering HCV infection, however both drugs still require combination treatment with PEG–IFNα and ribavirin, and are also prone to the appearance of drug resistance viruses.4,5 Moreover, the sale and distribution of telaprevir was discontinued in the United States by October 16th, 2014 (http://freepdfhosting.com/f75f8bac14.pdf), and boceprevir is also expected to be discontinued by December 2015 (http://www.fda.gov/downloads/drugs/drugsafety/drugshortages/ucm430818.pdf) because of available alternative treatments and the diminishing market demand. Although additional direct-acting antivirals are now on market or being developed, more specific, effective, and safe direct-acting antivirals are required.
RNA aptamers have been considered attractive alternatives to small molecule- and antibody-based drugs. RNA aptamers can be used in clinical areas due to their excellent specificity, high affinity, ease of large-scale synthesis by chemical methods, pharmaceutical amenability, and poor immunogenicity.6,7 Aptamers can be evolved by systematic evolution of ligands by exponential enrichment (SELEX), an iterative selection method, and can bind target proteins with high affinity and specificity via the formation of complementary three-dimensional structures.8,9 DNA or RNA aptamers that target HCV proteins such as NS3,10,11,12,13 NS5B,14,15,16,17,18 E1E2,19 or viral RNA20,21 have previously been reported. However, most of these have only been tested in a cell culture system and were not tested in animal models. Previously, we have shown that a 29 nucleotide length 2'-F modified RNA aptamer against the HCV non-structural protein 5B (NS5B) could efficiently inhibit HCV replication of both 1b and 2a genotypes, and suppress HCV infectious virus particle formation without the generation of escape mutant viruses and cellular toxicity in a cell culture system.18 Moreover, we have delivered a therapeutically amenable amount of the aptamer in vivo, to liver tissue in mice.18 However, it should be examined whether the aptamer has the desired pharmacokinetic properties for its pharmaceutical application.
There are three major hurdles of drug metabolism and pharmacokinetics (DMPK) in the systemic administration of oligonucleotides including aptamers, such as metabolic instability, rapid renal filtration and elimination of non–protein-bound oligonucleotides, and rapid biodistribution from the plasma compartment into the tissues.22 To overcome these limitations of DMPK, the aptamer should be modified and optimized by various in vitro and in vivo methods, and pharmacokinetic analysis is important to accomplish these modifications and optimization.23 Pharmacokinetic analysis is essential to describe the movement and action of a drug in the body compartments such as blood, interstitial bodily fluid, and tissue/organs. For instance, noncompartmental analysis for pharmacokinetics can provide drug exposure, bioavailability, and total clearance, and compartmental analysis can describe and predict the concentration–time relationship using kinetic models.24
In the current work, we examine the ability of a cholesterol-conjugated 29 nucleotide-long 2'-F modified aptamer against HCV NS5B on internalization and inhibition of HCV replication in a cell culture system. In addition, using microarray, we investigate whether any undesirable alteration in the gene expression pattern may be induced by the treatment of the conjugated aptamer. Moreover, we assess the toxicity of the conjugated aptamer by a dose escalation study and 100 mg·kg−1 intravenous injection of the aptamer twice a day for 2 weeks in a chimeric mouse with a human liver (PXB-mouse). Finally, we evaluate the pharmacokinetics of this cholesterol-conjugated aptamer by noncompartmental and population analyses, and also compare intravenous (IV) with intraperitoneal (IP) administration in normal BALB/c mice.
Results
A cholesterol-conjugated aptamer efficiently internalized and inhibited HCV replication
To develop RNA aptamer against HCV NS5B18 as a therapeutic agent, it is important to achieve intracellular delivery to target organs and tissues using modifications that would allow the aptamer to persist in the body for prolonged systemic exposure. To this end, we conjugated a cholesterol moiety at the 5' end of the 2'-F modified 29 nucleotide long RNA aptamer against the HCV NS5B protein, since cholesterol conjugation of oligonucleotide molecules has been reported to increase the plasma half-life through association with plasma lipoproteins, and to promote hepatic cell uptake via receptor-mediated endocytosis.25,26,27 Moreover, we further modified this aptamer by attaching inverted d(T) to the 3' end in order to increase the stability of the aptamer through the prevention of degradation by cellular RNases28 (Figure 1a). Cholesterol and inverted d(T)-conjugated aptamers (chol-aptamers) were internalized into HCV 1b subgenomic replicon cells dose-dependently, and ~35 pg chol-aptamers (corresponding to ~2.1 × 107 aptamer copies) were detected per 1 μg total RNA at a concentration of 4 μmol/l chol-aptamer in treated HCV-replicon cells (Figure 1b). Huh-7 and HCV-replicon cells were treated with 4 μmol/l chol-aptamer to directly visualize internalized chol-aptamers through in situ hybridization (Figure 1c). Although not quantitatively measured, the chol-aptamer signal was readily detected in both cell lines, and more chol-aptamer was detected in HCV-replicon cells than in naive Huh-7 cells (Figure 1c, compare line 2 and 4). Moreover, the HCV NS5A protein signal was significantly reduced in the HCV-replicon cells showing chol-aptamer signal (Figure 1c, compare line 3 and 4). Of note, in contrast with no effects by the mutant chol-aptamers as control, chol-aptamers efficiently inhibited the replication of the HCV type 1b subgenomic replicon dose-dependently (Figure 1d) by up to ~80% at a concentration of 4 μmol/l. Taken together, these data suggest that chol-aptamers can efficiently internalize into HCV-replicon cells and inhibit HCV replication.
Figure 1.

Chol-aptamers efficiently internalize into Huh-7 and hepatitis C virus (HCV) replicon cells and inhibit HCV replication. (a) Structure of the cholesterol and inverted d(T)-conjugated aptamer. (b) Internalized chol-aptamers were detected using qRT-PCR. Averages of three independent measurements are shown with SD. (c) Huh-7 and HCV-replicon cells were incubated with 4 μmol/l chol-aptamer for 48 hours. Cells were then fixed and the chol-aptamers were detected using a PNA probe, and the HCV NS5A proteins were visualized with an anti-NS5A monoclonal antibody. (d) HCV subgenomic RNA levels in the chol-aptamer- or mutant (mt) chol-aptamer-treated cells were analyzed using qRT-PCR and expressed relative to the level in untreated cells. Averages of 3 independent measurements are shown with SD. *P < 0.05; **P < 0.01; ***P < 0.002.
Chol-aptamers do not induce cellular toxicity
To determine whether treatment of cells with the chol-aptamer induces unwanted changes in the global gene expression pattern, we studied the gene expression profile using two independent sets of oligonucleotide microarray experiments (Figure 2). Changes in the mRNA levels were analyzed by comparing cholesterol-, aptamer-, and chol-aptamer-treated samples with control (saline treated) samples. To find mRNAs that could be specifically affected by the chol-aptamer, we compared chol-aptamer-treated samples with cholesterol- or aptamer-treated samples, each RNA level of which was normalized to the mock-treated sample. When compared with cholesterol- or aptamer-treated samples, the chol-aptamer-treated sample did not induce any notable changes in the gene expression profile (Figure 2a–c). Furthermore, we could not observe any innate immune-related genes showing ≥2-fold changes compared with cholesterol-treated samples when using a P < 0.05 data set. Moreover, we validated microarray data using quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analysis of innate immune genes such as IFNA-2, IFNB, TLR-3, MDA5, RIG-I, PKR, OAS-1, and MX-1 (data not shown).18 Coincidently with the microarray data, the chol-aptamer did not affect cell viability up to a concentration of 5 mg/ml, compared to poly (I:C) transfection which induces innate immune gene response18 and thereof affects cell proliferation29 (Figure 2d). Consequently, these data suggest that the chol-aptamer neither induced any change in the cellular gene expression profile including innate immune-related genes nor in cell viability.
Figure 2.

Chol-aptamer effect on gene expression response and cell viability. Scatter plot analysis of normalized microarray expression data from Huh-7 cells treated with cholesterol, aptamer, or chol-aptamer (a–c). (d) Huh-7 cells were treated with increasing amounts of poly (I:C), cholesterol, or chol-aptamer. Two days later, a cell viability test was performed using Celltiter 96 Aqueous one solution reagent (Promega) and estimated by assessing the absorbance at 490 nm.
In vivo toxicity test of the chol-aptamer
To test the tolerability of the chol-aptamer in an in vivo model, we used a uPA+/+/SCID-based chimeric mouse with a healthy and functional liver that has been 70–90 percent repopulated by human hepatocytes (PXB-mice).30 Chol-aptamers were intravenously administered twice a day with an exponentially-increasing injection dose for 7 days (Figure 3a, Upper). During injection periods, there were no abnormalities in the general condition of the mice, such as appearance, activity, body temperature, and perianal region, and no apparent change was observed in body weight (Figure 3a, Bottom, and Supplementary Table S1). When 100 mg·kg−1 chol-aptamer were intravenously injected twice a day for 2 weeks (Figure 3b, Upper), we also did not observe any abnormalities in the general condition of the mice or a change in body weight (Figure 3b, Bottom, and Supplementary Table S2). These results, including the in vitro toxicity data, suggest that the chol-aptamer has suitable properties for in vivo application in respect to in vivo safety.
Figure 3.

In vivo tolerability test of chol-aptamers in PXB-mice. (a) To determine the tolerability of chol-aptamers, mice were intravenously injected twice a day (BID) with an exponentially-increasing dose for 7 days. The group composition and injection schedule is indicated (Upper). The first day of administration was set as day 0. At each administration, individual body weights were examined (Bottom). (b) To evaluate the tolerability of the chol-aptamers, mice were intravenously injected with 100 mg·kg−1 chol-aptamer twice a day for 2 weeks. The group composition and injection schedule is indicated (Upper). The first day of administration was set as day 0. At each administration, individual body weights were also observed (Bottom). Numbers 101 to 105 indicated individual mouse.
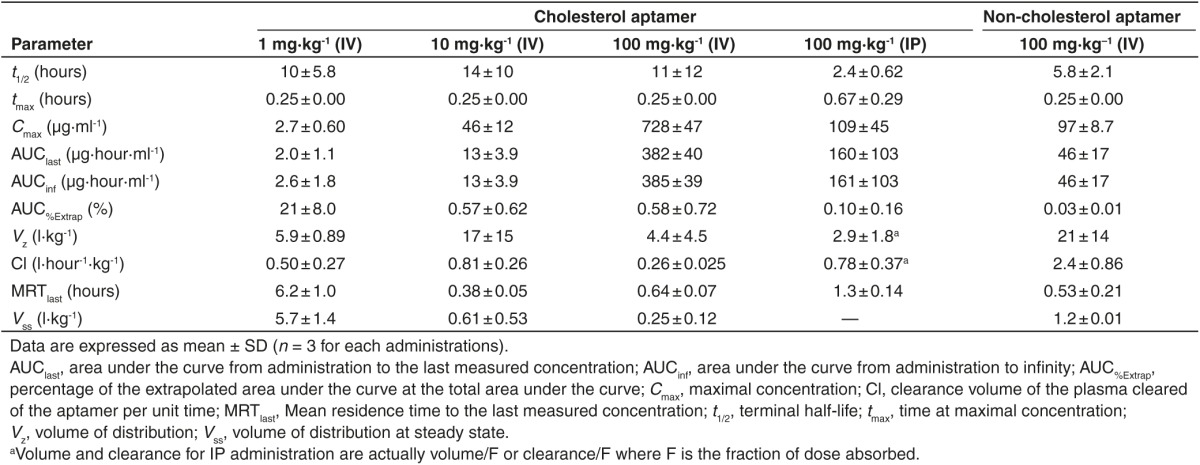
Noncompartmental pharmacokinetic analyses
Since PXB-mice have abnormal cholesterol and lipoprotein levels in their blood compared with normal mice (data not shown), they are not appropriate for studying pharmacokinetics of cholesterol-conjugated oligonucleotides, which use plasma lipoproteins to increase the plasma half-life and for hepatic cell uptake.25,26,27 Therefore, in order to evaluate in vivo pharmacokinetic properties of the chol-aptamer, 1, 10, and 100 mg·kg−1 chol-aptamer, or 100 mg·kg−1 non-cholesterol-conjugated aptamer were administered to male BALB/c mice intravenously through the tail vein or 100 mg·kg−1 chol-aptamer were intraperitoneally injected. Using the destructive sample technique, venous blood samples were collected immediately before administration (0 hour) and at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours after administration. Figure 4a,b shows the average plasma concentration–time profiles for the chol-aptamer after IV and IP administration in mice. Table 1 shows the pharmacokinetic parameters of the aptamer that were calculated by noncompartmental methods. In the linear regression between the log transformed total dose and AUCinf, the slope was 1.103 (95% CI = 0.8528–1.354) for the chol-aptamer IV group. As shown in Figure 4c, the 95% CIs of the slopes were close to 1, suggesting dose proportionality. These linear pharmacokinetics were also demonstrated by finding that the areas under the plasma concentration–time curves from time 0 to infinity, normalized by dose (AUCinf/dose) for the dose of 1, 10, and 100 mg·kg−1 chol-aptamer, were 2.6 ± 1.8, 1.3 ± 0.39, and 3.9 ± 0.39 hour·μg·ml−1·mg−1, respectively (P = 0.072). The Tmax after IV administration was 0.25 hours with both cholesterol-conjugated and non-cholesterol-conjugated administration, however the Tmax after IP administration (0.67 ± 0.29 hours) increased 2.7 times relative to IV. The Cmax after IV administration was 6.7 times higher than after IP administration (728 ± 47 versus 109 ± 45 μg·ml−1), and the chol-aptamer showed a seven to eight times higher Cmax than the non-cholesterol-conjugated aptamer (728 ± 47 versus 97 ± 8.7 μg·ml−1). The half-lives after IV administration of the chol-aptamers were about 10 to 14 hours, and half-life of non-cholesterol-conjugated aptamer (5.8 ± 2.1 hours) was approximately half of that of the chol-aptamer. Interestingly, the half-life after IV administration was approximately five to six times longer than after IP administration (2.4 ± 0.62 hours). AUClast after IV administration (382 ± 40 hour·μg·ml−1) was two to three times larger than after IP administration (160 ± 103 hour·μg·ml−1), indicating that drug exposure to the body was two to three times larger when the aptamer was administered by IV route, compared with IP route. AUClast of the chol-aptamer was eight times larger than that of the non-cholesterol-conjugated aptamer (Table 1). Relative bioavailability of non-cholesterol-conjugated administration over cholesterol-conjugated administration was 8.51% (Figure 4a). The absolute bioavailability of the aptamer was calculated as 40.52% for IP administration (Figure 4b).
Figure 4.

Noncompartmental pharmacokinetic analyses of the chol-aptamer. (a) The time course of plasma aptamer concentrations following IV administration at a dose of 1, 10, and 100 mg kg−1 chol-aptamers (Chol), or 100 mg kg−1 non-cholesterol aptamer (Non-chol). (b) The time course of plasma aptamer concentrations following IP administration at a dose of 100 mg·kg−1 chol-aptamer was compared with IV administration. Data are presented as the mean ± SD (n = 3 for each administration). (c) The relationship between the area under the curve from administration to infinity (AUCinf; µg·hour·ml−1) and the total dose (mg) of the chol-aptamer after log transformation. CI, confidence interval.
Table 1. Noncompartmental pharmacokinetic parameters of cholesterol and non-cholesterol aptamer following intravenous (IV) or intraperitoneal (IP) administration in mice.
Population pharmacokinetic analyses for the chol-aptamer
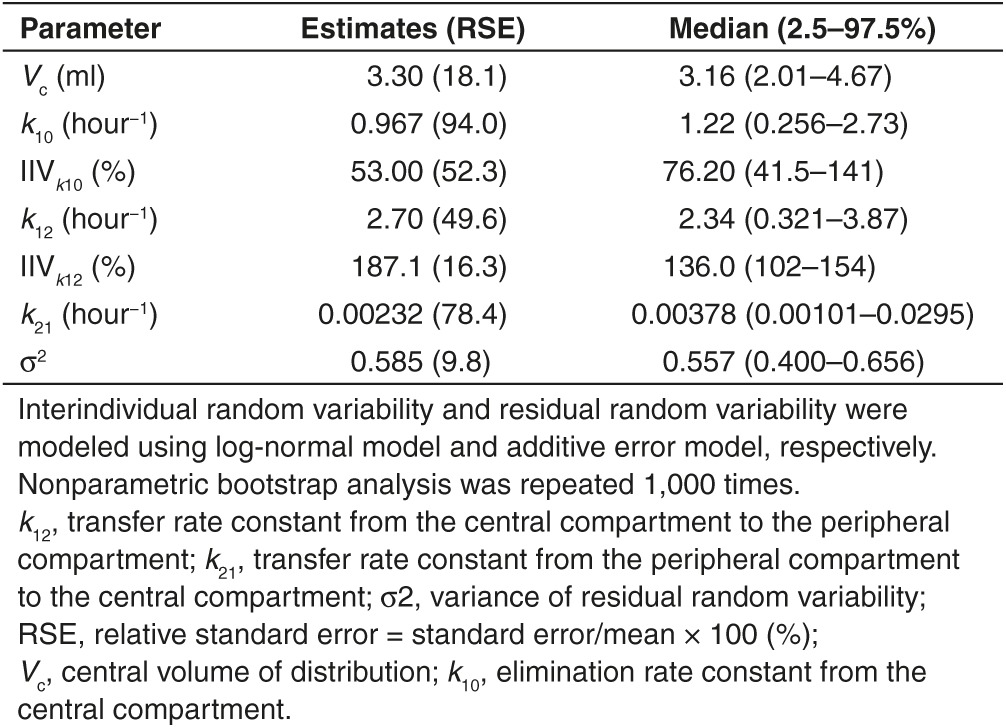
The pharmacokinetic characteristics of the chol-aptamer were best characterized using a two-compartment linear model (Figure 5a). Goodness-of-fit and conditional weighted residuals are depicted in Figure 5b,c, respectively. Population pharmacokinetic parameter estimates and median parameter values (2.5–97.5%) of the nonparametric bootstrap replicates of the final models for the chol-aptamer are summarized in Table 2. The dose was determined based on the body weight of each individual. The inclusion of body weight as a covariate did not contribute additional information to explain pharmacokinetic variability based on improvement of the objective function value in hierarchical models, model convergence, and diagnostic graphics. Therefore, no covariate was included in the final population pharmacokinetic model. Importantly, the available data for this investigation contained a relatively small number of subjects and a limited age range, thus, formal hypothesis (significance) testing for covariate effects did not find any significant covariates. The percentage of data distributed outside the 95% prediction intervals of the predictive check is 1.39%, and the visual predictive check plots are shown in Figure 5d.
Figure 5.

Population pharmacokinetic analyses of the chol-aptamer. (a) Diagram of the population pharmacokinetic model of the RNA aptamer. IV, intravenous administration; A(1), drug amount in the central compartment; A(2), drug amount in the peripheral compartment; k10, elimination rate constant from the central compartment; k12, transfer rate constant from the central compartment to the peripheral compartment; k21, transfer rate constant from the peripheral compartment to the central compartment. (b) Goodness-of-fit plots for the final model. Observed (LNDV, natural log of data) versus population predicted (PRED) concentrations (left panel). LNDV versus individual predicted (IPRED) concentrations (right panel). Dashed line is a line of identity. (c) The conditional weighted residuals (CWRES) as a function of time for the final population pharmacokinetic models of the chol-aptamer. The dashed line is a line of identity. (d) Predictive checks of the aptamer in the plasma based on 1,000 datasets for IV administration. A small portion of data (1.39%) was distributed outside the 95% prediction intervals, indicating that the final pharmacokinetic model for both formulations were adequate to describe the time-courses of the aptamer plasma concentrations. The gray filled area represents the model's 95% prediction interval.
Table 2. Population pharmacokinetic parameter estimates, interindividual variability (IIV, %CV), and median parameter values (2.5–97.5%) of the nonparametric bootstrap replicates of the final pharmacokinetic model of the chol-aptamer after intravenous administration.
Discussion
In the present study, we chemically conjugated cholesterol and inverted d(T) to the previously described 29 nucleotide, 2'-F-modified aptamer against HCV NS5B18 for in vivo application, and characterized its pharmaceutical properties. The modified aptamers are efficiently internalized and inhibit genotype 1b HCV subgenomic RNA replication in human liver cells (Figure 1), without induction of cellular toxicity or alteration of gene expression profiling (Figure 2). Similar to the cell culture system, chol-aptamers do not induce any abnormalities in mice intravenously injected twice-daily using the following two protocols: (i) starting with 0.5 mg·kg−1, the administration dose of chol-aptamer was exponentially increased for 7 days (Figure 3a); (ii) 100 mg·kg−1 chol-aptamer was administered for 2 weeks (Figure 3b). These results strongly suggest that the cholesterol-conjugated aptamers inhibit HCV replication without nonspecific binding to cellular proteins or induction of innate immunity.
In order to be used as a therapeutic agent, the aptamer should have characteristics such as remaining in the plasma compartment for an extended period of time. Furthermore, the aptamer must be modified to increase metabolic stability, slow distribution from the central compartment, and slow renal filtration and elimination.23 In the present study, we evaluated the pharmacokinetics of the cholesterol-conjugated aptamer against HCV NS5B using noncompartmental and population pharmacokinetics. The difference in pharmacokinetics between the IV and IP route was compared, and the efficiency of cholesterol-conjugation of the aptamer was evaluated by comparison to a nonconjugated aptamer. The half-life of the cholesterol-conjugated aptamer was longer than that of the non-cholesterol-conjugated aptamer, and in accordance, cholesterol conjugation reduced the clearance (cholesterol-conjugated versus non-cholesterol-conjugated, 0.26 ± 0.025 versus 2.4 ± 0.86 l·hour−1·kg−1) by appoximately nine times (Figure 4 and Table 1). Steady state refers to the dynamic equilibrium between the overall intake and elimination of a drug after regular dosing.24 The steady-state concentration of antisense oligonucleotides can be predicted on the basis of half-life only, and the steady state or near steady-state concentration can be reached when three to five times the half-life is passed after administration.22 Here, the predicted time to attain the steady state was approximately 33–55 and 17–29 hours for the cholesterol-conjugated and non-cholesterol-conjugated aptamer, respectively, according to the half-lives of each aptamer after IV administration of 100 mg·kg−1 (cholesterol-conjugated versus non-cholesterol-conjugated, 11 ± 12 versus 5.8 ± 2.1 hours). It is thought that cholesterol conjugation of an aptamer can induce delay in the time taken to reach a steady state, but it can also extend the time that the aptamer remains in the plasma, shown here by the cholesterol-conjugated aptamer having a longer half-life (cholesterol-conjugated versus non-cholesterol-conjugated, 11 ± 12 versus 5.8 ± 2.1 hours) and less clearance (cholesterol-conjugated versus non-cholesterol-conjugated, 0.26 ± 0.025 versus 2.4 ± 0.86 l·hour−1·kg−1) than the non-cholesterol-conjugated aptamer after IV administration of 100 mg·kg−1. In addition, the cholesterol-conjugated aptamer showed fast k12 (2.7 hour−1) and slow k21 (0.0023 hour−1) population pharmacokinetics, which strongly suggests that the aptamer is likely slowly released once it is rapidly transferred from the central compartment (e.g., plasma) to peripheral compartments (e.g., tissue and organ) (Figure 5 and Table 2). According to the comparison of AUClast between the cholesterol-conjugated (382 ± 40 μg·hour·ml−1) and non-cholesterol-conjugated aptamer (46 ± 17 μg·hour·ml−1), and the relative bioavailability of the non-cholesterol-conjugated aptamer over the cholesterol-conjugated aptamer (8.51%), cholesterol conjugation enhanced aptamer exposure to the body (Table 1).
Dose proportionality is a desirable property as it makes predicting the effects of dose adjustments easier.31 It may be mainly affected by dose range, but not by species or size.32 The cholesterol-conjugated aptamer showed dose proportionality in the ANOVA of dose-normalized AUCinf values and the power model, meaning that increases in the administered dose of the aptamer are accompanied by proportional increases in the measurements of exposure. In agreement, the Cmax increased approximately 16 times according to a 10 times increase in the IV dose of the cholesterol-conjugated aptamer (2.7 ± 0.60, 46 ± 12, and 728 ± 47 hour·μg·ml−1 for 1, 10, and 100 mg·kg−1, respectively) (Figure 4 and Table 1).
As mentioned above, the dosing method, which can result in the plasma concentration remaining for an extended period of time, may be beneficial in aptamer administration. Several studies have described pharmacokinetic characteristics of aptamers by nonintravenous routes such as subcutaneous, intravitreal, and IP administration.33,34,35,36 In general, drugs administered by non-intravenous route have a longer half-life than that by IV administration.37 Drugs administered by subcutaneous route show lower plasma concentrations and shorter elimination time than IV drugs because of slow absorption rate from the administered site and low bioavailability,23 and IP administration is regarded as the same with similar reasons. However, aptamer has longer half-life when given by IV versus subcutaneous36 or IP administration.34 Moreover, here, IV administration of the cholesterol-conjugated aptamer showed a longer half-life and a smaller clearance than IP administration (Table 1). IV administration route extended the remaining time of the aptamer in plasma and IP route has smaller aptamer exposure to the body regarding the smaller AUClast and bioavailability. Therefore, we can hypothesize that a cholesterol conjugated-aptamer should be administered via intravenous route to ensure it remains in the plasma compartment for an extended period of time.
In conclusion, we have demonstrated the pharmacokinetic properties of a cholesterol-conjugated aptamer using a two-compartment model, and have shown the dose proportionality. Cholesterol conjugation may extend the time that the aptamer remains in the plasma, thus enhancing aptamer exposure to the body. In addition, an IV administration route of a cholesterol-conjugated aptamer is more suitable than an IP route, due to the aptamer remaining in the plasma for a longer period time. Therefore, cholesterol-conjugated RNA aptamer against the HCV NS5B could be pharmaceutically useful for in vivo therapeutic purposes especially via IV administration route.
Materials and Methods
Cells. Huh-7 human hepatoma cells were cultured in Dulbecco's modified Eagle medium (HyClone, Thermo Fisher Scientific, South Logan, UT) with 10% fetal bovine serum. For the generation of a HCV replicon cell line, the HCV genotype 1b subgenomic replicon construct, pFKI389neo/NS3–3'/5.1, carrying two cell-culture adaptive mutations in NS3 and one in NS5A,38 was used. HCV replicon RNA was synthesized by in vitro transcription with the AseI and ScaI-digested replicon plasmid, and used to transfect Huh-7 cells as described previously.10,39 The HCV 1b subgenomic replicon cell line was generated using previously-described methods.40
Chemical synthesis of the aptamer. The chemically synthesized cholesterol- and inverted d(T)-conjugated HCV NS5B aptamer was purchased from ST Pharm Co., LTD (Seoul, Korea). The sequence of the aptamer is 5′-UUGAACGAUUGGUAGUAGAAUAUCGUCAG-3′, where 2′-deoxy-2′-fluoropyrimidine nucleotides are denoted by italics. The sequence of the mutant aptamer is 5′-UUGAACGAUUAAGAUGAUGGUAUCGUCAG-3′, where italics indicate 2′-deoxy-2′-fluoropyrimidine nucleotides.
Real-time PCR. Real-time PCR was performed as previously described.41 For the analysis of the HCV replicon RNA in the aptamer- or mutant aptamer-administered HCV 1b replicon cells, total RNA was isolated and reverse transcribed with a 3' primer specific for the negative strand of HCV cDNA (5′-CGTAACACCAACGGGCGCGCCATG) or a random primer for 18S cDNA. The resultant cDNAs were subsequently amplified using a real-time PCR premix (Solgent, Daejeon, South Korea) with primers specific for the neomycin resistance marker gene of the HCV replicon construct. PCR was performed using the Rotor-Gene (Corbett, Valencia, CA) and the SYBR green PCR method. The threshold levels obtained from the HCV assay were adjusted to the threshold levels found in the 18S PCR to correct for minor variations in cDNA loading. For the detection of the chol-aptamer, total RNA was reverse transcribed with the 5′-GTGTCGGTGTGTGTGTGTGTGT-3′ reverse primer using MultiScribe Reverse Transcriptase (Applied Biosystems, Carlsbad, CA) according to the manufacturer's protocol. PCR was performed using the Rotor-Gene (Corbett) and KAPA SYBR FAST Universal 2× qPCR Master Mix (#KK4602; Kapa Biosystems, Woburn, MA) with the 5′-TTGAACGATTGGTAGT-3′ (italics indicate LNA nucleotides) forward primer and the aforementioned reverse primer.
Fluorescence in situ hybridization. In situ hybridization was performed as previously described42 with some modifications. Briefly, HCV-replicon or Huh-7 cells (104 per chamber) were seeded onto Lab-Tek II chamber slides (Nunc, part of Thermo Scientific, Rochester, NY). The following day, 4 μmol/l chol-aptamer was added to the HCV-replicon or naive Huh-7 cells. After 48 hours, the cells were fixed with 4% paraformaldehyde solution for 15 minutes at room temperature. The slides were washed twice with DEPC-PBS for 3 minutes and PBS containing 0.5% tween-20 solution was added for 10 minutes at room temperature to permeabilize the cells. After washing twice with PBS for 3 minutes, hybridization buffer {50% (vol/vol) formamide, 5× SSC, 500 μg/μl yeast tRNA, and 1× Denhardt's solution in DEPC-treated water} was added to the slides and incubated for 30 minutes at 48 °C in a humidified chamber. The hybridization buffer was removed and fresh hybridization buffer containing 5 pmoles biotin-conjugated PNA probe (Panagene, Daejeon, Korea) was added and incubated for 1 hour at 48 °C in a humidified chamber. The slides were then washed three times with 0.1× SSC solution at 52 °C for 10 minutes and once for 5 minutes with 2× SSC at room temperature. After washing, the slides were treated with 3% (vol/vol) H2O2 to block endogenous peroxidases, washed three times for 5 minutes per wash with TNT buffer {0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, 0.5% (vol/vol) tween-20}, and blocked with TNB buffer {0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, 0.5% (wt/vol) blocking reagent, 0.5% (wt/vol) BSA} for 1 hour at room temperature. To simultaneously visualize the chol-aptamer and the NS5A HCV protein, an anti-NS5A (#MAB8694; Millipore, Billerica, MA) antibody was added for 1 hour at room temperature. The slides were then washed with TNT buffer three times for 5 minutes per wash and treated with Alexa Fluor 488 donkey anti-mouse IgG (H+L) (#A-21202, Invitrogen, Carlsbad, CA) for 30 minutes. To amplify the aptamer signals, slides were treated with the TSA BIOTIN SYSTEM (#NEL704, PerkinElmer, Santa Clara, CA), mounted with a mounting solution (Slowfade gold antifade reagent with DAPI, Invitrogen), and observed using a fluorescent microscope (Carl Zeiss, Thornwood, NY).
Microarray analysis. Huh-7 cells were seeded in 100 mm plates at a density of 2 × 106 cells/plate. The following day, 4 μmol/l cholesterol, aptamer, or chol-apatmer was added for 48 hours. The total RNA was then purified, amplified, and labeled with Cy3-dCTP (Mock-treated sample) or Cy5-dCTP (cholesterol, aptamer, or chol-aptamer-treated sample) using an Agilent Low RNA Input Linear Amplification kit PLUS, according to the manufacturer's protocol. Cy3- or Cy5-labeled cDNA was hybridized to the Agilent Human Whole Genome 4x44K Microarray kit (Agilent, Santa Clara, CA). Scanning and image analysis was performed using the Agilent DNA microarray scanner and Feature Extraction software. The data was analyzed using the Agilent Gene Spring software.
Cell toxicity assay. Huh-7 cells were seeded in 96-well plates at a density of 2 × 104 cells/well and incubated overnight at 37 °C. Triplicate wells were treated with cholesterol, chol-aptamer (range from 51.2 pg to 500 μg/ml) or transfected with poly (I:C) (range from 100 pg to 10 μg/ml) (Invivogen, San Diego, CA) using lipofectamin 2000 (Invitrogen). Cell toxicity assays were performed 48 hours after aptamer treatment using Celltiter 96 Aqueous one solution reagent (Promega, Madison, WI) according to manufacturer's instruction. Cell viability was estimated by assessing the absorbance at 490 nm. All experiments were performed in triplicate.
Animal toxicity assay. The animal toxicity assay was performed with SCID-Alb/uPA mice that were transplanted with human hepatocytes. All experiments were carried out by PhonixBio (Higashi-Hiroshima, Japan)
Animals. The present study was reviewed and approved (No. 2010-14-033) by the Institutional Animal Care and Use Committee (IACUC) of the Asan Institute for Life Sciences, Asan Medical Center (Seoul, Korea). The committee abides by the dictates of the Institute of Laboratory Animal Resources guide.43 Eighteen-week-old male BALB/C mice (weight: mean ± SD, 25.55 ± 0.65 g; range 24.2–26.8 g) were used in the study, and obtained from Orient Bio (Sungnam, Korea). Mice were allowed to acclimate to their new environment for at least 1 week before study initiation. Mice were housed with a controlled light-dark cycle (light on between 8:00 AM and 8:00 PM), an ambient temperature of 21–22 °C, and unlimited access to standard laboratory mouse diet and water.
Aptamer administration, blood sample acquisition, and measurement of aptamer concentration in animals. The cholesterol-conjugated aptamer (1, 10, and 100 mg·kg−1, n = 3 at each dose) was administered intravenously through the tail vein of nine mice, intraperitoneally (100 mg·kg−1) in three mice, and the non-cholesterol-conjugated aptamer (100 mg·kg−1) was administered intravenously in three mice. Venous blood samples were collected immediately before administration (0 hour) and at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours after administration. Samples were collected in tubes coated with ethylenediaminetetraacetic acid (BD microcontainer tubes with K2EDTA, BD Biosciences, San Jose, CA) and centrifuged for 30 minutes at 252× g. The plasma was stored at −80 °C until the assay. For the detection of aptamers in mouse blood, 1 μl plasma was directly reverse transcribed using MultiScribe reverse transcriptase (Applied Biosystems, Carlsbad, CA) according to the manufacturer's protocol. The aptamer concentrations in the plasma samples were then analyzed by real-time quantitative PCR analysis using the Rotor-Gene (Corbett) and Kapa SYBR fast universal 2× qPCR master mix (Kapa Biosystems) as previously mentioned.
Noncompartmental analyses of pharmacokinetics. The pharmacokinetic parameters were calculated by noncompartmental methods (WinNonlin Professional 5.2; Pharsight Corporation, Mountain View, CA). The area under the curve from the time of administration to the last measured concentration (AUClast) was estimated by linear trapezoidal integration (linear interpolation). The area under the curve from administration to infinity (AUCinf) was calculated as the sum of AUClast + Clast/λZ, in which Clast is the last measured concentration and λZ is the apparent terminal rate constant estimated by unweighted linear regression for the linear portion of the terminal log concentration–time curve. The half-life of the terminal phase (t1/2) was calculated using t1/2 = 0.693/λz. The maximal concentration (Cmax) and the time to reach Cmax (tmax) after administration of the aptamer were determined from the observed data. Summary statistics were determined for each parameter. To evaluate dose proportionality, dose-normalized AUCinf values of each cholesterol-conjugated aptamer IV administered group were compared using one-way analysis of variance.31 If the differences among the cholesterol-conjugated aptamer IV administered groups are statistically insignificant, we are able to conclude that pharmacokinetics showed dose proportionality. In addition, we used a power model and a confidence interval (CI) criteria approach as follows31:
where dose proportionality implies that β1 = 1 in equation (1) and PK denotes a pharmacokinetic variable (AUCinf in this study).
Absolute bioavailability (F) of IP administration of the cholesterol-conjugated aptamer was calculated from the ratio of the areas under the plasma aptamer concentration curve after IP and IV administration, respectively, indexed to their respective dose:
Relative bioavailability of the non-cholesterol-conjugated aptamer administration over the cholesterol-conjugated aptamer administration was calculated from the ratio of the areas under the plasma aptamer concentration curve after non-cholesterol-conjugated and cholesterol-conjugated aptamer administration, and indexed to their respective dose:
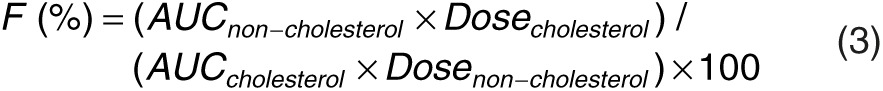 |
Population pharmacokinetic analyses for the cholesterol-conjugated aptamer. A total of 72 plasma concentration data of the cholesterol-conjugated aptamer from nine mice, which was administered intravenously, were analyzed by mixed effects modeling using NONMEM 7.3.0 (ICON Development Solutions, Dublin, Ireland). The pharmacokinetic parameters were estimated with NONMEM subroutine ADVAN3 using the first order conditional estimation method with interaction terms. The parameters for a specific subject were described by equation (4):
 |
where PTV is the typical value of the parameter and ηi is a normally distributed variable with zero-mean. The residual error model was characterized by the additive error model as described by equation (5):
where Cobs and Cpred are the observed and predicted plasma concentrations of the aptamer, respectively, and ɛ is a normally distributed variable with zero-mean.
Various compartmental and error models were assessed and guided by a graphical assessment of optimum fit properties and statistical significance criteria. A p-value of 0.05, representing a decrease in objective function value of 3.84 points, was considered statistically significant (chi-square distribution, degrees of freedom = 1). Standard diagnostic plots including the observed values of the dependent variable versus the individual predicted values or the population predicted values and the individual weighted residuals or conditional weighted residuals versus time were used for the diagnosis of optimum fit capabilities. Body weight was considered as a covariate, and covariate screening was performed. The R software (version 3.0.3; R Foundation for Statistical Computing, Vienna, Austria) was used for graphical model diagnostics.
For the final pharmacokinetic model, a nonparametric bootstrap analysis was performed as an internal model validation method, using the software package Wings for NONMEM (WfN; Nick Holford, Version 733, Auckland, New Zealand). Briefly, 1,000 bootstrap replicates were generated by random sampling from the original data set with replacement. The final model parameter estimates were compared with the median parameter values and the 2.5–97.5 percentiles of the nonparametric bootstrap replicates from the final model. Visual predictive check was performed by simulating 1,000 iterations and by comparing the simulated prediction interval to the original data.
SUPPLEMENTARY MATERIAL Table S1. In vivo tolerability test of chol-aptamers in PXB-mice related to Figure 3a. Table S2. In vivo tolerability test of chol-aptamers in PXB-mice related to Figure 3b.
Acknowledgments
This study was supported by grants from the National Research Foundation of Korea by the Ministry of Science, ICT and Future Planning (No. 2012M3A9B6055200).
Supplementary Material
References
- Alter, MJ (2007). Epidemiology of hepatitis C virus infection. World J Gastroenterol 13: 2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levrero, M (2006). Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 25: 3834–3847. [DOI] [PubMed] [Google Scholar]
- Fried, MW, Shiffman, ML, Reddy, KR, Smith, C, Marinos, G, Gonçales, FL Jr et al. (2002). Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 347: 975–982. [DOI] [PubMed] [Google Scholar]
- Kieffer, TL, Kwong, AD and Picchio, GR (2010). Viral resistance to specifically targeted antiviral therapies for hepatitis C (STAT-Cs). J Antimicrob Chemother 65: 202–212. [DOI] [PubMed] [Google Scholar]
- Kieffer, TL, Sarrazin, C, Miller, JS, Welker, MW, Forestier, N, Reesink, HW et al. (2007). Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology 46: 631–639. [DOI] [PubMed] [Google Scholar]
- Kaur, G and Roy, I (2008). Therapeutic applications of aptamers. Expert Opin Investig Drugs 17: 43–60. [DOI] [PubMed] [Google Scholar]
- Que-Gewirth, NS and Sullenger, BA (2007). Gene therapy progress and prospects: RNA aptamers. Gene Ther 14: 283–291. [DOI] [PubMed] [Google Scholar]
- Ellington, AD and Szostak, JW (1990). In vitro selection of RNA molecules that bind specific ligands. Nature 346: 818–822. [DOI] [PubMed] [Google Scholar]
- Tuerk, C and Gold, L (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249: 505–510. [DOI] [PubMed] [Google Scholar]
- Hwang, B, Cho, JS, Yeo, HJ, Kim, JH, Chung, KM, Han, K et al. (2004). Isolation of specific and high-affinity RNA aptamers against NS3 helicase domain of hepatitis C virus. RNA 10: 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa, F, Funaji, K, Fukuda, K and Nishikawa, S (2004). In vitro selection of RNA aptamers against the HCV NS3 helicase domain. Oligonucleotides 14: 114–129. [DOI] [PubMed] [Google Scholar]
- Nishikawa, F, Kakiuchi, N, Funaji, K, Fukuda, K, Sekiya, S and Nishikawa, S (2003). Inhibition of HCV NS3 protease by RNA aptamers in cells. Nucleic Acids Res 31: 1935–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umehara, T, Fukuda, K, Nishikawa, F, Sekiya, S, Kohara, M, Hasegawa, T et al. (2004). Designing and analysis of a potent bi-functional aptamers that inhibit protease and helicase activities of HCV NS3. Nucleic Acids Symp Ser 48: 195–196. [DOI] [PubMed] [Google Scholar]
- Bellecave, P, Andreola, ML, Ventura, M, Tarrago-Litvak, L, Litvak, S and Astier-Gin, T (2003). Selection of DNA aptamers that bind the RNA-dependent RNA polymerase of hepatitis C virus and inhibit viral RNA synthesis in vitro. Oligonucleotides 13: 455–463. [DOI] [PubMed] [Google Scholar]
- Bellecave, P, Cazenave, C, Rumi, J, Staedel, C, Cosnefroy, O, Andreola, ML et al. (2008). Inhibition of hepatitis C virus (HCV) RNA polymerase by DNA aptamers: mechanism of inhibition of in vitro RNA synthesis and effect on HCV-infected cells. Antimicrob Agents Chemother 52: 2097–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biroccio, A, Hamm, J, Incitti, I, De Francesco, R and Tomei, L (2002). Selection of RNA aptamers that are specific and high-affinity ligands of the hepatitis C virus RNA-dependent RNA polymerase. J Virol 76: 3688–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, LA, Clancy, LE, Rawlinson, WD and White, PA (2006). High-affinity aptamers to subtype 3a hepatitis C virus polymerase display genotypic specificity. Antimicrob Agents Chemother 50: 3019–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, CH, Lee, YJ, Kim, JH, Lim, JH, Kim, JH, Han, W et al. (2013). Inhibition of hepatitis C virus (HCV) replication by specific RNA aptamers against HCV NS5B RNA replicase. J Virol 87: 7064–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, D, Meng, X, Yu, Q, Xu, L, Long, Y, Liu, B et al. (2013). Inhibition of hepatitis C virus infection by DNA aptamer against envelope protein. Antimicrob Agents Chemother 57: 4937–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, K, Umehara, T, Fukuda, K, Kuno, A, Hasegawa, T and Nishikawa, S (2005). A hepatitis C virus (HCV) internal ribosome entry site (IRES) domain III-IV-targeted aptamer inhibits translation by binding to an apical loop of domain IIId. Nucleic Acids Res 33: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton, S, Romero-López, C and Berzal-Herranz, A (2013). RNA aptamer-mediated interference of HCV replication by targeting the CRE-5BSL3.2 domain. J Viral Hepat 20: 103–112. [DOI] [PubMed] [Google Scholar]
- Levin, AA, Yu, RZ and Geary, RS. Basic principles of the pharmacokinetics of antisense oligonucleotide drugs. In: Crooke ST (ed.). Antisense Drug Technology: Principles, Strategies and Applications. 2nd edn. CRC Press, Boca Raton, 2007. [Google Scholar]
- Bouchard, PR, Hutabarat, RM and Thompson, KM (2010). Discovery and development of therapeutic aptamers. Annu Rev Pharmacol Toxicol 50: 237–257. [DOI] [PubMed] [Google Scholar]
- Gabrielsson, J and Weiner, D. Pharmacokinetic & Pharmacodynamic Data Analysis. Swedish Pharmaceutical Press, Stockholm, 2007. [Google Scholar]
- de Smidt, PC, Le Doan, T, de Falco, S and van Berkel, TJ (1991). Association of antisense oligonucleotides with lipoproteins prolongs the plasma half-life and modifies the tissue distribution. Nucleic Acids Res 19: 4695–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy, JM, Lewis, SD, Kurz, M, Boomer, RM, Thompson, KM, Wilson, C et al. (2004). Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm Res 21: 2234–2246. [DOI] [PubMed] [Google Scholar]
- Krützfeldt, J, Rajewsky, N, Braich, R, Rajeev, KG, Tuschl, T, Manoharan, M et al. (2005). Silencing of microRNAs in vivo with ‘antagomirs'. Nature 438: 685–689. [DOI] [PubMed] [Google Scholar]
- Takei, Y, Kadomatsu, K, Itoh, H, Sato, W, Nakazawa, K, Kubota, S et al. (2002). 5'-,3'-inverted thymidine-modified antisense oligodeoxynucleotide targeting midkine. Its design and application for cancer therapy. J Biol Chem 277: 23800–23806. [DOI] [PubMed] [Google Scholar]
- Yoneda, K, Sugimoto, K, Shiraki, K, Tanaka, J, Beppu, T, Fuke, H et al. (2008). Dual topology of functional Toll-like receptor 3 expression in human hepatocellular carcinoma: differential signaling mechanisms of TLR3-induced NF-kappaB activation and apoptosis. Int J Oncol 33: 929–936. [PubMed] [Google Scholar]
- Mercer, DF, Schiller, DE, Elliott, JF, Douglas, DN, Hao, C, Rinfret, A et al. (2001). Hepatitis C virus replication in mice with chimeric human livers. Nat Med 7: 927–933. [DOI] [PubMed] [Google Scholar]
- Hummel, J, McKendrick, S, Brindley, C and French, R (2009). Exploratory assessment of dose proportionality: review of current approaches and proposal for a practical criterion. Pharm Stat 8: 38–49. [DOI] [PubMed] [Google Scholar]
- Takizawa, Y, Nishimura, H, Morota, T, Tomisawa, H, Takeda, S and Aburada, M (2004). Pharmacokinetics of TJ-8117 (Onpi-to), a drug for renal failure (I): Plasma concentration, distribution and excretion of [3H]-(-)epicatechin 3-O-gallate in rats and dogs. Eur J Drug Metab Pharmacokinet 29: 91–101. [DOI] [PubMed] [Google Scholar]
- Drolet, DW, Nelson, J, Tucker, CE, Zack, PM, Nixon, K, Bolin, R et al. (2000). Pharmacokinetics and safety of an anti-vascular endothelial growth factor aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharm Res 17: 1503–1510. [DOI] [PubMed] [Google Scholar]
- Lee, YJ, Han, SR, Kim, NY, Lee, SH, Jeong, JS and Lee, SW (2012). An RNA aptamer that binds carcinoembryonic antigen inhibits hepatic metastasis of colon cancer cells in mice. Gastroenterology 143: 155–65.e8. [DOI] [PubMed] [Google Scholar]
- McCauley, TG, Kurz, JC, Merlino, PG, Lewis, SD, Gilbert, M, Epstein, DM et al. (2006). Pharmacologic and pharmacokinetic assessment of anti-TGFbeta2 aptamers in rabbit plasma and aqueous humor. Pharm Res 23: 303–311. [DOI] [PubMed] [Google Scholar]
- Talbot, LJ, Mi, Z, Bhattacharya, SD, Kim, V, Guo, H and Kuo, PC (2011). Pharmacokinetic characterization of an RNA aptamer against osteopontin and demonstration of in vivo efficacy in reversing growth of human breast cancer cells. Surgery 150: 224–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland, M and Tozer, TN. Kinetics following an extravascular dose. Clinical Pharmacokinetics and Pharmacodynamics. 4th edn. Lippincott Williams & Wilkins, Baltimore, 2011. pp. 159–182. [Google Scholar]
- Krieger, N, Lohmann, V and Bartenschlager, R (2001). Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J Virol 75: 4614–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, KS, Lim, JH, Kim, JH, Myung, H and Lee, SW (2006). Inhibition of the replication of hepatitis C virus replicon with nuclease-resistant RNA aptamers. J Microbiol Biotechnol 16: 1634–1639. [Google Scholar]
- Lohmann, V, Körner, F, Koch, J, Herian, U, Theilmann, L and Bartenschlager, R (1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285: 110–113. [DOI] [PubMed] [Google Scholar]
- Lee, CH, Kim, JH, Kim, HW, Myung, H and Lee, SW (2012). Hepatitis C virus replication-specific inhibition of microRNA activity with self-cleavable allosteric ribozyme. Nucleic Acid Ther 22: 17–29. [DOI] [PubMed] [Google Scholar]
- Silahtaroglu, AN, Nolting, D, Dyrskjøt, L, Berezikov, E, Møller, M, Tommerup, N et al. (2007). Detection of microRNAs in frozen tissue sections by fluorescence in situ hybridization using locked nucleic acid probes and tyramide signal amplification. Nat Protoc 2: 2520–2528. [DOI] [PubMed] [Google Scholar]
- NRC. Guide for the Care and Use of Laboratory Animals. National Academies Press: Washington, DC, 1996. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.