

Abstract
Recent successful clinical trials with recombinant adeno-associated viral vectors (rAAVs) have led to a renewed interest in gene therapy. However, despite extensive developments to improve vector-manufacturing processes, undesirable DNA contaminants in rAAV preparations remain a major safety concern. Indeed, the presence of DNA fragments containing antibiotic resistance genes, wild-type AAV, and packaging cell genomes has been found in previous studies using quantitative polymerase chain reaction (qPCR) analyses. However, because qPCR only provides a partial view of the DNA molecules in rAAV preparations, we developed a method based on next-generation sequencing (NGS) to extensively characterize single-stranded DNA virus preparations (SSV-Seq). In order to validate SSV-Seq, we analyzed three rAAV vector preparations produced by transient transfection of mammalian cells. Our data were consistent with qPCR results and showed a quasi-random distribution of contaminants originating from the packaging cells genome. Finally, we found single-nucleotide variants (SNVs) along the vector genome but no evidence of large deletions. Altogether, SSV-Seq could provide a characterization of DNA contaminants and a map of the rAAV genome with unprecedented resolution and exhaustiveness. We expect SSV-Seq to pave the way for a new generation of quality controls, guiding process development toward rAAV preparations of higher potency and with improved safety profiles.
Keywords: DNA contaminants, next generation sequencing, quality control, rAAV
Introduction
The recent encouraging outcomes of clinical trials using vectors derived from recombinant adeno-associated viruses (rAAVs)1,2 have helped to promote gene therapy for the treatment of genetic and acquired diseases. As these advanced-therapy medicinal products head toward commercialization, exhaustive quality control (QC) must be performed to ensure their efficiency and safety. However, the production of rAAVs involves components of both cellular and viral origin, and despite extensive downstream purification, rAAV production results in a heterogeneous product that is particularly complex to characterize. In addition to bona fide therapeutic particles, a variety of process impurities can be found in the final rAAV product, including: empty viral capsids, replication-competent AAV particles, chemicals, lipids, proteins, and nucleic acids.3,4,5,6,7 Among the latter category, contaminating DNA sequences pose a significant safety hazard because they might encode proteins or regulatory RNAs and even trigger immune toxicity themselves' via TLR9 activation.8,9 To limit the oncogenic and infectious risk, the Food and Drug Administration recommendations are that the level of residual cell-substrate DNA should be below 10 ng per dose and a median DNA size of 200 bp or lower.10 Although several recent developments have been made to improve rAAV production and purification, DNA contamination remains a major concern.
A broad range of studies have reported the presence of DNA contaminants in rAAV preparations, identified as fragments of: (i) the bacterial backbone of the vector plasmid carrying antibiotic resistance genes6,11,12; (ii) helper viruses, such as Adenovirus, Herpesvirus, or Baculovirus7,13; (iii) wild-type AAV rep/cap sequences6,14,15,16; and (iv) genomic DNA originating from the packaging cells.6,7 Whether these contaminating nucleic acid sequences are actually packaged into rAAV particles remains unclear, but some of these sequences can be transferred after vector administration in vivo where they can persist for months, as previously shown in dogs and nonhuman primates in our laboratory.11 Finally, truncated rAAV genomes, resulting from incomplete encapsidation have been previously described,17,18 and may reduce vector potency.
While the presence of DNA contaminants in clinical grade rAAV batches is undesirable,19 their relative abundance can be estimated from quantitative PCR (qPCR) data. This method, however, suffers from a number of limitations and flaws, including: (i) the need to determine targets representing each contaminating sequence and to develop target-specific assays; (ii) inconsistencies in rAAV genome titration, which vary among laboratories and target regions20; (iii) limited coverage of DNA contaminants, particularly for the genomic DNA of packaging cells; and (iv) the inability to assess the presence of rearranged rAAV genomes. As technologies evolve, vector analytics must move forward to improve the characterization of DNA molecules in rAAV batches, including an advanced genomic identity of vector genomes.
To this end, we developed SSV-Seq, for next-generation sequencing (NGS) of single-stranded DNA viruses, together with ContaVect, a bioinformatic tool dedicated to QC of virus/vector preparations from NGS datasets.
As a proof of concept, we applied SSV-Seq to a single-stranded serotype 8 rAAV-expressing green fluorescent protein (GFP), manufactured by transient transfection in HEK-293 cells, followed by three different “state-of-the-art” GMP-compliant purification processes. Although our data were consistent with those from qPCR, we were able to exhaustively quantify DNA contaminants longer than 250 bp without the need for any indirect standard comparisons. Additionally, we identified unexpected contaminants and obtained a high-definition map of the rAAV vector genome.
SSV-Seq could be applied as an in-process analytic to guide upstream and downstream process development towards rAAV preparations of higher potency, but one might also expect this method to improve knowledge of rAAV vector biology. Finally, we believe that SSV-Seq responds to the need expressed by regulatory bodies for improved vector analytics standards when releasing clinical grade rAAV vectors.
Results
SSV-Seq workflow for rAAV vector preparations
DNA contaminant characterization of virus-derived advanced-therapy medicinal products for gene therapy is a critical QC test required for clinical trials and future market authorizations. The reference method for evaluating DNA contaminants in final rAAV products is qPCR, whereas rAAV genome identity is determined by Sanger sequencing. However, both methods are intrinsically insufficient to provide an extensive and accurate overview of the populations of DNA molecules, whether they are parts of the therapeutic fraction or are considered contaminants. To this end, SSV-Seq was designed as a powerful and reliable method based on NGS that provides in-depth characterization of DNA molecules in rAAV products.
The experimental workflow of SSV-Seq is presented in Figure 1, and extensive experimental details are provided in the Supplementary Results I section. Before DNA extraction, encapsidated DNA can be enriched by digesting DNase-sensitive nucleic acids using an optimized nuclease treatment (Figure 1b, Supplementary Figure S1). The efficiency of the DNAse digestion could be evaluated in-process by spiking irrelevant DNA from bacteriophage λ beforehand. Then, whole DNA is extracted, including the single-stranded virion DNA, and converted into dsDNA by random priming to generate a template compatible with NGS library preparation (Figure 1c). Because this step is critical, controls were developed to verify its efficiency (Supplementary Figure S2) and the absence of selection bias toward the rAAV genome (Supplementary Figure S3). Then, the DNA samples are sheared, and an Illumina-compatible NGS library is prepared using a custom protocol (Figure 1d). To note, the small nucleic acid fragments (>250 bp), either generated during sonication or initially present in the vector preparations, are eliminated during the protocol due to repeated washing steps (Supplementary Figure S4). Finally, the samples are paired-end sequenced with an Illumina HiSeq platform, and the data are processed through a dedicated bioinformatic pipeline (ContaVect) designed to perform quantitative and qualitative analyses (Figure 1e, Supplementary Table S3).
Figure 1.

Overview of SSV-Seq protocol. (a) A quantity of 2 × 1011 vector genomes of a purified rAAV preparation are required as input. (b) To reduce the amount of nonencapsidated DNA, eventually, a DNase digestion step can be performed, with a mix of highly efficient DNases (Baseline-ZERO and Plasmid-Safe DNases). (c) After DNA extraction, total DNA is denatured, and the second strand is synthesized by random priming with the high-fidelity Escherichia coli DNA Pol I, followed by a purification step. (d) The dsDNA template is sheared by sonication into 200–300 bp fragments, which are subsequently end-repaired and A-tailed to allow for the ligation of adaptors compatible with Illumina sequencing. One of the two adaptors contains a short DNA barcode, also called an “index”, which is different for each experimental sample. Finally, an optimized PCR amplification is performed. All of the library preparation steps are checked by chip electrophoresis. (e) A qPCR-based quantification of next-generation sequencing libraries is performed prior to pooling for cluster generation on flow cell in the presence of nonindexed ϕ-X DNA. High-throughput sequencing is achieved on an Illumina HiSeq platform (Rapid Run 2 × 101 bp). Finally, ContaVect performs automated bioinformatic analyses, including preprocessing of reference and sequencing reads, attribution of reads to a reference sequence and postprocessing, resulting in several simple reports.
Experimental evaluation of SSV-Seq
To challenge the SSV-Seq method, we analyzed a rAAV vector derived from serotype 8 that carried the synthetic expression cassette CMVp-eGFP-hygroTK-bGHpA. The recombinant vector was produced by transient transfection of HEK-293 cells and was subsequently purified by cesium chloride density gradient (CsCl), affinity chromatography (AVB), or ion exchange chromatography (IEX) (Supplementary Figure S5). The three purified rAAV stocks were subsequently characterized by current reference methods, including qPCR, for the quantification of DNA contaminants (Supplementary Results II, Supplementary Figure S6, Supplementary Table S1). For each rAAV vector stock originating from each purification process used, we prepared two NGS libraries, in which irrelevant phage λ DNA was spiked. Of these two libraries, only one was treated with our optimized DNase mix to remove nonencapsidated DNA (Supplementary Table S2, Supplementary Figure S7).
In addition, we processed a negative control and an internal reference normalizer along with the rAAV samples. The negative control exclusively consisted of phage λ DNA, to assess environmental contamination during sample handling. The internal normalizer control was amix of DNA molecules in proportions that are usually found in rAAV preparations, as described in previous studies6,7,11,14,15 (Supplementary Table S2, Supplementary Figure S7). This mix included: (i) a fragment of the vector plasmid containing the rAAV genome (ITR2-CMVp-eGFP-hygroTK-bGHpA-ITR2); (ii) the bacterial backbone from the vector plasmid; (iii) the pDP8 helper plasmid used for the production of rAAV particles; and (iv) the genomic DNA of the packaging HEK-293 cells containing the E1 region of the Ad5 genome21 (Supplementary Table S2). The internal normalizer was subsequently used to normalize sequencing coverage for qualitative analysis.
To evaluate the reproducibility of SSV-Seq, each sample was analyzed by two independent technical replications, and the experimenters remained blinded throughout all of the wet laboratory experiments and bioinformatic analyses. We obtained a balanced number of reads between samples (6,340,719 to 9,658,441), with a reasonable average Phred quality (34.74 to 36.52), as reported in Supplementary Table S2.
Distribution of DNA contaminants identified by NGS correlates with qPCR data
Reads were assigned by ContaVect to one of the following reference sequences: (i) the rAAV 2/8 CMVp-eGFP-hygroTK-bGHpA genome; (ii) the bacterial backbone of the vector plasmid; (iii) the entire pDP8 helper plasmid; (iv) fragments of the Ad5 genome integrated into the HEK-293 packaging cell line genome; and (v) the human genome primary assembly GRCh38. To avoid the misattribution of reads originating from phage DNA, the reference sequences of phage λ (J02459.1) and ϕ-X (J02482.1) were also provided to ContaVect. The parameters were optimized using an in silico-generated artificial dataset mimicking the estimated sequence composition of an rAAV CMVp-eGFP-hygroTK-bGHpA vector preparation (Supplementary Results III, Supplementary Table S4). The optimized parameters, detailed in the configuration files (http://dx.doi.org/10.5061/dryad.fs4cp), were used to obtain the raw distribution of reads in the references for the experimental and control samples (Supplementary Table S5). Based on the number of reads obtained in the negative control, we defined a positivity threshold per run and per reference, to avoid false-positive detection due to environmental contamination. Although there were reads in the negative control for all of the references, the read count was always higher in the experimental samples, except for the Ad5 sequence, which was virtually undetectable. The raw data also indicated that the DNase treatment was effective because the read count for phage λ in DNase-positive samples was less than the internal normalizer values after DNAse treatment (Supplementary Table S5).
To reflect the contaminants present in the rAAV preparation, we excluded unmapped reads or reads mapped to bacteriophage genomes and then calculated the relative representation of the remaining reads (Table 1). The maximal difference between two replicates was 1.10% for the rAAV genome, 1.12% for the vector plasmid backbone, 0.03% for the helper plasmid, and 0.07% for the human genome, emphasizing the reproducibility of SSV-Seq.
Table 1. Percentages of DNA populations in rAAV preparations obtained by next-generation sequencing and inferred from qPCR.
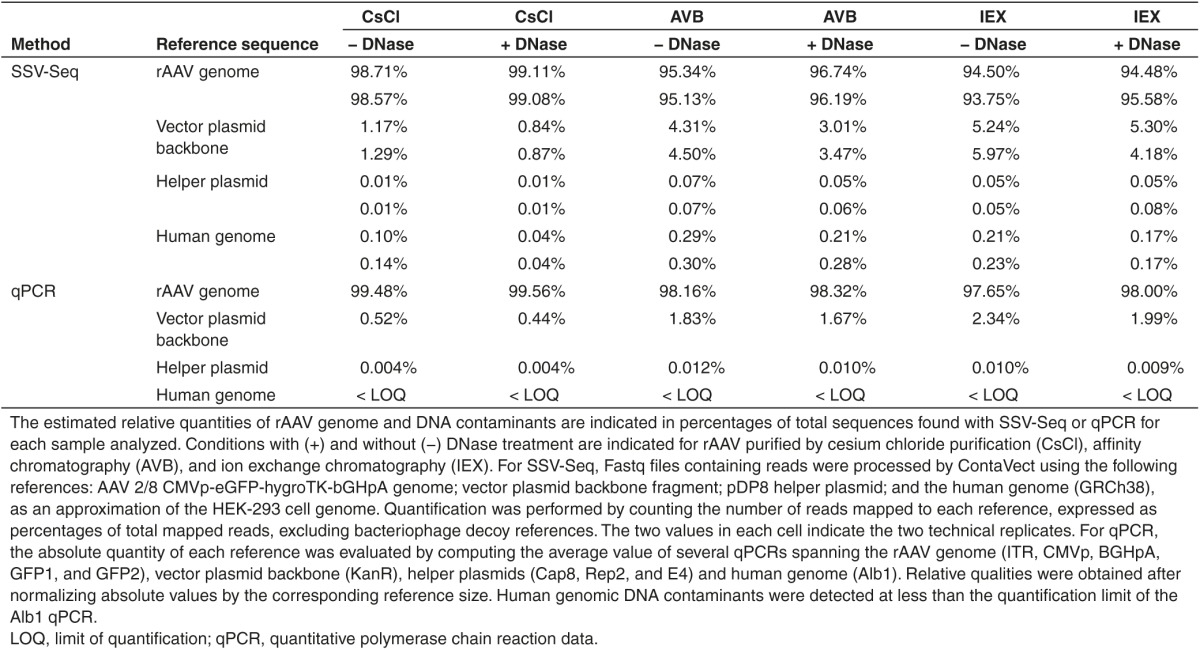
Regardless of the rAAV purification process (CsCl, AVB, or IEX), we obtained a large majority of reads matching the rAAV reference genome (93.75–99.11%), followed by a lower quantity matching the vector plasmid backbone (0.84–5.97%) and an even lower amount matching the helper plasmid (0.01–0.08%) or the human genome (0.04–0.30%). In these experiments, the rAAV vector contained less DNA contamination when purified by CsCl compared to both chromatographic methods, although it should not be considered a general rule until more vector preparations are analyzed. Finally, the DNase-treated samples showed a reduction of DNA contaminants of up to 1.5-fold for plasmids and 3.1-fold for human DNA. These findings suggest that although some of the DNA contaminants were accessible to DNase, most of them were likely to be encapsidated or tightly associated with the capsid.
Compared to SSV-Seq, qPCR does not result in direct relative proportions of the DNA contaminants because only a subset of the reference sequences is quantified. To compare these two methods, the copy numbers of each target obtained by qPCR were normalized according to the size of the corresponding targeted reference (i.e., rAAV genome, vector plasmid backbone, helper plasmid, and human genome). Such conversion is questionable, however, because the percentages of DNA contaminants are affected by the choice of the target in the rAAV genome used for qPCR. To smooth out the inter-qPCR variability, we quantified several targets per reference and calculated an average normalized percentage (Figure 2, Table 1).
Figure 2.

Correlation of the percentages of DNA populations in rAAV preparations obtained by next-generation sequencing (NGS) and inferred from quantitative polymerase chain reaction (qPCR) data. The data obtained for the three references (rAAV genome, backbone of the vector plasmid, and helper plasmid) detected in both NGS and qPCR are clustered in gray squares crossed by diagonal lines, indicating the perfect correlation between the two methods. The x- and y-axes are symmetrical, represented on log scales and truncated between 0.1–0.2 and 10–90 to highlight the intersample variability. Each point corresponds to the average percentage of two technical replicates for both NGS and qPCR. The correlation between the methods was evaluated with the nonparametric Spearman's test because of the small sample size (18 pairs) and the absence of information about the distribution of the measured variables (Spearman's correlation coefficient, two-tailed P value, 95% confidence interval).
The qPCR data indicated the presence of a higher proportion of the rAAV genome than found using SSV-Seq (average +2.1%), mirrored by a smaller number of DNA contaminants (Table 1). All of the qPCR spanning the rAAV genome yielded very similar results, except for the widely used “universal” ITR2 qPCR, which led to higher values, as usually observed in our laboratory (Supplementary Table S1). This observation is likely partially responsible for an over-estimation of the rAAV genome in our qPCR data. However, despite these limited variations, we obtained a significant correlation with the NGS results (Spearman's correlation coefficient = 0.9938, P < 0.0001) (Figure 2). Altogether, our NGS data obtained by SSV-Seq were consistent with the qPCR findings and yielded reproducible results.
Advanced analysis of the human genomic DNA contaminants
SSV-Seq generated an enormous amount of data at single-nucleotide resolution, which opened up new possibilities for exploring the qualitative and quantitative features of rAAV preparations. Thus, we further analyzed the origin of reads spanning the human genome. The normalized densities of reads per chromosome suggested an overall random distribution for the rAAV samples, within a twofold range (Figure 3a). Interestingly, we found two over-represented loci: (i) mitochondrial DNA (mtDNA) in rAAV preparations purified by CsCl; and (ii) specific DNA sequences from chromosome 15 in particles purified by AVB.
Figure 3.

Distribution of DNA contaminants from human chromosomes. (a) Density of reads mapped per chromosome and mitochondrial DNA (mtDNA) obtained after normalization to the read count of the internal normalizer, which contained sonicated DNA extracted from HEK-293 cells. A value of 1 indicates a random distribution, 2 indicates twofold enrichment, and 0.5 indicates twofold depletion. Each point is the average value of the two technical replicates. The “Other” category aggregates results obtained for 169 regions of the GRCh38 primary assembly that are not assembled into chromosomes. (b) Depth of coverage along the mitochondrial D-loop (human genome GRCh38 MT: 16,078—16,561) for rAAV purified by CsCl and for the negative control. These data were confirmed by a D-loop-specific qPCR. Values in copies/ng of DNA are indicated for the corresponding samples on the right of the coverage graph, and the positions of the qPCR primers are represented below the graphs by black arrows. (c) Depth of coverage over a gene locus from chromosome 15 for rAAV purified with AVB columns and for the negative control. The locus is not disclosed due to confidentiality concerns.
The sequences of the mtDNA found in the rAAV preparations purified by CsCl were highly specific for the D-loop, which is a triple-stranded DNA region found in the major noncoding region of mtDNA22 (Figure 3b) that is insensitive to DNase I.23 The D-loop contamination was reduced by our optimized DNase cocktail treatment, indicating that mtDNA was somehow copurified along with the rAAV particles during CsCl purification but was probably not encapsidated. Without DNase treatment, the D-loop represented approximately 1.3% of the reads mapped to the human genome but only 0.0015% of all mapped reads (Supplementary Table S6). This low level of contamination in the CsCl-purified preparations was confirmed by D-loop-specific qPCR, as indicated on the right side of Figure 3b.
In AVB-purified rAAV batches, we identified a high number of reads mapped to chromosome 15, concentrated on the exons of a unique gene (the precise nature of which is confidential information). As opposed to the mtDNA contamination, the read counts of the DNA from chromosome 15 remained stable after the optimized DNase treatment, suggesting an encapsidated contaminant (Figure 2b). Further investigations led us to identify this sequence as a cDNA carried by a rAAV preparation previously purified on the same AVB-Sepharose column, indicating insufficient sanitization of the affinity matrix between manufacturing campaigns. We estimated these contaminating rAAVs to represent approximately 1 out of 2,000 bona fide particles (Supplementary Table S6).
These two examples illustrate the ability of SSV-Seq to identify unexpected or rare DNA populations in rAAV batches. We believe that this approach will allow for rational improvement of current rAAV manufacturing processes by enabling the routine implementation of in-process SSV-Seq by development teams and/or by identifying new relevant targets for qPCR analysis.
High-definition genomic identity of the rAAV genome
In addition to the quantitative analyses, we were able to study important functional features of the rAAV genomes, including the single-nucleotide variants (SNVs) and the enrichment in specific sequences that could unmask vector genomes heterogeneity. Beyond the limitations of the current reference method (Sanger sequencing), we obtained a high-definition genomic identity due to a tremendous sequencing depth over the rAAV genome, i.e., > 200,000 reads/base.
Figure 4 represents the depth of coverage of the experimental samples along the rAAV genome at single-nucleotide resolution (Figure 4a), compared with the plasmid control of the internal normalizer (black line) and the in silico control (gray-shaded area). The artificial in silico control indicates the accuracy of ContaVect along the entire rAAV genome. Although the depths of coverage of the three experimental samples were more scattered than the in silico control, they followed the same trend as the plasmid control from the normalizer. This finding indicates that the variability of sample coverage was due to selection/amplification biases during the SSV-Seq protocol, rather than an under/over-representation of rAAV genome fragments in preparations. Therefore, rAAV genome was homogeneously packaged in our experimental samples.
Figure 4.

Sequencing coverage and percentage of single nucleotide variants along the rAAV genome. (a) Sequencing coverage along each base of the rAAV CMVp-eGFP-hygroTK-bGHpA genome. To compare samples independently of their sequencing depth, a normalized depth of coverage was computed by counting the number of reads aligned to each base (×1,000), divided by the sum of coverage for all bases mapped along the rAAV genome. Lines correspond to the average normalized coverage of the two technical replicates for the rAAV preparations, purified by CsCl (red), IEX (green), AVB (blue), and the internal normalizer control (black), without DNase treatment. The gray area below the graph represents the normalized coverage of the in silico-generated control. The shoulders at the extremities correspond to the range of artificial fragmentation specified in the program that generates the artificial Fastq datasets (250–450 bases). (b) Cumulative percentage of alternative base A (red), C (blue), T (green), and G (brown) compared with the reference sequence, i.e., single-nucleotide variants. When several variants were found at the same nucleotide position, variant contributions were stacked. SNVs are represented on the graph if they were found in at least half of all of the experimental samples. (c) Map and length of the rAAV genome, represented to scale below the graphs, with coordinates in base pairs. CMVp, cytomegalovirus promoter; eGFP, enhanced green fluorescent protein CDS; HygroTK, hygromycin-thymidine kinase fusion CDS; IRES, internal ribosome entry site; ITR, inverted terminal repeat; pA, bovine growth hormone polyadenylation signal.
In addition, we analyzed the distribution of SNVs along the recombinant genome. Compared to the reference sequence of the rAAV genome, in experimental samples, we identified 162, 13, and 1 SNVs with frequencies greater than 1/1,000, 1/100, and 1/10, respectively (Figure 4b). However, these SNVs were also found in the control rAAV cassette from the internal normalizer (Supplementary Figure S8), indicating that the sequence variability in the rAAV genome was not due to de novo mutations arising during vector production. Therefore, controlling the sequence of the AAV vector plasmids prior to production with a resolution greater than what is possible by Sanger sequencing may help to improve the quality of rAAV vectors.
Finally, we investigated the reverse- and copackaging of the vector plasmid backbone by realigning all of the sequencing reads along the entire vector plasmid, i.e., the rAAV genome and plasmid backbone references fused in a single sequence (Figure 5a). Although the large difference in coverage between rAAV and the backbone is consistent with previous findings (Supplementary Table S2), we found approximately 1.4-fold more reads aligned on AAV inverted terminal repeats (ITRs) (Supplementary Table S7), suggesting the existence of reads overlapping the right and left junctions between the rAAV genome and the plasmid backbone. We focused on the reads supporting such junctions because they indicate reverse packaging of the plasmid backbone (triggered by ITRs in cis) or copackaging with the rAAV genome (Figure 5b,c). We did not find any false positives in the in silico control, for which no junction between rAAV ITR and the plasmid backbone had been generated. In contrast, reads (or read pairs) supporting backbone/ITR junctions were obtained in all of the experimental samples, ranging from 0.02 to 0.12% of all of the mapped reads for the left ITR and from 0.05 to 0.29% for the right ITR. Although the phenomenon appeared to be limited, to our knowledge, this is the first attempt to precisely quantify the extent of reverse and/or copackaging of the vector plasmid backbone into rAAV particles.
Figure 5.

Sequencing depth along the rAAV vector plasmid and visualization of ITR/backbone junctions. All of the sequencing reads were realigned on a single vector plasmid reference sequence composed of the rAAV genome and the plasmid backbone, as indicated in the inner circle. Each lane of the circular histograms corresponds to the average values obtained for the two technical replicates of the in silico control (1, black) and for the rAAV preparations purified by IEX (2, green), AVB (3, blue), and CsCl (4, red), without DNase treatment. (a) The depth of coverage for each position was normalized to the total number of nucleotides aligned along the full plasmid reference. Panels (b) and (c) represent enlarged images of the left and right junctions between the rAAV genome and plasmid backbone, respectively. The number of reads overlapping at least 20 nt on each side of both junctions was evaluated for each sample using a dedicated bioinformatic tool. The numbers of reads and corresponding proportions are indicated for both technical replicates.
As demonstrated through these examples, SSV-Seq can provide information regarding the genomic identity of a viral/vector genome in purified preparations at a much higher resolution than Sanger sequencing, the current reference.
Discussion
The lack of characterization of DNA contaminants was one of the six major points raised by the European Medicines Agency in 2012 when it granted marketing authorization for Glybera (AAV2/1-CMV-LPLS447X) under exceptional circumstances.19,24 In addition, the US Food and Drug Administration recently published a statement encouraging researchers to develop “robust, accurate and consistent testing methodologies” to characterize rAAV vector preparations.25,26 Meanwhile, a number of clinical trials with AAV vectors are moving toward later phases, further reinforcing the need for an upgrade of the methods for evaluating DNA contamination.
In this study, we described a new strategy based on NGS of single-stranded DNA viruses (SSV-Seq) for the characterization of vector genome integrity and DNA contaminants in rAAV preparations (Figure 1). Each step of the protocol, including bioinformatics, was thoroughly validated using dedicated controls to be applicable in QC laboratories (Supplementary Results I and III).
When applied to a rAAV serotype 8-encoding GFP and hygro-TK, the SSV-Seq-generated results were consistent with the current reference method (qPCR) (Figure 2) but also reported unexpected contaminants (Figure 3). For example, we found mtDNA contaminants in CsCl-purified rAAV preparations, the presence of which was confirmed by qPCR (Figure 3a). A recent qPCR screening of more than 10 other rAAV preparations produced in the laboratory identified a variable amount of D-loop in all of them, independent of the serotype or the purification process (data not shown). Although it would require further investigation, it is likely that the over-representation of this region is due to its resistance to DNAse23 generally employed during rAAV-manufacturing processes.
We also obtained a high-definition map of the vector genome, indicating: (i) overall homogeneous encapsidation from the left ITR to the right ITR (Figure 4a); (ii) the absence of de novo SNVs introduced during vector production (Figure 4b); and (iii) the presence of a limited number of ITR/vector plasmid backbone junctions (Figure 5). In this study, we chose an optimally sized rAAV (4.7 kb) carrying a synthetic reporter expression cassette, which was ideal for this proof of concept but which does not reflect the biological variability of rAAV encapsidation mechanisms. However, in the future, SSV-Seq could provide a better understanding of these complex mechanisms by analyzing a range of over- and under-sized rAAV genomes as well as vector plasmids containing various DNA elements (i.e., promoters, ITR sequences, cDNA, polyA tails, over-sized plasmid backbones, insulators, stuffer sequences, etc.).
Nevertheless, SSV-Seq has inherent limitations compared with qPCR, including: (i) an extended amount of time required for sample preparation and analysis (~10 versus ~2 days for qPCR); (ii) a higher cost of reagents and instrumentation; and (iii) the absence of turnkey solutions for data analysis. In addition, NGS libraries can be easily contaminated by exogenous DNA, which might affect the results of bioinformatic analyses, as reported in previous studies.27,28 To limit the impact of such contamination, we established strict guidelines for library preparation and processed a negative control consisting of bacteriophage λ DNA along with the experimental samples. Finally, DNA sequences can be unevenly amplified during PCR steps, depending on the sequence composition and secondary structures, leading to quantitative bias in NGS data.29 To circumvent this issue, the data can be normalized to a control consisting of sequences expected in the experimental samples, such as our internal normalizer, which contained fragments of the plasmids and the HEK-293 cell genome used for vector production. Altogether, SSV-Seq can provide information regarding the DNA species in rAAV preparations with unprecedented definition and exhaustiveness, but appropriate controls must be performed to avoid misleading conclusions, as already emphasized in other NGS-based strategies.27,30
SSV-Seq is not intended to replace qPCR for routine vector characterization. Instead, it should be used as an in-process and end-point QC step to guide R&D teams toward rAAV preparations containing fewer DNA contaminants. Our method could also be helpful in comparing the standard method to produce rAAV vector based on transient transfection with more recent and less characterized processes, such as stable mammalian producer cell lines31 and Sf9 insect cells infected with baculoviruses.32 In addition, SSV-Seq could be used to identify specific targets for routine qPCR analyses based on their representativeness or to detect specific contaminants.
Finally, given the concerns expressed by regulatory bodies regarding nucleic acid contaminants, NGS-based methods, such as SSV-Seq, should be included as an informative QC test for clinical-grade AAV preparations. In addition, we recommend that datasets be publicly released via an open-access repository for external review and transparency.
Materials and methods
Quality system and good experimentation practices. A quality management system has been implemented to cover all of the activities in INSERM UMR 1089, including the management of research teams and the vector core. This system has been approved by Lloyd's Register Quality Assurance to meet the requirements of international Management System Standards ISO 9001:2008.
We followed good experimentation practices throughout the SSV-Seq protocol. One experimenter and one observer were involved in all of the experiments to verify the proper manipulation of the samples. In addition, when validating the conformity of the samples before sequencing, the observer assigned a random identifier to the samples before starting the protocol so that the experimenter was blinded until disclosure of the QC results. Similarly, the sequencing core technicians and the bioinformatician were also blinded until the final disclosure of the results.
rAAV vector production and purification. The pSSV9-derived vector plasmid contains enhanced GFP (eGFP) and hygromycin-thymidine kinase fusion protein (HygroTK) coding sequences, separated by an EMCV IRES, under the control of the cytomegalovirus (CMV) promoter. The construct ends with a bovine growth hormone (bGH) polyadenylation signal and is flanked by ITR sequences originating from AAV serotype 2.
The rAAV CMVp-eGFP-hygroTK-bGHpA was produced as previously described by Ayuso et al.33 Briefly, HEK-293 cells were cotransfected with the vector plasmid and the pDP8 helper plasmid, which contained AAV2 rep, AAV8 cap, and adenovirus helper genes (E2A, VA RNA, and E4). After harvesting of both the cells and the culture supernatant, the crude bulk was split in three parts, and rAAV particles were purified from each subset using three different GMP-compatible methods: (i) IEX; (ii) affinity chromatography (AVB Sepharose High Performance, GE Healthcare, Little Chalfont, UK)32; and (iii) double cesium chloride (CsCl) gradient ultracentrifugation.33 In-process benzonase digestion was performed only during AVB- and CsCl-based purification. Finally, the three rAAV batches were concentrated by tangential flow filtration (TFF, GE Healthcare). The concentrated vectors were formulated in Dulbecco's phosphate-buffered saline (Lonza, Verviers, Belgium) containing 0.001% Pluronic F-68 (Sigma-Aldrich, St. Louis, MO). All of the vectors were produced and purified at INSERM UMR 1089 Vectors Production Center (Nantes, France), except for AVB purification, which was performed at Genethon (Evry, France). Details of the purification methods are provided in Supplementary Figure S5 and in Supplementary Methods.
Preparation of SSV-Seq negative control and internal normalizer control. The internal normalizer control was prepared by mixing DNA sequences in proportions that are usually found in rAAV preparations. The vector plasmid was digested by restriction endonucleases to release the rAAV genome (including ITRs) from the plasmid backbone. Both fragments were separated by agarose gel electrophoresis, extracted from the gel and purified using NucleoSpin Gel and a PCR Clean-up kit (Macherey-Nagel, Düren, Germany). The pDP8 helper plasmid was linearized by restriction endonuclease and was purified as mentioned above. The HEK-293 cell genome was sheared in 6 kb fragments using g-TUBE (Covaris, Woburn, MA) according to the manufacturer's recommendations. Finally, the control was prepared by mixing 2 × 1011 copies of vector genome, 1 × 1010 copies of vector plasmid backbone, 4 × 109 copies of helper plasmid, and 400 pg of HEK-293 sheared DNA (~122 copies). The negative control consisted of an amount of phage λ DNA corresponding to 2 × 1011 copies of the vector genome (484 ng). Both samples were processed following the same protocol as the rAAV vectors but without DNase treatment.
rAAV DNA extraction. Total rAAV DNA was extracted from 2 × 1011 full rAAV particles (i.e., DNase-resistant rAAV genomes quantified by ITR2 qPCR, Supplementary Table S9) in the presence of phage λ (24.2 ng). Where indicated, the samples were treated with 10 U of Baseline ZERO endonuclease and 40 U of Plasmid-Safe exonuclease (Epicentre, Madison, WI) for 2 hours at 37 °C in Baseline ZERO buffer, supplemented with 1 mmol/l of ATP in a final volume of 200 µl. The reaction was stopped by the addition of 3 mmol/l ethylenediaminetetraacetic acid and 30 minutes of incubation at 75 °C. Then, all of the samples were treated with 0.5 mg of proteinase K (Macherey Nagel) and 10 U of RNase A (Qiagen, Venlo, Limburg, the Netherlands) for 3 hours at 55 °C and for 15 minutes at 37 °C, respectively. Finally, the rAAV DNA was extracted using Gentra Puregene Blood kit (Qiagen) according to the manufacturer's recommendations.
Second-strand synthesis. First, the extracted DNA was heated for 5 minutes at 95 °C and then was quenched on ice. A mix containing 58 µmol/l of random hexamers (NEB, Ipswich, MA), 2 mmol/l of each dNTP and 10 U of DNA polymerase I (NEB) was added to the cold samples in a final volume of 50 µl. Randomly primed DNA synthesis was then performed by a ramp of 0.1 °C/second until 37 °C, followed by 1 hour of incubation at 37 °C. The reaction was stopped with 0.1 mmol/l ethylenediaminetetraacetic acid.
NGS library preparation. The NGS library was prepared according to a protocol adapted from Kozarewa et al.34 Briefly, for each library, 200 ng of the double-stranded DNA was sonicated into fragments using Bioruptor (Diagenode, Seraing, Belgium). The average size of the fragments was 300 bp. The fragmentation conditions consisted of low intensity (160 W) and 12 cycles of 30 seconds ON/90 seconds OFF. After fragmentation, DNA ends were repaired with T4 DNA polymerase (15 U), T4 PNK (50 U), and Klenow DNA polymerase (5 U) in the presence of 10 mmol/l of each dNTP in a final volume of 55 µl (NEB). Then, a single deoxyadenosine was added to the 3' end of each blunted DNA end using 15 U of Klenow Fragment DNA polymerase (3′-5′ exo-) and 1 mmol/l of dATP, for 30 minutes at 37 °C (NEB). Adaptor ligation was performed using 0.5 µmol/l of preannealed adapters compatible with Illumina TrueSeq Universal P5 and P7 (see the sequences and details in Supplementary Table S8) and T4 DNA ligase (10,000 U) (NEB) for 15 minutes at room temperature.
The samples were amplified independently with PfuUltra II Fusion Hotstart DNA polymerase (Agilent Technologies, Santa Clara, CA) using P5-F (5′-AATGATACGGCGACCACCG-3′) and P7-R primers (5′-CAAGCAGAAGACGGCATAC-3′) (Sigma-Aldrich). The amplification program was 2 minutes at 95 °C; followed by 15 cycles of denaturation at 95 °C for 20 seconds, annealing at 60 °C for 20 seconds and elongation at 72 °C for 15 seconds. The run was ended by final elongation at 72 °C for 3 minutes.
The samples were purified with 1.6× SPRIselect (Beckman Coulter, Indianapolis IN) after each step of the protocol, following the manufacturer's instructions. A final double purification with 1× SPRIselect was performed after PCR amplification to eliminate adapter dimers before sequencing. During these washing steps, DNA fragments smaller than 200 bp were eliminated. The distribution of DNA fragment size was verified by the Agilent 2100 Bioanalyzer system using High sensitivity DNA chips (Agilent Technologies), according to the manufacturer's guidelines.
NGS sequencing. Samples were quantified using KAPA Library Quantification Kits (Kapa Biosystems, Wilmington, MA) according to the manufacturer's instructions and were pooled in equimolar quantities. PhiX Control v3 DNA (1–5%; Illumina, San Diego, CA) was added to the libraries, which were subsequently sequenced with a HiSeq 1500 platform (Illumina) using rapid-run paired-end mode (2*101 bp) at the genomics and bioinformatics core facility of Nantes (INGB, Nantes, France).
Bioinformatics. The reference sequences of the rAAV genome, vector plasmid backbone, helper plasmid and adenovirus 5 (Ad5) sequence are available in fasta format and annotated GenBank format from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.fs4cp. The genome of the HEK-293 cells was approximated by the last build of the human genome (GRCh38 primary assembly) from Ensembl, while the genomes of bacteriophage λ (J02459.1) and coliphage ϕ-X174 (J02482.1) were retrieved from the European Nucleotide Archive.
An in silico control was generated to mimic a real NGS library and to determine the prediction accuracy of the mapping software using Fastq Control Sampler, a custom C program. The open source code for the software is freely available with its documentation at https://github.com/a-slide/fastq_control_sampler. Paired Fastq files were generated from the rAAV genome (14,000,000 reads), vector plasmid backbone (420,000 reads), helper plasmid (5,000 reads), Ad5 sequence (10 reads), human genome (29,524 reads) and a randomly generated sequence (200,000 reads), with the following parameters: size of the randomly generated reference sequence: 100,000; size of the reads: 101; lower sonication size of the fragments: 250; upper sonication size of the fragments: 450; maximal PHRED quality: 40; minimal PHRED quality: 30; frequency of errors in sequence strings: 0.01; and maximum number of tries to generate a valid read pair: 100; pairs not selected in repeat regions. A detailed report of reads generation is supplied in the Supplementary Material.
Raw BCL data for the samples were demultiplexed with CASAVA (Illumina, San Diego) according to their barcodes and were stored in independent files. Fastq files were analyzed with ContaVect using the configuration file. The program generated standard genomic files (Bam, Bed, and Bedgraph) as well as comprehensive text reports. The Fastq and raw output files are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.fs4cp. The program is still under active development, but the version used for the analyses performed in this study (v0.2) is freely available with extensive user and developer documentation at https://github.com/a-slide/ContaVect/tree/v0.2.
SNVs in the rAAV genome were retrieved from the BAM files containing reads aligned on rAAV with a custom program rather than with a classical program, such as samtools-mpileup, because the read depth was far too great (> 200,000) for the classical (but more robust) callers. The source code for this java program is available on GitHub at https://github.com/lindenb/jvarkit/wiki/MiniCaller. Essentially, this program uses the java library for BAM (htsjdk) to load a set of BAM files and scans all of the bases in the reference genome from 5′ to 3′; for each position, it detects the proportion of bases in each sample and prints a summary in VCF format; no quality score is calculated. The VCF files were subsequently parsed and analyzed using an ipython notebook, available at http://nbviewer.ipython.org/github/a-slide/iPython-Notebook/blob/master/Notebooks/VCF_analysis.ipynb. The VCF and CSV files are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.fs4cp.
Graphical representations and statistics. Graphs were generated using PRISM 5 software (GraphPad, La Jolla, CA), except for Figure 5, which was created using Circos 0.67 (http://circos.ca/). The vector graphics pictures were postprocessed with Inkscape 0.48 (https://inkscape.org) for aesthetic purposes (alignment, legends, fonts, etc.). Raw tables containing the data are provided directly in the manuscript (Figure 2) or are available at the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.fs4cp (Figures 3, 4, and 5). When appropriate, statistical analyses were performed with PRISM 5 using nonparametric tests due to the limited number of samples. The details of the statistical tests are indicated directly in the figure or table legend.
SUPPLEMENTARY MATERIAL Figure S1. Comparative efficiency of DNase on DNA spiked in rAAV production. Figure S2. Controil of efficient second strand synthesis. Figure S3. Selection bias induced by second strand synthesis. Figure S4. Distribution of DNA fragment sizes after NGS library preparation. Figure S5. Overview of AAV 2/8-CMV-GFP-hTK-BGHpA vecors purification. Figure S6. Characterization of rAAV productions purity and titer. Figure S7. Overview of the protocol followed in this study. Figure S8. Percentage of single-nucleotide variants along rAAV genome for the plasmid control from the internal normalizer. Table S1. qPCR titration of rAAV and DNA contaminants. Table S2. Description of the samples analyzed by SSV-Seq. Table S3. Confusion matrix and mapping prediction rate of ContaVect determined without pre-processing of references. Table S4. Confusion matrix and mapping prediction rate of ContaVect determined with a pre-processing of references. Table S5. Distribution of contaminants in absolute number of reads. Table S6. Distribution of reads in a specific locus of chr15 and in the D-loop of mtDNA. Table S7. Comparative distribution of reads in AAV ITR extremeties with separated of merged AAV and vector backbone references. Table S8. Index Sequences. Table S9. Details of the QPCR and PCR Primers and amplification conditions.
Acknowledgments
We thank the Vectors production center of UMR 1089 (Nantes, Biogenouest, France) and the genomics and bioinformatics core facility of UMR 1087 (Nantes, Biogenouest, France) for their expert services. We are also grateful to Eduard Ayuso (VMD, PhD) for his critical reading and to Céline Vandamme for helping to maintain the blinding throughout the protocol. This work was supported by the “Association Française contre les Myopathies”, the “région Pays-de-la-Loire” (Emergence collective 2012, AGTI-1), and the “Commissariat général à l'investissement” (ANR-PGT Investissements d'avenir).
Supplementary Material
References
- Pierce, EA and Bennett, J (2015). The status of RPE65 gene therapy trials: safety and efficacy. Cold Spring Harb Perspect Med 5: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, K, Li, M, Zhong, L, Su, Q, Li, J, Li, S et al. (2014). Empty virions in AAV8 vector preparations reduce transduction efficiency and may cause total viral particle dose-limiting side-effects. Mol Ther Methods Clin Dev 1: 20139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, JM, Debelak, DJ, Reynolds, TC and Miller, AD (1997). Identification and elimination of replication-competent adeno-associated virus (AAV) that can arise by nonhomologous recombination during AAV vector production. J Virol 71: 6816–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, B, Duan, X, Chow, HY, Chen, L, Lu, H, Wu, W et al. (2014). Proteomics analysis of co-purifying cellular proteins associated with rAAV vectors. PLoS One 9: e86453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allay, JA, Sleep, S, Long, S, Tillman, DM, Clark, R, Carney, G et al. (2011). Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum Gene Ther 22: 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, GJ, Scotti, MM, Liu, J, Wang, L, Knop, DR and Veres, G (2011). Clearance and characterization of residual HSV DNA in recombinant adeno-associated virus produced by an HSV complementation system. Gene Ther 18: 135–144. [DOI] [PubMed] [Google Scholar]
- Martino, AT, Suzuki, M, Markusic, DM, Zolotukhin, I, Ryals, RC, Moghimi, B et al. (2011). The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood 117: 6459–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust, SM, Bell, P, Cutler, BJ, Ashley, SN, Zhu, Y, Rabinowitz, JE et al. (2013). CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest 123: 2994–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA Vaccines and Related Biological Products Advisory Committee (2012). FDA Briefing Document : Cell Lines Derived from Human Tumors for Vaccine Manufacture. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/UCM319573.pdf.
- Chadeuf, G, Ciron, C, Moullier, P and Salvetti, A (2005). Evidence for encapsidation of prokaryotic sequences during recombinant adeno-associated virus production and their in vivo persistence after vector delivery. Mol Ther 12: 744–753. [DOI] [PubMed] [Google Scholar]
- Wright, JF and Zelenaia, O (2011). Vector characterization methods for quality control testing of recombinant adeno-associated viruses. Methods Mol Biol 737: 247–278. [DOI] [PubMed] [Google Scholar]
- Noordman, Y, Lubelski, J and Bakker, AC (2013). Mutated rep encoding sequences for use in AAV production. http://www.google.com/patents/US20130023034.
- Nony, P, Chadeuf, G, Tessier, J, Moullier, P and Salvetti, A (2003). Evidence for packaging of rep-cap sequences into adeno-associated virus (AAV) type 2 capsids in the absence of inverted terminal repeats: a model for generation of rep-positive AAV particles. J Virol 77: 776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck, B, Murphy, SL, Smith, PH, Qu, G, Liu, X, Zelenaia, O et al. (2009). Undetectable transcription of cap in a clinical AAV vector: implications for preformed capsid in immune responses. Mol Ther 17: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H, Qu, G, Yang, X, Xu, R and Xiao, W (2011). Systemic elimination of de novo capsid protein synthesis from replication-competent AAV contamination in the liver. Hum Gene Ther 22: 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov, P, Chen, L, Dederich, D, Dong, B, He, J, Steinmann, KE et al. (2012). Native molecular state of adeno-associated viral vectors revealed by single-molecule sequencing. Hum Gene Ther 23: 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y, Ling, C, Song, L, Wang, L, Aslanidi, GV, Tan, M et al. (2012). Limitations of encapsidation of recombinant self-complementary adeno-associated viral genomes in different serotype capsids and their quantitation. Hum Gene Ther Methods 23: 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, LM, Christopher, DM, Giles, AR, Hinderer, C, Rodriguez, JL, Smith, JB et al. (2013). Lessons learned from the clinical development and market authorization of Glybera. Hum Gene Ther Clin Dev 24: 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso, E, Blouin, V, Lock, M, McGorray, S, Leon, X, Alvira, MR et al. (2014). Manufacturing and characterization of a recombinant adeno-associated virus type 8 reference standard material. Hum Gene Ther 25: 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis, N, Evelegh, C and Graham, FL (1997). Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology 233: 423–429. [DOI] [PubMed] [Google Scholar]
- Nicholls, TJ and Minczuk, M (2014). In D-loop: 40 years of mitochondrial 7S DNA. Exp Gerontol 56: 175–181. [DOI] [PubMed] [Google Scholar]
- Aiken, JM, Williamson, JL, Borchardt, LM and Marsh, RF (1990). Presence of mitochondrial D-loop DNA in scrapie-infected brain preparations enriched for the prion protein. J Virol 64: 3265–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Medicines Agency (2012). Glybera : EPAR - Public assessment report. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002145/WC500135476.pdf.
- Wilson, JM (2015). A call to arms for improved vector analytics! Hum Gene Ther Methods 26: 1–2. [DOI] [PubMed] [Google Scholar]
- Gavin, DK (2015). FDA statement regarding the use of adeno-associated virus reference standard materials. Hum Gene Ther Methods 26: 3. [DOI] [PubMed] [Google Scholar]
- Strong, MJ, Xu, G, Morici, L, Splinter Bon-Durant, S, Baddoo, M, Lin, Z et al. (2014). Microbial contamination in next generation sequencing: implications for sequence-based analysis of clinical samples. PLoS Pathog 10: e1004437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence, M, Hatzis, C and Brash, DE (2014). Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS One 9: e97876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird, D, Ross, MG, Chen, WS, Danielsson, M, Fennell, T, Russ, C et al. (2011). Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 12: R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogné, B, Snyder, R, Lindenbaum, P, Dupont, JB, Redon, R, Moullier, P et al. (2014). NGS library preparation may generate artifactual integration sites of AAV vectors. Nat Med 20: 577–578. [DOI] [PubMed] [Google Scholar]
- Thorne, BA, Takeya, RK and Peluso, RW (2009). Manufacturing recombinant adeno-associated viral vectors from producer cell clones. Hum Gene Ther 20: 707–714. [DOI] [PubMed] [Google Scholar]
- Smith, RH, Levy, JR and Kotin, RM (2009). A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol Ther 17: 1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso, E, Mingozzi, F, Montane, J, Leon, X, Anguela, XM, Haurigot, V et al. (2010). High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther 17: 503–510. [DOI] [PubMed] [Google Scholar]
- Kozarewa, I and Turner, DJ (2011). 96-plex molecular barcoding for the Illumina Genome Analyzer. Methods Mol Biol 733: 279–298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.