

Abstract
C/EBP homologous protein (Chop) has been shown to have altered expression in patients with idiopathic pulmonary fibrosis (IPF), but its exact role in IPF pathoaetiology has not been fully addressed. Studies conducted in patients with IPF and Chop−/− mice have dissected the role of Chop and endoplasmic reticulum (ER) stress in pulmonary fibrosis pathogenesis. The effect of Chop deficiency on macrophage polarization and related signalling pathways were investigated to identify the underlying mechanisms. Patients with IPF and mice with bleomycin (BLM)-induced pulmonary fibrosis were affected by the altered Chop expression and ER stress. In particular, Chop deficiency protected mice against BLM-induced lung injury and fibrosis. Loss of Chop significantly attenuated transforming growth factor β (TGF-β) production and reduced M2 macrophage infiltration in the lung following BLM induction. Mechanistic studies showed that Chop deficiency repressed the M2 program in macrophages, which then attenuated TGF-β secretion. Specifically, loss of Chop promoted the expression of suppressors of cytokine signaling 1 and suppressors of cytokine signaling 3, and through which Chop deficiency repressed signal transducer and activator of transcription 6/peroxisome proliferator-activated receptor gamma signaling, the essential pathway for the M2 program in macrophages. Together, our data support the idea that Chop and ER stress are implicated in IPF pathoaetiology, involving at least the induction and differentiation of M2 macrophages.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a severe health problem worldwide and has one of the poorest prognoses.1 Despite extensive studies, the mechanisms underlying IPF pathoaetiologies remain poorly understood. As a result, current therapies for this devastating disorder have been largely unsuccessful,2,3 resulting in an average survival time of only 3–5 years following diagnosis. Macrophages are the most abundant inflammatory cells after neutrophil degranulation during lung injury.4 In particular, macrophages are hallmarked by their plasticity and diversity, in which they can be activated either by interferon-gamma and lipopolysaccharides with manifestations of a classically activated phenotype (M1)5 or by IL-4 and IL-13 with characteristics of an alternatively activated phenotype (M2).6 Importantly, IPF patients are predominantly infiltrated by M2 macrophages during the course of disease development and progression.7 Evidence in animals also suggests that M2 macrophages could be a novel target for the prevention and treatment of pulmonary fibrosis.8,9,10 Indeed, upon activation, M2 macrophages produce profibrotic mediators such as transforming growth factor β (TGF-β) and platelet-derived growth factor to induce myofibroblast activation.11
C/EBP homologous protein (Chop) is a marker for endoplasmic reticulum (ER) stress following the phosphorylation of eukaryotic initiation factor 2 α and the upregulation of ATF-4.12 Although Chop has been recognized as being involved in the pathogenesis of diabetes,13 neurodegenerative disorders,14 renal dysfunction15 and experimental colitis,16 implying the pathogenesis of IPF, the mechanism has yet to be fully elucidated. Interestingly, a recent study indicated that ER stress may modulate the activation of M2 macrophages,17 and we further noted that macrophages originating from the lungs of patients with IPF exhibited significantly upregulated Chop expression. These observations prompted us to hypothesize that Chop modulates M2 macrophages in favour of pathological processes during the course of IPF development. Therefore, in the present report, we induced pulmonary fibrosis in mice and then assessed the impact of Chop deficiency on disease development. Remarkably, the loss of Chop significantly attenuated bleomycin (BLM)-induced pulmonary fibrosis and markedly reduced M2 macrophages in the lung. In line with this observation, depletion of macrophages in the lung or adoptive transfer of wild-type (WT) M2 macrophages into the Chop-deficient lung after macrophage depletion almost completely abolished the protective effect. Mechanistic studies showed that Chop regulates signal transducer and activator of transcription 6 (STAT6) phosphorylation and peroxisome proliferator-activated receptor gamma (PPAR-γ) expression, promoting the M2 program in macrophages. Together, our studies suggest that targeting Chop-related ER stress could be a viable strategy for the prevention and treatment of pulmonary fibrosis in clinical settings.
Results
Pulmonary fibrosis manifests altered Chop expression and ER stress
We first sought to examine the expression of Chop in the lungs of patients with IPF. Interestingly, Chop expression in lung sections from normal subjects was quite low (Figure 1a, left panel), whereas high levels of Chop were detected in IPF patient-derived lung sections (Figure 1a, right panel). In particular, patients with IPF were characterized by significant M2 macrophage infiltration, manifesting in the overexpression of Chop as evidenced by the costaining of Chop along with CD206, a marker for M2 macrophages (Figure 1a).
Figure 1.

Analysis of Chop expression and ER stress in patients with IPF and in BLM-induced mice. (a) Representative results for coimmunostaining of Chop and CD206, an M2 macrophage marker in the lung sections from patients with IPF and healthy subjects. The nuclei were stained blue by DAPI, and the images were taken under original magnification ×400. A total of eight patients with IPF and six control subjects were analyzed. (b) Western blot analysis of Chop expression in the lung of mice 21 days after BLM induction. (c–e) Analysis of additional ER stress markers: p-eIF2α (c), Atf4 (d) and Bip (e) in the lung of mice 21 days after BLM induction. Twelve mice were analyzed in each group. Bar graphs indicate the differences (mean ± SEM) between BLM- and saline-treated mice. **P < 0.01; ***P < 0.001. Atf4, activating transcriptional factor 4; BLM, bleomycin; ER, endoplasmic reticulum; IPF, idiopathic pulmonary fibrosis; p-eIF2α, phosphorylated eukaryotic initiation factor 2 α.
To confirm the above data, B6 mice were treated with BLM for 21 days followed by analysis of Chop expression in the lung homogenates. A twofold higher Chop expression was detected in BLM-induced mice as compared with control mice (Figure 1b); its upstream molecules, phosphorylated eukaryotic initiation factor 2 α (Figure 1c) and activating transcriptional factor 4 (Figure 1d) were increased as well. Another ER stress marker, Bip, was also significantly increased (Figure 1e). Collectively, these data support that ER stress that occurs during the development of pulmonary fibrosis.
Loss of Chop attenuates BLM-induced lung injury and fibrosis
Based on the above observations, we next sought to address the impact of Chop-related ER stress on pulmonary fibrosis in which Chop−/− and WT mice were induced with BLM, as above, and lung injury and fibrosis were assessed after 21 days of induction. Significantly attenuated lung injury and pulmonary fibrosis were noted in the Chop−/− mice as shown by the H&E, Sirius red, and Masson's trichrome staining (Figure 2a, left panel). In particular, the severity of pulmonary fibrosis was substantially lower, as shown by the lower Ashcroft scores (Figure 2a, right panel). Indeed, western blot analysis of lung homogenates revealed a significant reduction of collagen I (Figure 2b) and fibronectin expression (Figure 2c). Interestingly, both WT and Chop−/− mice were observed to undergo significant weight loss on day 7 of BLM induction, although a temporal increase in body weight was observed after this point. However, Chop−/− mice exhibited significantly less weight loss at all examined time points (Figure 2d). Together, our data support that loss of Chop protects mice against BLM-induced lung injury and fibrosis.
Figure 2.

Comparison of the severity of lung fibrosis between Chop−/− and WT mice 21 days after BLM induction. (a) Histological analysis for the severity of lung fibrosis in mice after BLM induction. Left panel: representative results for H&E (Top), Sirius red (center), and Masson staining (bottom). The inset shows higher magnification for a particular location. Right panel: a bar graph showing the semiquantitative Ashcroft scores for the severity of fibrosis. Images were taken under original magnification ×200. b, c Western blot analysis of fibrotic markers collagen I (b) and fibronectin (c). Left panel: a representative western blot result. Right panel: a bar graph showing the mean data of all mice analyzed in each group. (d) Body weight change during the course of BLM-induced fibrosis. Each bar represents mean ± SEM of 12 mice analyzed. *P < 0.05; **P < 0.01; ***P < 0.001. BLM, bleomycin; Coll, collagen I; Fib, fibronectin; WT, wild type.
Chop deficiency represses TGF-β signalling
Given the role that TGF-β plays in the progression of pulmonary fibrosis, we examined TGF-β1 expression in the lung. Interestingly, Chop−/− mice displayed substantially lower levels of TGF-β1 expression after BLM induction (Figure 3a). Similarly, ELISA analysis of mature TGF-β1 in the bronchoalveolar lavage fluids (BALF) revealed a onefold lower TGF-β1 secretion in BLM-induced Chop−/− mice than in control mice (Figure 3b). These results prompted us to examine TGF-β1 downstream molecules. Indeed, significantly higher levels of p-Smad2 and p-Smad3 were detected in BLM-induced WT mice, although there was no difference in total Smad2/3 (Figure 3c). Immunostaining revealed that airway epithelial cells were the major source of TGF-β1 before BLM induction and was predominantly localized in the infiltrated macrophages after BLM induction as determined by F4/80 costaining (Figure 3d).
Figure 3.

Loss of Chop attenuates TGF-β signalling after BLM induction. (a) Western blot analysis of TGF-β1 expression in the lung homogenates. Left panel: representative western blot results. Right panel: a bar graph showing the data of all mice studied. (b) ELISA results for TGF-β1 levels in the BALF. (c) Results for western blot analysis of TGF-β downstream Smad2 and 3 activities. Left panel: a representative western blot result for Smad2/3, p-smad2, and p-smad3 in the lung homogenates. Right panel: a bar graph showing the results of all mice examined. (d) Coimmunostaining of TGF-β1 and F4/80 in the lung sections. The images were taken under original magnification ×400. Each bar represents the mean ± SEM of 12 mice studied. **P < 0.01; ***P < 0.001. BALF, bronchoalveolar lavage fluids; BLM, bleomycin; TGF-β, transforming growth factor β.
Chop deficiency attenuates the M2 program in macrophages
To dissect the mechanisms by which Chop deficiency represses TGF-β1 secretion, we examined lung sections after BLM induction to characterize the cells with altered Chop expression. Similar to the data from patients with IPF (Figure 1a), Chop was predominantly overexpressed by the infiltrated macrophages (Figure 4a), although immunohistochemical staining revealed a moderate increase in Chop expression in airway epithelial cells. However, its relative intensity was significantly lower than in infiltrated macrophages (see Supplementary Figure S1). Of note, arginase-1, a marker for M2 macrophages, was highly expressed in all F4/80+ cells (Figure 4a), indicating an M2 phenotype for those macrophages. Indeed, a sixfold increase in arginase-1 expression was characterized in the lung homogenates of BLM-induced WT mice, while Chop deficiency attenuated BLM-induced arginase-1 expression by 1.6-fold (Figure 4c). Similarly, RT-PCR analysis of two additional M2 markers, YM1 (Figure 4d) and Found in Inflammatory Zone-1 (FIZZ1; Figure 4e), showed consistent results.
Figure 4.

Chop is predominantly overexpressed by the infiltrated M2 macrophages in the lung following BLM induction. (a,b) Results for coimmunostaining of Chop and F4/80 (a), and arginase-1 and F4/80 (b) in BLM-induced lung sections. Chop was significantly overexpressed in infiltrated macrophages, and type II alveolar epithelial cells also resulted in increased Chop expression in BLM-induced mice, while arginase-1 was predominantly localized within the infiltrated macrophages. (c) Results for arginase-1 expression in the lung homogenates. Left panel: a representative western blot result. Right panel: a bar graph showing the expression levels of arginase-1 of all mice examined for each group. (d) Real-time PCR results for analysis of YM1 expression in the lung. (e) Real-time PCR analysis of FIZZ1 expression in the lung. Each bar represents the mean ± SEM of 12 mice examined. *P < 0.05; **P < 0.01. Arg-1, arginase-1; BLM, bleomycin; FIZZ1, Found in Inflammatory Zone-1.
The aforementioned results suggest that Chop deficiency attenuates the induction of M2 macrophages, thereby repressing TGF-β1 secretion. To address this question, bone marrow-derived macrophages (BMDMs) were generated from WT and Chop−/− mice and then subjected to IL-4 stimulation, as described. Remarkably, flow cytometry analysis revealed a significantly lower number of M2 macrophages in Chop−/− BMDMs as manifested by CD206 expression (Figure 5a), and similar results were observed by RT-PCR analysis of YM1 (Figure 5b) and FIZZ1 expression (Figure 5c). Consistently, ELISA analysis of culture supernatants from IL-4-stimulated WT BMDMs showed significantly higher levels of TGF-β1 (Figure 5d). To further address the impact of Chop expression on the induction of M2 macrophages, we examined Chop temporal expression changes in BMDMs during IL-4 stimulation. Chop was almost undetectable in BMDMs before stimulation, but a steady increase was noted upon IL-4 stimulation, and the highest levels of expression were detected after 12 hours of IL-4 stimulation (Figure 5e). Overall, these data support that Chop modulates the induction of M2 macrophages and promotes the production of TGF-β. Notably, unlike its impact on M2 macrophages, Chop deficiency did not seem to affect the induction of M1 macrophages, as we failed to detect perceptible differences in the number of CD11b+F4/80+ macrophages following lipopolysaccharides stimulation (Figure 5f), suggesting that Chop might selectively modulate the M2 phenotype in macrophages.
Figure 5.

Loss of Chop inhibits the macrophage M2 program. (a) Flow cytometry analysis of CD206 expression in BMDMs following IL-4 stimulation. Left panel: representative results for flow cytometry analysis. Right panel: a bar graph showing the data with three replications. (b) Real-time PCR analysis of YM1 expression in IL-4-stimulated BMDMs. (c) Real-time PCR results for FIZZ1 in BMDMs after IL-4 stimulation. (d) ELISA analysis of TGF-β1 production in BMDMs following IL-4 stimulation. (e) Temporal Chop expression changes in BMDMs during the course of IL-4 stimulation. (f) Flow cytometry analysis of BMDMs following lipopolysaccharides stimulation. Each bar represents the mean ± SEM of at least six mice. *P < 0.05; **P < 0.01; ***P < 0.001. BMDMs, bone marrow-derived macrophages; FIZZ1, Found in Inflammatory Zone-1.
M2 macrophages are required for Chop deficiency-mediated protection
To further investigate whether the protective effect observed in Chop−/− mice was dependent on the induction of M2 macrophages, we first compared the differences in disease severity after macrophage depletion. Clodronate liposome was intratracheally administered to deplete macrophages in the lung, and the mice administered only liposomes (control vehicle) served as the controls. Indeed, macrophages were almost undetectable in the BALF of clodronate liposome-treated mice, and a threefold reduction for the total cell number was also noted as compared with liposome-treated mice (Figure 6a). Next, WT and Chop−/− mice were induced with BLM as above to induce pulmonary fibrosis 1 day after administration of clodronate liposome. As expected, WT and Chop−/− mice displayed comparable disease severity as manifested by the similar histological changes and Ashcroft scores (Figure 6b). Western blot analysis further demonstrated comparative lung expression levels of fibronectin and collagen I between WT and Chop−/− mice (Figure 6c).
Figure 6.

Loss of Chop protects mice from BLM-induced pulmonary fibrosis dependent on its effect on the macrophage M2 program. (a) Clodronate liposome efficiently depleted the macrophages in the lung; macrophages were almost undetectable in the BALF of clodronate liposome-treated mice along with a significant reduction for the number of total cells. (b) Depletion of macrophages restored Chop−/− mice with manifestations similar as WT mice after BLM induction, as evidenced by the comparable histological changes and Ashcroft scores. Left panel: representative results for H&E, Sirius red, and Masson staining and an inset picture was employed to show the indicated area at higher magnification. Right panel: a bar graph showing the semiquantitative Ashcroft scores for the severity of fibrosis. (c) Macrophage depleted WT and Chop−/− mice manifested comparable levels of collagen I and fibronectin expression in the lung after BLM induction. (d) Adoptive transfer of WT macrophages into Chop−/− mice restored their susceptibility to BLM-induced pulmonary fibrosis. Similar as above, the left panel presents representative results for H&E, Sirius red, and Masson staining, and the right panel displays the semiquantitative Ashcroft scores relevant to the severity of fibrosis. (e) western blot results for analysis of collagen I and fibronectin expression in the lung homogenates of above WT and Chop−/− mice with adoptive transfer studies. All images were taken under original magnification ×200, and eight mice were included in each study group. *P < 0.05; **P < 0.01; and ***P < 0.001. BALF, bronchoalveolar lavage fluids; BLM, bleomycin; Coll I, collagen I; Fib, fibronectin; WT, wild type.
Next, BMDMs originated from WT mice were treated with IL-4 for the induction of M2 macrophages, and equal number of IL-4-induced BMDMs was adoptively transferred through intratracheal injection into clodronate liposome-treated WT and Chop−/− mice at day 7 of BLM induction as described. Both WT and Chop−/− mice with adoptively transferred BMDMs developed severe lung injury and fibrosis following BLM induction. Remarkably, no perceptible difference in terms of disease severity was noted between WT and Chop−/− mice, as shown by the comparable histological changes and Ashcroft scores (Figure 6d), which was further confirmed by the observation of similar expression levels of fibronectin and collagen I between WT and Chop−/− mice (Figure 6e). Collectively, those data support that the loss of Chop protects mice against BLM-induced lung injury and fibrosis depends on the induction of M2 macrophages.
Loss of Chop enhances suppressors of cytokine signaling 1/3 (SOCS1/3) activity to repress STAT6/PPAR-γ signalling
STAT6/PPAR-γ signalling has been suggested to be critical for an optimal and sustained M2 program upon IL-4 or IL-13 stimulation.18 We, therefore, examined the impact of Chop deficiency on STAT6/PPAR-γ signalling in IL-4-stimulated BMDMs. No significant difference was detected in terms of total STAT6 between WT and Chop−/− BMDMs following IL-4 stimulation, and phosphorylated STAT6 (p-STAT6) was undetectable in both types of BMDMs before stimulation. However, high levels of p-STAT6 were detected after 0.5 hour of IL-4 stimulation, after which, p-STAT6 underwent a steady decrease and became undetectable following 12 hours of stimulation. A similar trend was noted for the Chop−/− BMDMs, but the p-STAT6 levels were substantially lower and even undetectable after 1 hour of IL-4 stimulation (Figure 7a). In line with these results, a steady increase in PPAR-γ expression was characterized in WT BMDMs upon IL-4 stimulation. The highest expression was noted following 12 hours of stimulation. By contrast, no perceptible change in PPAR-γ expression was observed in Chop−/− BMDMs during the course of IL-4 stimulation, and its expression after 12 hours of IL-4 stimulation was threefold lower than that of WT BMDMs (Figure 7a).
Figure 7.

The impact of Chop deficiency on IL-4-stimulated STAT6/PPAR-γ signalling and SOCS1 and 3 expressions in macrophages. (a) Loss of Chop attenuated IL-4-induced STAT6/PPAR-γ signalling. Left panel: a representative western blotting results for STAT6, p-STAT6, and PPAR-γ at different time points of IL-4 stimulation. Right panel: figures showing the data with three replications. (b) Chop deficiency enhanced SOCS1 and 3 expression during the course of IL-4 stimulation. Left panel: representative western blotting results for SOCS1 and SOCS3 at indicated time points of IL-4 simulation. Right panel: figures showing the data with three replications. (c) Chop deficiency did not affect MAPK (p38, ERK1/2, and JNK) and PI3K signalling. *P < 0.05; **P < 0.01; ***P < 0.001. PPAR-γ, peroxisome proliferator-activated receptor gamma; p-STAT6, phosphorylated STAT6; SOCS1 and SOCS3, suppressors of cytokine signaling 1 and suppressors of cytokine signaling 3; STAT6, signal transducer and activator of transcription 6.
To dissect the mechanisms by which Chop deficiency attenuates STAT6/PPAR-γ signalling, we examined the impact of Chop deficiency on the expression of SOCS1 and 3, which are inhibitors of STAT6 in macrophages.19 Only low levels of SOCS1 and 3 were detected in WT BMDMs, and no significant changes were observed following IL-4 stimulation. In sharp contrast, substantially higher levels of SOCS1 and 3 were detected in Chop−/− BMDMs, and IL-4 induced a steady increase for both SOCS1 and 3 with the highest expression detected following 12 hours of IL-4 stimulation (Figure 7b). Of note, MAPK and PI3K signalling are also implicated in macrophage M2 programming,20 but did not seem to be involved in the Chop-mediated M2 program, because we failed to detect a significant difference in terms of the phosphorylated form of p38, JNK, ERK1/2 and, PI3K between WT and Chop−/− BMDMs (Figure 7c). Together, our data support that Chop deficiency enhances SOCS1 and 3 expression, which then represses STAT6/PPAR-γ signalling to attenuate the M2 program in macrophages.
Discussion
In this report, we conducted studies in patients and animals to dissect the impact of Chop-related ER stress on the development of pulmonary fibrosis. We demonstrated that IPF patients and mice with onset of BLM-induced pulmonary fibrosis experienced altered Chop expression and ER stress. In particular, Chop was predominantly overexpressed by the infiltrated macrophages. As a result, the loss of Chop protected mice against BLM-induced pulmonary fibrosis. Mice deficient in Chop were characterized by a reduced TGF-β production along with attenuated Smda2/3 signalling following BLM induction. Mechanistic studies showed that Chop deficiency enhanced the expression of SOCS1 and 3 and then repressed the activity of STAT6/PPAR-γ signalling to attenuate the M2 program in macrophages. Thereafter, the loss of Chop reduced the production of TGF-β. As a result, WT and Chop−/− mice manifested with a similar disease severity following BLM induction once macrophages were depleted or WT M2 macrophages were adoptively transferred into the lungs after depletion of endogenous macrophages. These results not only provided novel insights into the understanding of IPF pathoaetiology but also provided evidence supporting that targeting Chop-related ER stress could be a viable strategy for the prevention and treatment of pulmonary fibrosis in clinical settings.
Although a great deal of effort has been recently devoted to dissect the pathoaetiologies underlying pulmonary fibrosis, the exact molecular mechanisms remained poorly understood. The lack of those related information significantly hampered the development of novel and effective therapies against this devastating disorder, reducing the survival rate for these patients to less than 5 years upon diagnosis. There is emerging evidence that ER stress is present in the lungs of patients with familial and sporadic IPF. Specifically, ER stress markers, such as ATF-6, ATF-4, and Chop, were significantly elevated in type II alveolar epithelial cells originated from IPF lungs.21 In particular, targeted induction of ER stress in type II alveolar epithelium exacerbated BLM-induced lung fibrosis along with increased apoptosis of alveolar epithelial cells and greater numbers of fibroblasts in the lungs.22 In line with these studies, we also detected ER stress in the type II alveolar epithelial cells of mice following BLM induction. Together, those data support that ER stress is implicated in the pathogenesis of pulmonary fibrosis, but the mechanisms by which ER stress leads to pulmonary fibrosis are yet to be elucidated.
To address the above question, we first examined inflammatory cells in IPF lungs and mouse lungs after BLM induction and found that pulmonary fibrosis was featured by a predominant macrophage infiltration. The most exciting discovery in this report is that those infiltrated macrophages also resulted in ER stress as evidenced by the elevated expression of Chop and other ER stress markers such as eIF-2α, activating transcriptional factor 4, and Bip. More importantly, those macrophages with altered ER stress showed an M2 phenotype as evidenced by CD206 coimmunostaining. These results support the notion that altered Chop expression along with ER stress may modulate the generation of M2 macrophages, which then trigger the pathological processes of pulmonary fibrosis. In support of this notion, Chop has been suggested as affecting the generation of M2 macrophages during atherosclerosis17 and the neutralization of CCL17, an M2-associated chemokine, abolishing the lethal effect in mice caused by BLM-induced lung fibrosis.23 In particular, we demonstrated that mice deficient in Chop were significantly protected from BLM-induced lung injury and fibrosis, although the protective effect was abolished once macrophages were depleted or WT M2 macrophages were adoptively transferred into the lung.
Because TGF-β is an essential factor for generating and maintaining a fibrotic milieu, we next conducted immunostaining of the lung sections to analyze TGF-β expression. Interestingly, TGF-β was only detected in epithelial cells under physiological conditions but was predominantly localized in infiltrated macrophages after BLM induction, suggesting that BLM induces pulmonary fibrosis through enhancing macrophage infiltration along with increased TGF-β secretion. Indeed, BLM induced a 43-fold increase in TGF-β production in the BALF, whereas Chop deficiency attenuated BLM-induced TGF-β production by onefold. Consistently, TGF-β downstream signalling was significantly inhibited in Chop−/− mice, as shown by the decreased levels of p-Smad2 and p-Smad3. To address how the loss of Chop attenuates TGF-β production, we next sought to examine the impact of Chop deficiency on the macrophage M2 program. Given that IL-4 has been shown to be the most powerful inducer of M2 macrophages,24 IL-4 was employed to stimulate both WT and Chop−/− macrophages followed by the analysis of phenotypical differences. In line with our expectation, flow cytometry analysis revealed that Chop deficiency significantly inhibited the IL-4-induced production of M2 macrophages as shown by the decreased number of CD206+ cells. Similarly, RT-PCR analysis of YM1 and FIZZ1, two additional M2 markers, further confirmed that loss of Chop attenuated IL-4 induced the expression of YM1 and FIZZ1 in macrophages. Importantly, studies, including ours, have also shown that Chop modulates BLM-induced epithelial apoptosis. Therefore, it is likely that Chop-mediated epithelial apoptosis serves as the initiating factor, and Chop-regulated macrophage recruitment and M2 program are critical causative factors during the course of BLM-induced pulmonary fibrosis.
The last important question is how Chop expression promotes the M2 program in macrophages. Previous studies have revealed that Th2 cytokines (e.g., IL-4 or IL-13) can stimulate STAT6 phosphorylation, which then directly induces macrophage expression of M2 genes.25 However, nuclear receptor PPAR-γ activity seems to also be required for full implementation of the M2 program.26 These observations prompted us to focus on the impact of Chop deficiency on STAT6/PPAR-γ signalling. Indeed, the loss of Chop markedly inhibited IL-4-induced STAT6 phosphorylation as shown by the significantly lower levels of p-STAT6 detected in Chop−/− BMDMs as compared with WT BMDMs. Consistently, IL-4 induced a steady increase of PPAR-γ expression in WT BMDMs but did not result in a perceptible change in PPAR-γ expression in Chop−/− BMDMs. To further confirm those data, we examined the effect of Chop deficiency on SOCS1 and 3 expression because they inhibit STAT6 signalling in macrophages.19 Notably, Chop−/− macrophages resulted in significantly higher levels of SOCS1 and SOCS3 than WT macrophages upon IL-4 stimulation. All together, our results suggest that the loss of Chop promotes SOCS1 and 3 expression in macrophages and then suppresses STAT6/PPAR-γ signalling to attenuate the induction of M2 macrophages.
Previous studies have suggested a negative feedback network between STAT6 phosphorylation and SOCS1 expression in macrophages upon IL-4 stimulation.27 Because Chop deficiency attenuates STAT6 phosphorylation, it would therefore be logical to observe increased SOCS1 expression in Chop−/− BMDMS following IL-4 stimulation. Interestingly, IL-4 was only shown to induce a rapid de novo SOCS1 expression in WT macrophages,20 whereas BMDMs deficient in Chop were found to result in enhanced expression for SOCS3 and SOCS1 following IL-4 stimulation. Chop has been characterized as the only inhibitor of the C/EBP family, in which it can form a heterodimer with C/EBPβ to repress its transcriptional activity.28 Because C/EBPβ is a crucial transcription factor for SOCS3 expression,29 the loss of Chop would release C/EBPβ from the inhibitory heterodimer with Chop, promoting SOCS3 transcription.
In conclusion, we demonstrated evidence indicating that altered Chop expression along with ER stress is a characteristic manifestation during the course of pulmonary fibrosis. Therefore, mice deficient in Chop are protected from BLM-induced lung injury and fibrosis. Mechanistic studies showed that Chop is involved in the pathogenesis of pulmonary fibrosis by regulating the generation of M2 macrophages and TGF-β signalling. Specifically, the loss of Chop promotes expression of SOCS1 and 3, which then represses STAT6/PPAR-γ signalling, thereby attenuating the induction of M2 macrophages. Together, our data show that targeting Chop and ER stress could be a viable strategy for the prevention and treatment of pulmonary fibrosis in clinical settings.
Materials and Methods
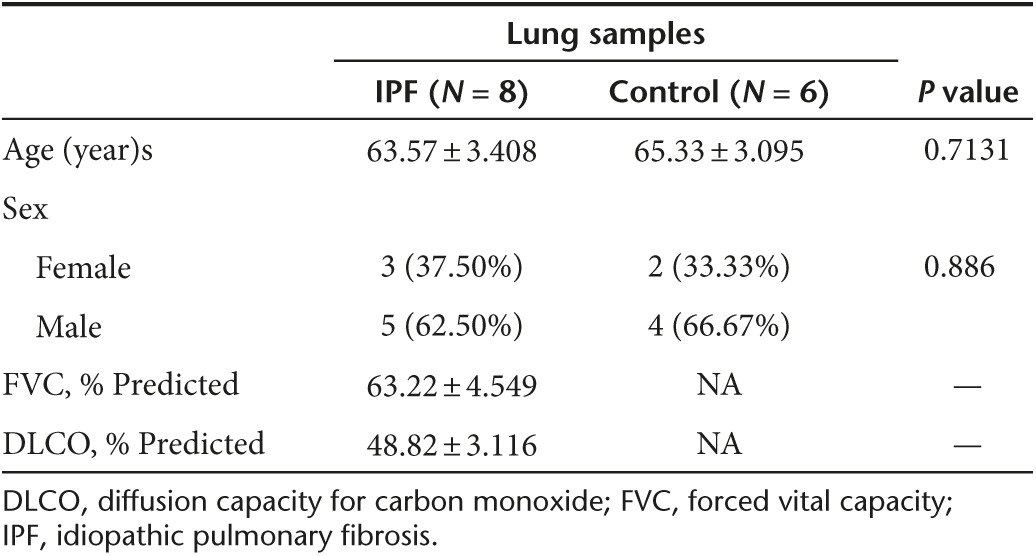
Human samples. Lung tissues from patients with non-small cell lung cancer (NSCLC, n = 6), patients with IPF (n = 8) were collected in the Tongji hospital after receiving informed consent. IPF was diagnosed according to the ATS/ERS consensus diagnostic criteria.30 The studies were approved by the Human Assurance Committee of Tongji Hospital. Clinical data and pulmonary function tests are provided in Table 1.
Table 1. Characteristics of subjects for lung samples.
Animals. Chop-knockout (Chop−/−) mice were purchased from the Jackson's Laboratory (Bar Harbor, ME). WT C57BL/6 (B6) mice were purchased from the Animal Experimental Center of the Hubei province (Wuhan, China). All animals were housed in an specific pathogen-free animal facility at the Tongji Medical College under a 12:12 hours light/dark photocycle provided with free diet and water ad libitum. All experimental procedures were approved by the Animal Care and Use Committee at the Tongji Hospital.
Reagents and antibodies. Collagen type I was purchased from EMD Millipore (Rockford, IL) , and murine recombinant IL-4 was obtained from PeproTech (Rocky Hill, NJ), and lipopolysaccharides and paraformaldehyde were obtained from Sigma (St. Louis, MO). Clodronate liposome was obtained from FormuMax (Shanghai, China). Antibodies against activating transcriptional factor 4, fibronectin, and arginase 1 were purchased from Abcam (Cambridge, MA), and phosphorylated eukaryotic initiation factor 2 α, p-Smad2, p-Smad3, Stat6, JNK, p-JNK, PI3Kp85, p-PI3Kp85, P38, and p-P38 antibodies were obtained from Cell Signaling (Danvers, MA). Bip antibody was obtained from BD Bioscience (San Jose, CA), Chop, CD206, TGF-β, GAPDH, β-actin, p-Stat6, SOCS1 and SOCS3 antibodies originated from Santa Cruz Biotechnology (Santa Cruz, CA), and ERK1/2 and p-ERK1/2 antibodies were from R&D Systems (Minneapolis, MN). Anti-mouse F4/80-PE and CD206-FITC were from Biolegend (Santa Cruz, CA), and CD11b-PerCP/Cy5.5 was from BD Pharmingen (San Diego, CA).
BLM induction of pulmonary fibrosis. WT and Chop−/− mice (8–10 weeks old) were anesthetized with 1% pentobarbital sodium and then subjected to 2 U/kg BLM administration (Nippon Kayaku, Japan) in 30 μl of normal saline via an intratracheal route. Mice administered the same volume of normal saline served as controls, and the mice were sacrificed 21 days after BLM administration for the analysis of pulmonary fibrosis.
Preparation of BALF. BALF was collected by cannulating the trachea and lavaging the lung with 0.6 ml of sterile PBS using established techniques,31 and ~0.4 ml of BALF was routinely recovered from each animal.
Histological and immunohistochemical analysis. The left lung was inflated with 4% neutral buffered paraformaldehyde by 25 cm of H2O pressure for 1 minute and were then removed and placed in fresh 4% neutral buffered paraformaldehyde for 24 hours at room temperature, followed by paraffin embedding and histological analysis, as previously reported.32 Each successive field was individually assessed for the severity of interstitial fibrosis in a blinded fashion by two pathologists using the Ashcroft scoring system,33 and six mice were included in each group. For immunostaining, the frozen sections (10 μm) were probed with antibodies against TGF-β, Chop, arginase-1 and F4/80. For immunostaining of the blood smear and lung sections from patients with IPF, the slides were first probed with a mouse-derived CD206 antibody and a rabbit-originated Chop antibody and were then stained with an Alexa 594-labelled anti-mouse and an Alexa 488-conjugated anti-rabbit antibody (Invitrogen, San Diego, CA), as instructed.
ELISA. The levels of TGF-β in the BALF and culture medium were measured using a TGF-β1 ELISA kit (eBioscience, San Diego, CA) using established techniques.34
Culture and treatment of primary BMDMs. Primary BMDMs were obtained from male mouse as previously reported.35 Bone marrow cells first underwent lysis of red blood cells and were then resuspended in 50 ml of culture medium consisting of RPMI 1640, 10% foetal bovine serum, penicillin/streptomycin, and 30 ng/ml macrophage colony-stimulating factor (MCSF). The cells were next plated across in 35 × 15-mm tissue culture dishes and maintained at 37 °C with culture medium changed every 2 days. After 7 days, the differentiated macrophages were cocultured with IL-4 (10 ng/ml) for the indicated time to collect supernatants to assay TGF-β secretion and protein analysis.
Macrophage depletion and macrophage adoptive transfer studies. Clodronate liposome (40 µl) was administered for two successive days intratracheally 1 day before BLM induction, and the severity of pulmonary fibrosis was assessed after 21 days of BLM induction. Total cell and macrophage counts in the BALF were conducted using Wright–Giemsa-stained cytospins to confirm the depletion of macrophages after 4 days of clodronate liposome treatment. For adoptive transfer studies, BMDMs derived from WT mice were stimulated by IL-4 (10 ng/ml) for 12 hours as described and were then transferred by intratracheal injection into the lungs of clodronate liposome-treated WT and Chop−/− mice at a density of 1 × 106 cells/mouse (50 μl) at day 7 of BLM induction, and the mice were sacrificed for the analysis of pulmonary fibrosis after 2 weeks of adoptive transfer as above.
Western blot analysis. Lung tissues and cultured cells were homogenized in RIPA lysis buffer (Biyuntian, China). The proteins were subjected to western blotting with the indicated primary antibodies using established techniques.36,37
Quantitative RT-PCR analysis. Quantitative RT-PCR analysis was performed using the SYBR Premix Ex Taq (Takara Dalian, China) as reported.38 The relative expression for each target gene was normalized by β-actin. The following primers were used for each target gene: YM1 (5′-GGG CAT ACC TTT ATC CTG AG-3′, 5′-CCA CTG AAG TCA TCC ATG TC-3′); FIZZ1 (5′-TCC CAG TGA ATA CTG ATG AGA-3′, 5′-CCA CTC TGG ATC TCC CAA GA-3′); Chop (5′-CAT ACA CCA CCA CAC CTG AAA G-3′, 5′-CAT ACA CCA CCA CAC CTG AAA G-3′); and β-actin (5′-TGA CGT TGA CAT CCG TAA AGA CC-3′, 5′-CTC AGG AGG AGC AAT GAT CTT GA-3′).
Flow cytometry analysis. The cultured BMDMs were stimulated by lipopolysaccharides (0.5 μg/ml) or IL-4 (10 ng/ml) for 12 hours, followed by staining with anti-mouse F4/80-PE, CD11b-PerCP/Cy5.5, and CD206-FITC. After washes, the cells were analyzed by flow cytometry as previously described.39 Data analysis was performed using the FACS Express V3 software (De Novo Software, Glendale, CA) as instructed.
Statistical analysis. All statistical analyses were conducted using standard one-way analysis of variance with Dunnett's multiple comparison test (GraphPad Prism 5.lnk, GraphPad Software, San Diego, CA). The data are presented as the mean ± SEM. In all cases, P < 0.05 was considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Immunohistochemical analysis of Chop expression in the lung sections of WT mice after BLM induction.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81130014, 81428001 and 81470226), the European Foundation for the Study of Diabetes (EFSD)/Chinese Diabetes Society (CDS)/Lilly Program for Collaborative Diabetes Research between China and Europe, the Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R20), and the Innovative Funding for Translational Research from Tongji Hospital. The authors declare no competing financial interests.
Supplementary Material
Immunohistochemical analysis of Chop expression in the lung sections of WT mice after BLM induction.
References
- Kolb, M and Collard, HR (2014). Staging of idiopathic pulmonary fibrosis: past, present and future. Eur Respir Rev 23: 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu, G, Behr, J, Brown, KK, Egan, JJ, Kawut, SM, Flaherty, KR et al.; ARTEMIS-IPF Investigators* (2013). Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med 158: 641–649. [DOI] [PubMed] [Google Scholar]
- Shulgina, L, Cahn, AP, Chilvers, ER, Parfrey, H, Clark, AB, Wilson, EC et al. (2013). Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: a randomised controlled trial. Thorax 68: 155–162. [DOI] [PubMed] [Google Scholar]
- Wynn, TA (2011). Integrating mechanisms of pulmonary fibrosis. J Exp Med 208: 1339–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica, A and Mantovani, A (2012). Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122: 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, T and Natoli, G (2011). Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11: 750–761. [DOI] [PubMed] [Google Scholar]
- Ji, WJ, Ma, YQ, Zhou, X, Zhang, YD, Lu, RY, Sun, HY et al. (2014). Temporal and spatial characterization of mononuclear phagocytes in circulating, lung alveolar and interstitial compartments in a mouse model of bleomycin-induced pulmonary injury. J Immunol Methods 403: 7–16. [DOI] [PubMed] [Google Scholar]
- Murray, LA, Rosada, R, Moreira, AP, Joshi, A, Kramer, MS, Hesson, DP et al. (2010). Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS One 5: e9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, B, Jin, W, Xu, J, Liang, Z, Yao, J, Zhang, Y et al. (2014). Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J Immunol 193: 2801–2811. [DOI] [PubMed] [Google Scholar]
- Gharib, SA, Johnston, LK, Huizar, I, Birkland, TP, Hanson, J, Wang, Y et al. (2014). MMP28 promotes macrophage polarization toward M2 cells and augments pulmonary fibrosis. J Leukoc Biol 95: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, E, Ouyang, N, Hörbelt, M, Antus, B, Wang, M and Exton, MS (2000). Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol 204: 19–28. [DOI] [PubMed] [Google Scholar]
- Hengstermann, A and Müller, T (2008). Endoplasmic reticulum stress induced by aqueous extracts of cigarette smoke in 3T3 cells activates the unfolded-protein-response-dependent PERK pathway of cell survival. Free Radic Biol Med 44: 1097–1107. [DOI] [PubMed] [Google Scholar]
- Oyadomari, S, Koizumi, A, Takeda, K, Gotoh, T, Akira, S, Araki, E et al. (2002). Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109: 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, RM, Ries, V, Oo, TF, Yarygina, O, Jackson-Lewis, V, Ryu, EJ et al. (2005). CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem 95: 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, JR, Yao, FH, Zhang, JG, Ji, ZY, Li, KL, Zhan, J et al. (2014). Ischemia–reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Renal Physiol 306: F75–F84. [DOI] [PubMed] [Google Scholar]
- Namba, T, Tanaka, K, Ito, Y, Ishihara, T, Hoshino, T, Gotoh, T et al. (2009). Positive role of CCAAT/enhancer-binding protein homologous protein, a transcription factor involved in the endoplasmic reticulum stress response in the development of colitis. Am J Pathol 174: 1786–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, J, Riek, AE, Weng, S, Petty, M, Kim, D, Colonna, M et al. (2012). Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem 287: 11629–11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, S and Martinez, FO (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32: 593–604. [DOI] [PubMed] [Google Scholar]
- Stevenson, NJ, Addley, MR, Ryan, EJ, Boyd, CR, Carroll, HP, Paunovic, V et al. (2009). CCL11 blocks IL-4 and GM-CSF signaling in hematopoietic cells and hinders dendritic cell differentiation via suppressor of cytokine signaling expression. J Leukoc Biol 85: 289–297. [DOI] [PubMed] [Google Scholar]
- Whyte, CS, Bishop, ET, Rückerl, D, Gaspar-Pereira, S, Barker, RN, Allen, JE et al. (2011). Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol 90: 845–854. [DOI] [PubMed] [Google Scholar]
- Korfei, M, Ruppert, C, Mahavadi, P, Henneke, I, Markart, P, Koch, M et al. (2008). Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 178: 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom, JW (1987). The interaction of rDNA factor VIII, factor VIIIdes-797-1562 and factor VIIIdes-797-1562-derived peptides with phospholipid. Thromb Res 48: 439–448. [DOI] [PubMed] [Google Scholar]
- Belperio, JA, Dy, M, Murray, L, Burdick, MD, Xue, YY, Strieter, RM et al. (2004). The role of the Th2 CC chemokine ligand CCL17 in pulmonary fibrosis. J Immunol 173: 4692–4698. [DOI] [PubMed] [Google Scholar]
- Pechkovsky, DV, Prasse, A, Kollert, F, Engel, KM, Dentler, J, Luttmann, W et al. (2010). Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol 137: 89–101. [DOI] [PubMed] [Google Scholar]
- Wei, Q, Sha, Y, Bhattacharya, A, Abdel Fattah, E, Bonilla, D, Jyothula, SS et al. (2014). Regulation of IL-4 receptor signaling by STUB1 in lung inflammation. Am J Respir Crit Care Med 189: 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard, JI, Ricardo-Gonzalez, RR, Goforth, MH, Morel, CR, Subramanian, V, Mukundan, L et al. (2007). Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447: 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickensheets, H, Vazq\uez, N, Sheikh, F, Gingras, S, Murray, PJ, Ryan, JJ et al. (2007). Suppressor of cytokine signaling-1 is an IL-4-inducible gene in macrophages and feedback inhibits IL-4 signaling. Genes Immun 8: 21–27. [DOI] [PubMed] [Google Scholar]
- Batchvarova, N, Wang, XZ and Ron, D (1995). Inhibition of adipogenesis by the stress-induced protein CHOP (Gadd153). EMBO J 14: 4654–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, TX, Lin, G, LaPensee, CR, Calinescu, AA, Rathore, M, Streeter, C et al. (2011). C/EBPβ mediates growth hormone-regulated expression of multiple target genes. Mol Endocrinol 25: 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu, G, Collard, HR, Egan, JJ, Martinez, FJ, Behr, J, Brown, KK et al.; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis (2011). An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, S, Li, J, Ma, L, Li, K, Zhang, L, Wang, G et al. (2013). Blockade of dopamine D1-like receptor signalling protects mice against OVA-induced acute asthma by inhibiting B-cell activating transcription factor signalling and Th17 function. FEBS J 280: 6262–6273. [DOI] [PubMed] [Google Scholar]
- Zou, R, He, Y, Li, YQ, Han, M, Ma, ZF, Liu, XC et al. (2014). Telmisartan protects 5/6 Nx rats against renal injury by enhancing nNOS-derived NO generation via regulation of PPARγ signaling. Am J Transl Res 6: 517–527. [PMC free article] [PubMed] [Google Scholar]
- Ashcroft, T, Simpson, JM and Timbrell, V (1988). Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol 41: 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, J, Yu, Q, Yang, P, Rao, X, He, L, Fang, J et al. (2014). MBD2 regulates TH17 differentiation and experimental autoimmune encephalomyelitis by controlling the homeostasis of T-bet/Hlx axis. J Autoimmun 53: 95–104. [DOI] [PubMed] [Google Scholar]
- Schwegmann, A, Guler, R, Cutler, AJ, Arendse, B, Horsnell, WG, Flemming, A et al. (2007). Protein kinase C delta is essential for optimal macrophage-mediated phagosomal containment of Listeria monocytogenes. Proc Natl Acad Sci USA 104: 16251–16256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, P, Zhang, Y, Pang, J, Zhang, S, Yu, Q, He, L et al. (2013). Loss of Jak2 impairs endothelial function by attenuating Raf-1/MEK1/Sp-1 signaling along with altered eNOS activities. Am J Pathol 183: 617–625. [DOI] [PubMed] [Google Scholar]
- Ran, L, Yu, Q, Zhang, S, Xiong, F, Cheng, J, Yang, P et al. (2015). Cx3cr1 deficiency in mice attenuates hepatic granuloma formation during acute schistosomiasis by enhancing the M2-type polarization of macrophages. Dis Model Mech 8: 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S, Lv, JW, Yang, P, Yu, Q, Pang, J, Wang, Z et al. (2012). Loss of dicer exacerbates cyclophosphamide-induced bladder overactivity by enhancing purinergic signaling. Am J Pathol 181: 937–946. [DOI] [PubMed] [Google Scholar]
- Zhong, J, Yang, P, Muta, K, Dong, R, Marrero, M, Gong, F et al. (2010). Loss of Jak2 selectively suppresses DC-mediated innate immune response and protects mice from lethal dose of LPS-induced septic shock. PLoS One 5: e9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunohistochemical analysis of Chop expression in the lung sections of WT mice after BLM induction.