

Abstract
Background: A long-standing epidemiological puzzle is the reduced rate of rheumatoid arthritis (RA) in those with schizophrenia (SZ) and vice versa. Traditional epidemiological approaches to determine if this negative association is underpinned by genetic factors would test for reduced rates of one disorder in relatives of the other, but sufficiently powered data sets are difficult to achieve. The genomics era presents an alternative paradigm for investigating the genetic relationship between two uncommon disorders.
Methods: We use genome-wide common single nucleotide polymorphism (SNP) data from independently collected SZ and RA case-control cohorts to estimate the SNP correlation between the disorders. We test a genotype X environment (GxE) hypothesis for SZ with environment defined as winter- vs summer-born.
Results: We estimate a small but significant negative SNP-genetic correlation between SZ and RA (−0.046, s.e. 0.026, P = 0.036). The negative correlation was stronger for the SNP set attributed to coding or regulatory regions (−0.174, s.e. 0.071, P = 0.0075). Our analyses led us to hypothesize a gene-environment interaction for SZ in the form of immune challenge. We used month of birth as a proxy for environmental immune challenge and estimated the genetic correlation between winter-born and non-winter born SZ to be significantly less than 1 for coding/regulatory region SNPs (0.56, s.e. 0.14, P = 0.00090).
Conclusions: Our results are consistent with epidemiological observations of a negative relationship between SZ and RA reflecting, at least in part, genetic factors. Results of the month of birth analysis are consistent with pleiotropic effects of genetic variants dependent on environmental context.
Keywords: Schizophrenia, rheumatoid arthritis, genetic relationship, pleiotropy
Key Messages
The proportion of variance in liability attributable to common SNPs is 0.223 (s.e. 0.006) for schizophrenia and 0.194 (s.e. 0.007) for rheumatoid arthritis.
The MHC region is proportionally more important for rheumatoid arthritis than for schizophrenia; after excluding this region, the proportion of variance attributable to common SNPs is reduced to 0.212 (s.e. 0.006) for schizophrenia and 0.137 (s.e. 0.007) for rheumatoid arthritis, respectively.
The epidemiological observations of a negative relationship between SZ and RA reflects, at least in part, genetic factors.
Introduction
A long-standing epidemiological puzzle is the reduced prevalence of rheumatoid arthritis (RA) in those with schizophrenia (SZ) and vice versa. First identified nearly 80 years ago, it has been reported in nearly every observational study of the incidence of the two disorders. A meta-analysis1 of nine studies found that risk of RA in SZ subjects was less than 29% of the risk in the general population. Under-reporting of somatic disease in those with severe psychiatric conditions may be a contributing factor,2 but the prevalence of RA is not reduced in those with other psychiatric disorders.1 Surprisingly, the reduced prevalences are observed despite the high prevalence of smoking in SZ [odds ratio (OR) = 5.3],3 which is an established risk factor for RA in general population samples (OR > 2).4 Both disorders have similar lifetime risk (∼1%), a waxing and waning pattern of symptoms and increased mortality. On the other hand, there are also dissimilarities including age at onset (16–30 years in SZ vs 25–55 years in RA)5 and male:female ratio (nearly 3 females to 1 male for RA5 and 1 female to 1.4 males for SZ6). Sex differences were not considered in early studies of the SZ-RA relationship, but recent population-based studies from Sweden2 and Denmark7 accounted for age and sex differences and still reported reduced risks of RA in SZ compared with those without SZ.
Both SZ and RA have a strong genetic component to their aetiology. For both, heritabilities estimated from national hospital records (SZ 64%8 and RA 40%9) are lower than estimates from twin studies (SZ 81%10 and RA 60%11). RA has two distinct subtypes classified on the presence (seropositive) or absence (seronegative) of antibodies to citrullinated protein antigen. Approximately two-thirds12 of cases are seropositive, and of seronegative cases 4–11% have been estimated to reflect misdiagnosis (for example of ankylosing spondylitis) and 15–37% have been estimated to be undetected seropositive.13 Using national data from Sweden, the heritability of seropositive RA was estimated as ∼50% compared with ∼20% for seronegative RA.9
A number of hypotheses have been proposed to explain the SZ-RA protective relationship,14 including abnormal tryptophan metabolism,15 prostaglandin deficiency,16–18 an imbalance in corticosteroids,19 psychosocial factors14 or consequence of medication.1 Definitive evidence to support these hypotheses is lacking. There is evidence that factors influencing immune activation, including environmental insults such as infectious agents, are potential pathogenic mechanisms for both disorders.5,20 For example, both RA and SZ have been linked, albeit with some controversy, to increased rates of infection by viruses such as Epstein-Barr virus and the parasite Toxoplasma gondii (for a review see5). SZ is considered to be a neurodevelopmental disorder, and immune activation in early life may be of particular importance, consistent with perinatal risk factors21 such as infection and month of birth.22 RA is an autoimmune disorder, and it has been suggested that there is an autoimmune component to SZ.23,24 The autoimmune theory of SZ is supported by epidemiological evidence showing that whereas the relationship between SZ and RA is a negative one, the risk of many other autoimmune disorders is higher in SZ than in controls.7,25,26 An analysis of Danish national records showed a dose-response relationship between risk of SZ and hospitalizations for infection and autoimmune disorders, where three or more infections and an autoimmune disease were associated with an incidence rate ratio of 3.40 [95% confidence interval (CI) 2.91–3.94].27 As for all autoimmune disorders,28 the major histocompatibility complex (MHC) plays an important role in RA29,30 but with different alleles being associated with seropositive cases compared with seronegative cases.13 A role for the MHC in the aetiology of SZ has been proposed for decades,31 but the empirical evidence has been less consistent than for RA. The first large genome-wide association studies (GWAS) for SZ identified the MHC locus as the most strongly associated locus.32–34 Using the latest published GWAS results,35,36 the MHC locus is the only locus that reaches genome-wide significance for both SZ and RA. The most associated single nucleotide polymorphism (SNP) for RA is associated with SZ and vice versa; contrary to expectation, given the negative SZ-RA association, the associated alleles are the same for the two disorders, albeit stronger for RA than SZ (Box 1). This unexpected result may reflect the well-recognised complexity of the MHC region.37 The primary association for rheumatoid arthritis is within HLA-DRB1 in the class II MHC region, and has a large effect relative to the more modest and less clearly localized effect in schizophrenia. It may be the case that the role of HLA-mediated antigen recognition is simply different in the two diseases, playing a dominant role in rheumatoid arthritis and perhaps a more modest or absent role in schizophrenia. If the negative association between RA and SCZ reflects genetic factors, then it may be driven predominantly by non-HLA genetic factors that are related to immune activation rather than antigen recognition. Genome-wide significant variants explain 3.4% of the variance in liability to schizophrenia and 11.4% of the variance in liability of RA (Box 1). Genome-wide polygenic methods38,39 have estimated that for SZ, ∼23%40 of the variance in liability is attributable to common SNPs (or SNP heritability), and 14%41 to 18%39 for RA excluding the contribution from the MHC region (∼5%). These results imply that more associated loci will be identified for each disorder as sample size increases.
-
MHC locus. Odds ratios for the two most highly associated SNPs for SZ, rs115329265 (aka rs1233578, hg19:chr 6: 28 712 247 bp), RAF = 0.85 and for RA, rs9268839 (aka rs116633882, hg19:chr 6: 32 428 772 bp) RAF = 0.45. Both are located in the MHC region and the LD r2 of these SNPs is zero. We note that the RA allele tags the HLA DRB-1 allele, but that the SZ allele is not associated with any classical HLA allele, although it is reported36 to be in LD r2 = 0.32 with an eQTL SNP for HLA-A. The association P -values are listed above the error bars.
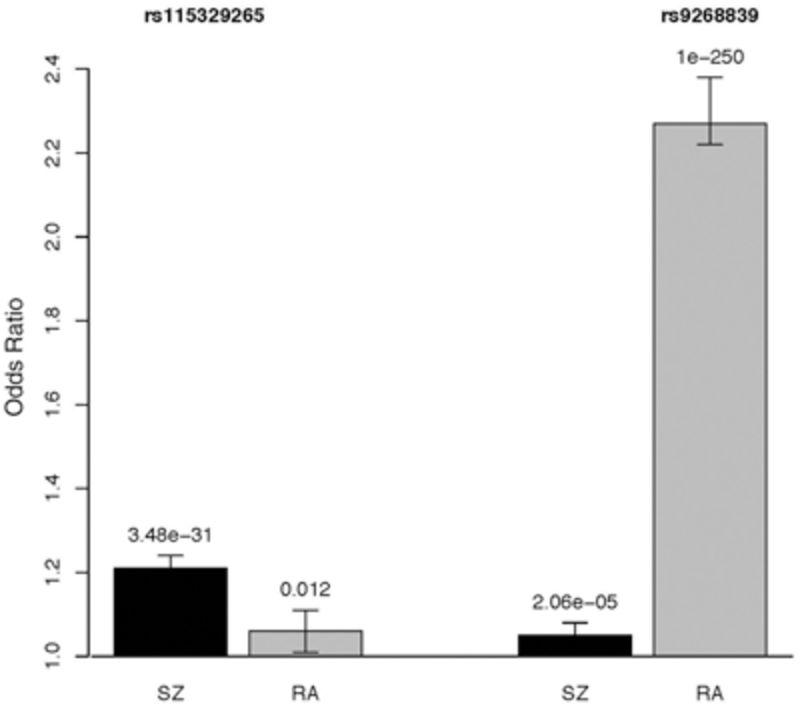
- Variance explained by genome-wide significant (GWS) loci, reported as associated at P < 5e-8.
- For SZ 128, statistically independent GWS loci are reported.36 Together these explain 3.4% of the variance in liability (calculated from reported RAF and OR using INDI-V71 assuming lifetime risk of 1%); all SNPs associated with P < 0.05 explain 7% of variance in out of sample prediction.36 Of these 128 SNPs, 102 could be matched to GWAS results for RA. The minimum P-value in RA of a SZ GWS locus was 0.004.
-
QQ plots: (i)- P-values from SZ GWAS for 101 RA GWS loci; (ii) P-values from RA GWAS for 102 SZ GWS loci.
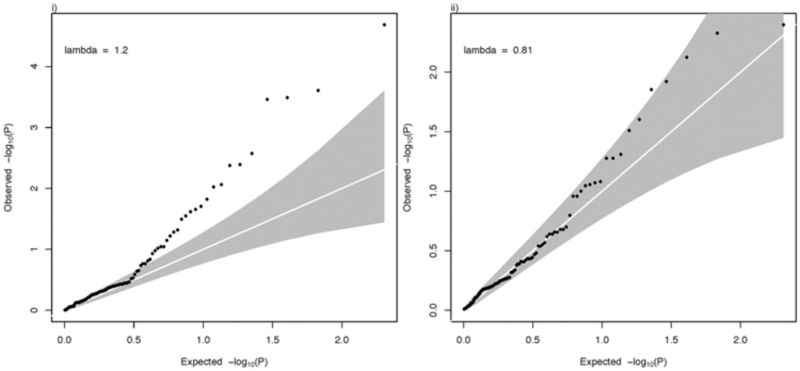
-
OR plots: (i) RA GWS loci (correlation 0.35, P = 4.0e-4, becomes non-significant if SNPs with the two largest OR are excluded); (ii) SZ GWS loci (correlation 0.24, P = 0.015, which becomes non-significant if the MHC SNP is excluded).
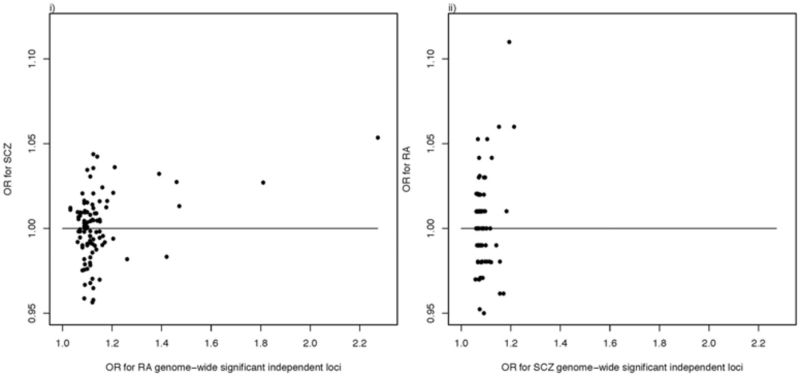
Given the substantial genetic contribution to both disorders, can the negative association between SZ and RA be attributed to genetic factors? This can be investigated from traditional epidemiological studies by measuring risk of RA in relatives of those with SZ compared with relatives of control subjects, and vice versa. As RA and SZ are relatively uncommon, very large cohorts of families with multiple family members measured for both disorders are needed, and this has not been achievable through the family study framework from which estimates of heritability are traditionally derived. National databases provide the only viable strategy to explore a genetic relationship through traditional epidemiological methods, but few countries have suitable national recording frameworks. A Danish national study25 compared 7704 persons in Denmark diagnosed with SZ and their parents with a sample of matched subjects and their parents. Contrary to a hypothesis of a negative genetic association, this study found significantly increased rates of RA in parents of those with SZ compared with parents of control subjects. A Swedish national study reported that first-degree relatives of schizophrenia patients were not at reduced risk of RA, but the risk for seronegative RA was significantly decreased in children and siblings of SZ probands [hazard ratio (HR) = 0.13; 95% CI 0.02–0.95, and HR = 0.67; 95% CI 0.50–0.91, respectively].2 These studies assumed that risk in first-degree relatives is only attributable to genetic factors, but sharing of environmental factors could also contribute.
The genomics era provides a new opportunity to investigate whether the SZ-RA relationship may be attributable to genetic factors, by determining whether common alleles conferring increased risk to SZ are protective against RA and vice versa. The hypothesis has been considered for candidate genes outside the MHC region, but no shared associations were found.42,43 Comparison of the latest GWAS results for SZ36 and RA35 shows evidence for more association of the genome-wide significant (GWS) loci from each disease in the other disease than expected by chance (Box 1). Across GWS SNPs, there is a positive correlation between the OR of the two disorders (Box 1), although this relationship is dominated by the positive correlation between SNPs in the MHC region described above.
Here we use linear mixed model methods, applied to genome-wide SNPs from case-control cohorts collected for GWAS, to explore the relationship between SZ and RA.44 By comparing additive genetic similarities between SZ and RA cases with their genetic similarities with controls, we quantify the relationship between the disorders by the SNP-genetic correlation.44,45 Since this approach uses unrelated cases and controls to estimate the SNP correlation, estimates are less likely to be confounded with shared environmental factors that can bias estimates from family studies or population studies where individuals are measured for both phenotypes. We use this framework to explore the genetic relationship between SZ and RA.
Methods
Data
Three RA and two SZ GWAS data sets (see Supplementary Table 1, available as Supplementary data at IJE online) were made available to us. Briefly, the Stahl et al. (‘Stahl’) RA sample comprises 5441 seropositive cases and 22 532 controls of European ancestry from six independent case-control cohorts.39,46 The Okada et al. (‘Okada’) RA sample has 3427 cases (1840 seropositive) and 6837 controls of European descent from five independent case-control cohorts,35 including the Corrona RA cohort. The Epidemiological Investigation of Rheumatoid Arthritis (EIRA) sample comprises 770 seronegative RA cases (EIRA seropositive cases from this cohort were already included in the Stahl sample). The Psychiatric Genomics Consortium (PGC) for Schizophrenia Wave 1 sample comprises data from 17 GWAS cohorts47 and a total of 9431 cases and 12 848 controls; and the Swedish (SWE) sample comprises 5193 case and 6391 controls48 in four cohorts defined by genotyping platform which were independent of Swedish samples in PGC. All sample sizes are those after sample quality control (QC).
All data sets were processed through similar QC and imputation pipelines47 using the CEU + TSI Hapmap Phase 3 data as the reference panel. We augmented these QC so that estimates of genetic variance would not be influenced by artefacts of genotyping.38 SNPs with an imputation r2 > 0.6 and an MAF > 0.01 in all cohorts were retained, resulting in 797 875 SNPs for analysis. Sex chromosome data were not available for all data sets and so were excluded. If any pair of individuals had an estimated similarity relationship coefficient49 >|0.05|, one person was excluded at random so that all SZ cases, RA cases and controls were unrelated. The final analysis data set consisted of 8064 seropositive RA cases (including 1131 RA cases of unknown status of which at least two-thirds12,13 are expected to be seropositive), 1197 seronegative RA cases and 26 737 controls plus and 12 793 SZ cases and 15 912 controls (Figure 1). Given accumulating evidence that seropositive and seronegative RA should be regarded as different clinical entities,13 we did not combine seropositive and seronegative RA cases.
Figure 1.

Sample sizes.
SNP heritabilityand SNP genetic correlation
The bivariate linear mixed model genomic relationship matrix (GRM) restricted maximum likelihood (GREML) approach44 implemented in GCTA50 was used to estimate SNP heritabilties, the SNP coheritability and the SNP correlation between the disorders. The standard error (s.e.) of each estimate was calculated by the delta method51 which has been shown to agree well with s.e. expected from normal distribution theory.52 We used the estimate and its s.e. to generate a Wald statistic to test hypotheses that SNP heritabilities were different from zero and that SNP correlations were less than zero; the directional hypothesis for the correlation is justified by the epidemiological data reported in the Methods section. The model of analysis estimates SNP heritabilities as the proportion of variance in case-control status attributable to genome-wide SNPs, but estimates of SNP heritabilites and coheritabilities are presented on the liability scale,38,44 assuming population lifetime risk of 1% for SZ, 0.7% for seropositive RA and 0.3% for seronegative RA, so that they can be compared with estimates from epidemiological data. We note that when genetic relationships between individuals are small, the relationship between disease and liability scale is approximately linear and so the estimated genetic correlation is independent of scale.44 A SNP correlation of zero is estimated if the genome-wide relationship between cases of one disorder is the same with the cases as with the controls of another disorder. A SNP correlation reflects the magnitude of the covariance term between the traits relative to the product of the standard deviations, and so can be high even when the covariance is low. A genome-wide SNP correlation could represent a uniform correlation across the genome or a weighted average of higher and lower correlations. Hence, we undertook genomic partitioning analyses which included multiple additive genetic random effects terms in the linear mixed model with multiple GRM constructed from non-overlapping SNP sets.45,49 Cohort and the first 20 principal components were included as covariates in all analyses. Sex was included as a covariate for SZ and in some analyses for RA. SNP heritabilities are presented on the liability scale. Follow-up analyses were conducted by sex and considering functional annotation.
Sensitivity analysis
We explored the sensitivity of our results and sought to exclude the possibility that genetic outliers could explain our results. We tested this by restricting the coefficient of similarity between any pair of individuals to be < |0.025|. As one individual from a pair was excluded at random, we constructed 20 randomly drawn samples with restricted ancestry, and drew 20 random samples of the same size from the sample with coefficient of similarity < |0.05|.
Benchmarking with epidemiological observations
From epidemiological studies we can obtain estimates of the population risk of SZ and RA, KSZ and KRA, respectively, and also for the probability of RA in those with SZ, KRA|SZ. We assume that the phenotypic liabilities of SZ (lSZ) and RA (lRA) are distributed as bivariate normal with mean 0, standard deviation 1 and correlation :
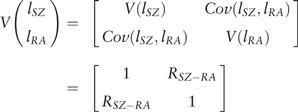 |
The variances and covariance of liabilities among those affected with SZ
where ksz = isz(isz–tsz) and reflects the proportional variance reduction as a consequence of ascertainment on SZ status,50,51 with mean , with dsz the height of the normal curve at the threshold tsz defined from . From KRA|SZ we define the normal distribution threshold53,54 for RA in those with SZ as
Solving the quadratic for gives
| (equation 1) |
Genotype x environment analysis
Our analyses led us to a postulate a hypothesis of genotype x environment interaction for SZ. Specifically we hypothesized that the SNP correlation would be less than 1 for coding and regulatory SNPs in a bivariate analysis in which the two traits are winter-born and non-winter born SZ cases and controls. We undertook a bivariate GREML analysis to test this hypothesis. Month of birth was only available for the SWE SZ sample, which comprised winter-born cases (born January to April22, n = 1511) and winter-born controls (n = 2036), as well as non-winter cases (n = 2962) vs non-winter controls (n = 3772); 199 individuals did not have month of birth recorded. To visualize the interaction, we identified 47318 SNPs associated with schizophrenia36 at P < 0.05 (the threshold that maximized out of sample prediction across multiple cohorts;36 the SNPs were quasi-independent with minor allele frequency > 0.05, pairwise linkage disequilibrium r2 < 0.25 in a 250-kb window). We identified the risk alleles of the SNPs that defined the odds ratio to be greater than 1. We undertook association analysis (logistic regression with 20 principal components, cohort and sex as covariates) in the Swedish sample that had season of birth recorded. We estimated the OR of the risk alleles and compared mean OR for SNPs annotated as C&R (coding/regulatory, the genomic region showing strong negative SNP correlation between SZ and RA, 2820 SNPs, 6%) and not coding/regulatory, testing the hypothesis H0: Mean OR for winter-born sample = Mean OR for other sample.
Results
SNP heritability and SNP genetic correlation
The estimated SNP heritability for RA seronegative cases was indistinguishable from zero (−0.006, s.e. 0.025, P = 0.98). Despite the smaller sample size for seronegative cases, the s.e. shows that it was powered to detect SNP heritability > 5%. Given there was no evidence of contribution to risk of common variants for this sample, detection of a genetic relationship between these cases and SZ was not possible. Hence, all reported analyses are for seropositive RA cases only. Given the major contribution of the MHC region in RA (which may violate underlying assumptions of GREML55,56), we undertook analyses using as the phenotype residuals after adjusting for the 550 SNPs (the number after pruning for SNPs with linkage disequilbrium r2 > 0.99) located within the MHC region (29–34Mb in chromosome 6) and for the other covariates. For SZ, the estimated SNP heritabilities were 0.223 (s.e. 0.006) including the MHC region and 0.212 (s.e. 0.006) after correcting for the MHC region. For RA, the estimated SNP heritabilities were 0.194 (s.e. 0.007) including the MHC region and 0.137 (s.e. 0.007), after correcting for the MHC region. The estimated SNP genetic correlations were −0.046 (s.e. 0.026) and −0.065 (s.e. 0.030) for including the MHC and after correcting for the MHC, respectively, which were significantly less than zero (P-values = 0.036 and 0.015, respectively) (Figure 2; Supplementary Table 2, available as Supplementary data at IJE online). We confirmed that the method to estimate P-values was robust, by checking the P-value from a likelihood ratio test comparing models with and without genome-wide SNP effects.
Figure 2.
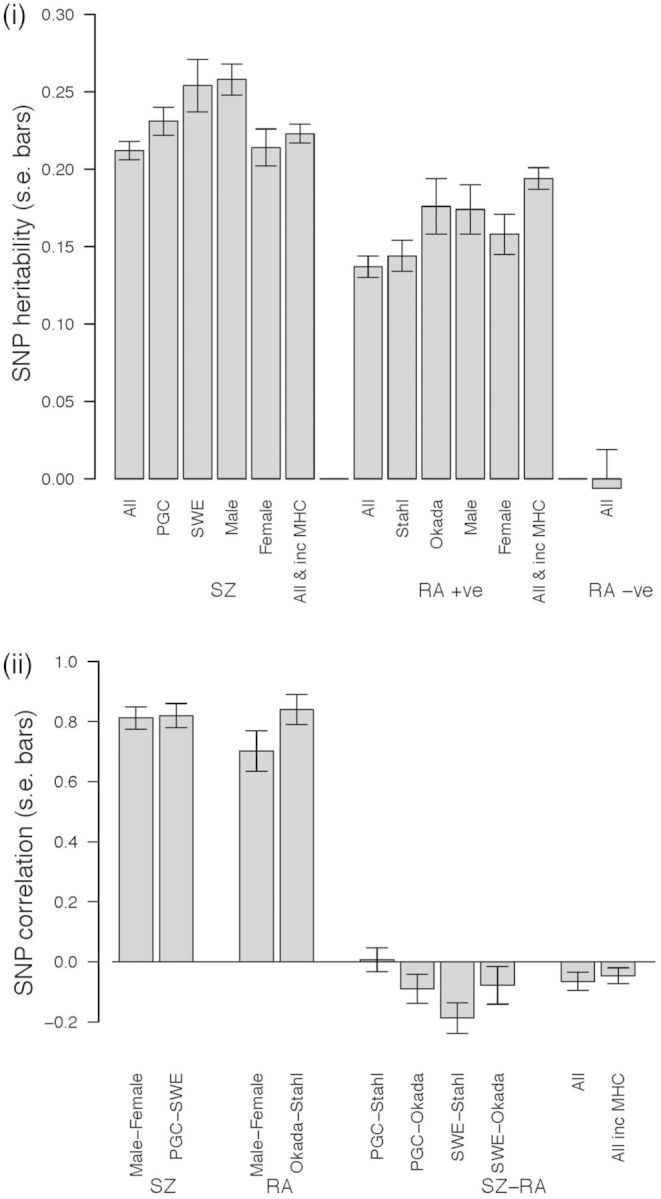
Genome-wide estimates: (i) SNP heritability; (ii) SNP correlation (using RA + ve only). Estimates are from analyses using as the phenotype residuals after adjusting SNPs in the MHC region unless otherwise stated.
To aid interpretation and comparisons, we also present the SNP genetic covariance or coheritability, the latter represents the relationship between the disorders on the same scale as the heritability (Supplementary Table 2). Subset analyses (PGC-SZ/Stahl-RA, PGC-SZ/Okada-RA, SWE-SZ/Stahl-RA and SWE-SZ/Okada-RA) showed negative genetic correlations estimated for all combinations except PGC-SZ/Stahl-RA (Figure 2, Supplementary Table 2). We explored the sensitivity of our results and sought to exclude the possibility that genetic outliers could explain our results (see Methods). We found that the SNP correlation between SZ and RA was significantly (P = 0.0049) more negative in 20 samples drawn from our data when ancestry was more restricted (similarity relatedness coefficient < |0.025|) [−0.054, standard deviation over replicates (s.d.) 0.002 vs −0.047 s.d. 0.001] (Supplementary Figure 1). This sensitivity analysis implies that the negative correlation is not driven by ancestry artefacts and provides confidence that SNP correlation between SZ and RA is negative.
Sex analyses
Given that risk of RA is higher in females and risk of SZ is higher in males, we undertook SZ/RA analyses stratified by sex (i.e. four-trait multivariate GREML, in which the four traits were SZ-male, SZ-female, RA-male, RA-female each matched by their sex-specific control set) to determine if SNP correlations (based on autosomal SNPs) were sex dependent. Sex information was missing for ∼11% of the RA sample who were excluded in the analysis. Based on reported male:female population ratios,5,6 we assumed the male and female baseline risks were 0.42% and 0.98% for seropositive RA, and 1.15% and 0.85% for SZ respectively. SNP heritabilities were significantly greater when estimated from males compared with females for SZ (male 0.258, s.e. 0.010, female 0.214, s.e. 0.012, P = 0.0053) but not for RA (male 0.174, s.e. 0.016, female 0.158, s.e. 0.013, P = 0.43) (Figure 2, Supplementary Table 2); these estimates must be interpreted with caution, recognizing that they are dependent on the lifetime risk of disease chosen for each sex. SNP correlations between sexes were high but were significantly different from 1 for both RA (P = 6.1e-06) and SZ (P = 2.4e-07, Supplementary Table 2). All SNP correlation point estimates are negative between male/female SZ/RA analyses.
Functional annotation analyses
Previous studies40,41,57 have demonstrated that contributions to SNP heritabilities are not distributed equally over the genome. We therefore set out to test if the SNP-correlation between SZ and RA was dependent on SNP annotation. We undertook genomic partitioning analyses in which multiple additive genetic random effects terms were considered in the linear mixed model,45,49 with multiple GRM each constructed from SNPs grouped by a functional annotation. Following Gusev et al.,57 SNPs were classified as being in coding/regulatory (in exons, 3’ UTR, 5’UTR, 1-kb region up- and downstream of transcription start and end site and noncoding RNA), DNase I hypersensitivity sites (DHS) and intronic or intergenic regions. SNPs with multiple annotations were allocated with hierarchical preference of coding/regulatory, over DHS and over intronic. For SZ, all annotations had estimates of SNP heritability that were significantly greater than zero (Figure 3; Supplementary Table 3, available as Supplementary data at IJE online), although the proportion of total variance allocated to intergenic SNPs was significantly less than expected given the proportion of all SNPs annotated to that group (Figure 3; Supplementary Table 3). For RA, the coding/regulatory and DHS annotations had SNP heritability estimates that were both significantly different from zero and were higher than expected based on the proportion of SNPs in those functional partitions (Figure 3; Supplementary Table 3). As a consequence in RA, and in contrast to SZ, neither intronic nor intergenic variants made a significant contribution to SNP heritability, a finding that is underscored by the fact that the proportion of all SNP heritability contributed by these types of variant is significantly less than expected based upon the proportion of SNPs of these classes. Given that in RA, SNP heritability is essentially restricted to SNPs in coding/regulatory and DHS regions, these are the only classes of SNP for which meaningful estimates of SNP correlation between SZ and RA can be made. We note that currently available DHS annotation may be biased towards cell types of relevance to RA compared with SZ. We estimated a significant and stronger genetic correlation for coding/regulatory region regions (−0.322, s.e. 0.115, P = 0.003) (Figure 3; Supplementary Table 3) than for the whole genome.
Figure 3.
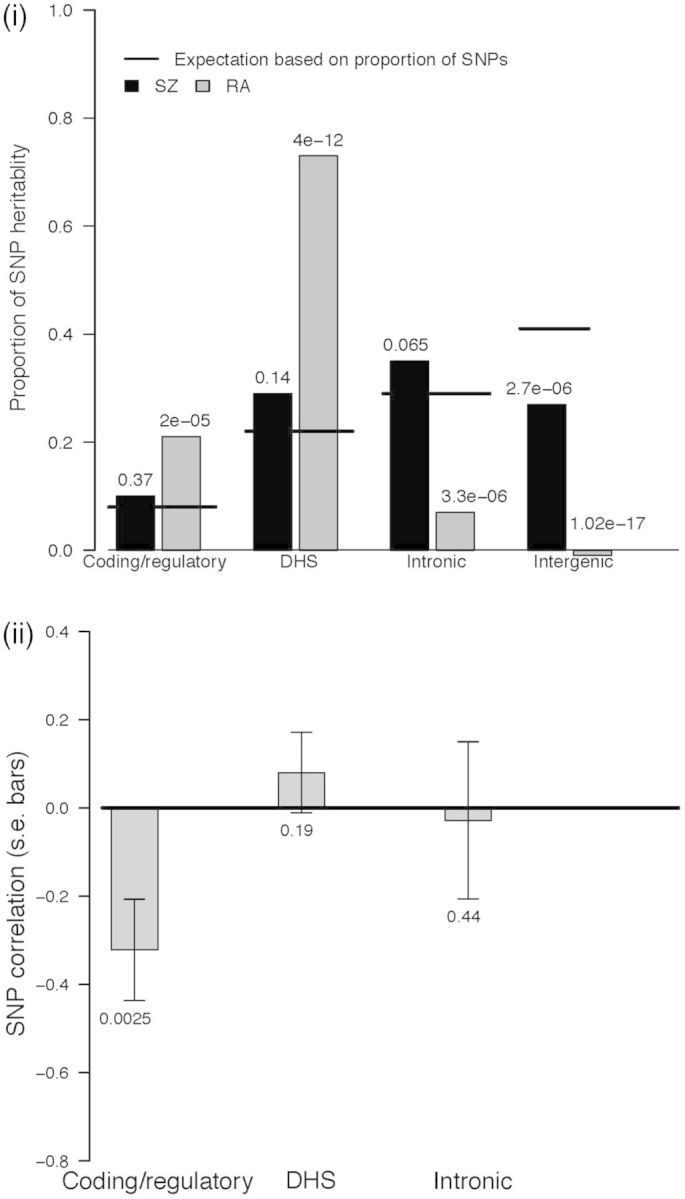
Genomic partitioning analyses; (i) Percentage of SNP heritability attributed to each functional annotation class for SZ and RA, compared with the percentage of SNPs attributed to each class. Adjacent to the bars are the P-values for H0: percentage of variation attributed to annotation class = percentage of SNPs attributed to the annotation class; (ii) SNP correlation between SZ and RA based on annotation. Estimates are from analyses using as the phenotype residuals after adjusting SNPs in the MHC region. When the MHC is included, the coding/regulatory correlation is −0.174, s.e. 0.071, P = 0.0075.
Immune related pathway analyses
Since RA is an autoimmune disease and since the epidemiological negative relationship between SZ and RA has contributed to the autoimmune hypothesis of SZ, we set out to test if the SZ-RA SNP correlation is more negative in SNPs in immune-related pathways. To avoid multiple testing, we selected a single immune gene set based on previous work using the Stahl GWAS results of Hu et al.58 Using gene expression from 223 murine immune cell types, they reported over-representation of RA-associated SNPs in genes expressed specifically in CD4+ effector memory T cells, with strongest over-representation in genes expressed in the subcutaneous lymph node subset named T.4Mem44h62l.LN.58 We selected the top 4000 genes expressed in the T.4Mem44h62l.LN cells to be in the ‘T4Mem’ set. The arbitrary threshold of 4000 genes generated an SNP set of about 10% of the total SNPs analysed. We tested if the T4Mem genes make an enriched contribution to SNP heritabilities and the SNP correlation, partitioning the coding/regulatory, DHS and intronic partitions into T4Mem and non-T4Mem classes (Supplementary Table 4, available as Supplementary data at IJE online). As expected, there was a significant enrichment for variance attributable to the T4Mem class for RA (28% of the SNP heritability compared with only 11.5% of SNPs, 2.5 fold enrichment, P = 2.0e-11). Interestingly, there was also significant enrichment of variance attributable to T4Mem regulatory SNPs in SZ (16% of SNP heritability, P = 2.4e-04). We did not find evidence that SNPs in this group of genes were more negatively correlated than those in the rest of the genome, although the patterns of correlations are difficult to interpret, and the size of the standard errors means that the sample is underpowered (Supplementary Table 4).
Benchmarking with epidemiological observations
We can benchmark our estimated SNP correlation between RA and SZ of −0.046 relative to expectation from epidemiological data. Using the meta-analysis result that the risk of RA in those with SZ is 29% of the risk in the general population,1 we estimate that this implies a phenotypic correlation between the disorders of −0.15 (equation 1; KSZ = 0.01 and KRA = 0.01, respectively, KRA|SZ = 0.29*KRA). More recent epidemiological studies2,7 imply a substantially smaller negative phenotypic correlation ( ∼−0.05). Phenotypic correlation is considered a reasonable benchmark for genetic correlation,59 and therefore our small negative estimate of the genetic correlation is consistent with the epidemiological data. However, the genetic relationship between SZ and RA is complex. As an autoimmune disease, the MHC region contains risk factors for RA which alone explain ∼5% of the variance in liability to RA, whereas the most significant individual SNP association for SZ, also in the MHC region, explains only ∼0.1% of variance in liability (ref60 and consistent with Table 1 with and without MHC region). These MHC risk alleles are positively correlated in SZ and RA, although clearly the effect sizes are very different. In analyses in which we first removed the contribution to variance of the MHC region, the magnitude of negative correlation between SZ and RA increased (−0.065, s.e. 0.015, P = 0.015), and increased further still when considering only SNPs in coding and regulatory regions of the genome (−0.322, s.e. 0.115, P = 0.003), indicating that regions of the genome other than the MHC region contribute to the epidemiological observations.
Genotype x environment analysis
Although RA is well recognized as an immune disorder, the contribution of immune activation to SZ is open to debate (see Introduction). To add further insight to the complex relationship between SZ and RA, we postulated that if there is any interplay in risk for SZ between environmental risk factors associated with immune challenge and genes that are relevant to the immune response, it is likely to occur at the coding and regulatory regions. This is based on the rationale that these are the sets of variants that both capture the SNP heritability of RA (i.e. are likely to be most enriched for genes with influences on immune activation) and are negatively correlated with SZ (i.e. are both immune activation and SZ relevant). If our hypothesis is correct, we predict that the apparent effect sizes in schizophrenia at these loci will be greater in those exposed to a relevant immune challenge than in those who are not. Consequently, we predict that the negative correlation between SZ and RA will be larger at these loci in cases exposed to an immune challenge. At present, our ability to test this G x E hypothesis is limited by availability of samples that are both genetically informative and recorded for environmental risks. We therefore sought a proxy for immune challenge in SZ.
A robust epidemiological finding in SZ research is that people with the disorder are more likely to be born in winter or spring than summer or autumn61 (odds ratio 1.07, 95% CI 1.05–1.08, estimated from a meta-analysis of 27 studies62). Winter/spring birth is also associated with recognized immune-mediated disorders including RA.63 Candidate exposures underlying this finding include seasonally varying factors such as prenatal vitamin D, or maternal/fetal exposure to infections. Both factors can impact on the immune system.64–66 In the absence then of direct measures of immune activation, we used season of birth as our proxy measure; in doing so, we are aware this proxy measure is only likely to be weakly correlated with exposure and hence its use will adversely affect power.
Month of birth was only available for the SWE SZ sample. We selected quasi-independent SNPs associated at P < 0.05 and minor allele frequency > 0.05 (47 318 SNPs) from the largest published schizophrenia meta-analysis.36 We next divided them into coding/regulatory (2820 SNPs, 6%) and non coding/regulatory sets. We undertook a bivariate GREML analysis of the SWE datasets in which the two traits were winter-born (January to April) vs non-winter born SZ cases and controls; this division was justified by studies using Swedish data.22 The correlation between winter/non-winter born was significant for coding/regulatory SNPs (0.56, s.e. 0.14, P = 0.0009) but not so for other SNPs (0.95, s.e. 0.05, P = 0.15) (Supplementary Table 5, available as Supplementary data at IJE online). To visualize this interaction, and to demonstrate that effects sizes of the coding/regulatory SNP set are increased in the winter-born cohort, we present the mean association OR for the different season by annotation classes estimated in the SWE SZ sample (Figure 4). We confirmed that results were robust to the P-value threshold used for selection of associated SNPs (Supplementary Table 6, available as Supplementary data at IJE online) and we checked the seasonal trend by using sliding window definitions of 4-month season definitions (Supplementary Table 7, available as Supplementary data at IJE online). The results are consistent with our G x E hypothesis of immune-related disruption proxied by winter birth, in concert with risk variants in coding/regulatory regions increasing risk of schizophrenia.
Figure 4.

Relationship between season of birth and association odds ratios of SNPs associated with SZ at P < 0.05 identified in the GWAS of the Psychiatric Genomics Consortium.
P1 H0: OR Winter C&R SNPs = OR for Non-Winter C&R SNPs. HA: OR Winter C&R SNPs > OR for Non-Winter C&R SNPs. P2 H0: OR Winter C&R SNPs = OR for Winter All SNPs except C&R. HA: OR Winter C&R SNPs > OR for Winter All SNPs except C&R. C&R = Coding and Regulatory (2820 SNPs). All SNPs except C&R = All SNPs except Coding and Regulatory (44498 SNPs). Winter-born cases (N = 1511) and controls (N = 2036) were born January to April; N = 2962 for non-winter cases and N = 3772 for non-winter controls.
Strengths and limitations
The strength of our methodological approach to explore the genetic relationship between SZ and RA is that it is based on genome-wide genotype data and uses independently collected data for the two diseases studied. While under review, two studies have investigated the relationship between SZ and RA based partially67 or fully68 on GWAS summary statistics; one reported a non-significant small positive relationship67 and the other a non-significant small negative relationship.68 Use of genotype data is computationally more demanding but is considered more definitive than methods based on summary statistics,57 and such an approach is needed when the relationship between the disorders is benchmarked by a weak phenotypic correlation of −0.05 to −0.15. The results here indicate that there is a subset of genetic variants that are risk alleles for SZ and protective for RA, and vice versa. Furthermore, as sample sizes increase, more fine-scaled partitioning of variance by annotation and genomic region will become possible and specific variants involved will be identified. Our analyses indicate that these pleiotropic loci are located in the coding/regulatory regions of the genome, hinting at directly functional variants. Pleiotropy could help explain why common risk variants are maintained in the population, since selection operates on the net effect of fitness.
As limitations, we first note that cases and controls for each disorder were not screened for the other disorder. However, since both disorders are uncommon, the impact is expected to be small. Second, we were unable to investigate the relationship between SZ and seronegative RA because we found no evidence of SNP heritability for this case group. An analysis of seronegative RA is of particular interest since a recent epidemiological study found that decreased risk of RA in relatives of those with SZ was only apparent for seronegative RA.2 The stringent QC needed for case-control GREML analyses may remove true signals38 and this may play a role in the seronegative samples available here. Nonetheless, a lower SNP heritability for seronegative RA vs seropositive RA is consistent with recent estimates of heritability based on family data,9 and also consistent with smaller OR for seronegative genetic associations than for seropositive associations.13,69 A SNP correlation of 0.98, s.e. 0.165, was estimated in a Han Chinese sample;69 we note that the proportion of seronegative cases in this study was high (519/952 = 55%) and any misclassification could serve to inflate the correlation.70 Third, the non-availability of sex chromosome data meant that we could only explore sex differences based on autosomal SNPs. Fourth, we did not have the power to break down the signal attributed to coding/regulatory regions into more finely defined functional categories. Last, given the limitations on the data available to test the G x E hypothesis, alternative explanations for our results cannot be excluded.
Discussion
In summary, we have applied a mixed linear model method to estimate the genetic correlation between RA and SZ. Epidemiological evidence has demonstrated decreased prevalence rates of RA in SZ cases, consistent with a phenotypic correlation of liabilities of up to −0.15. We show that there is a small but significant negative correlation across the genome and the signal is stronger for SNPs annotated as coding and regulatory. Given that RA is an immune-related disorder and that a role for immune activation has long been hypothesized for SZ, a negative genetic correlation could imply that variants in immune response pathways have different roles in different tissues and/or in response to different challenges. The immune activation hypothesis of SZ is partly founded on an increased risk for SZ associated with month of birth. Our hypothesis that increased effect sizes for SZ-associated SNPs in the coding/regulatory SNP set for a winter-born case-control set was supported by our analyses, although other explanations for these results may be possible. Most importantly, if the complexity of SZ is to be unraveled, then data sets that are informative for both genetic and environmental risk factors are essential. Since SZ is an adult-onset disorder, and yet perinatal and childhood experience, especially infections, are known environmental risk factors, then prospective gathering of data in nationally accessible repositories is needed.
Funding
This work was supported by the Australian Research Council [grant number DE130100614 to S.H.L.], the National Health and Medical Research Council [grant numbers 613602, 1078901 to N.R.W; 1047956 to N.R.W., S.H.L. and B.J.M., 1053639 to E.M.B]; the Arthritis Foundation to S.R., the Doris Duke Foundation to S.R.; the National Institutes of Health [grant numbers 1R01AR063759–01A1, 1U01HG0070033, 5U01GM092691–04 to S.R.; R01 MH077139 for the Sweden SZ Study to P.F.S.]. The Swedish SZ study was also funded by the Karolinska Institutet, Karolinska University Hospital, and the Swedish Research Council. The RA dataset from Vanderbilt University Medical Center's BioVU is supported by institutional funding and by the Vanderbilt CTSA grant ULTR000445 from NCATS/NIH. Other funding acknowledgements can be found in the primary publications from each study, as references. Statistical analyses were carried out on the Genetic Cluster Computer (http://www.geneticcluster.org) hosted by SURFsara, and financially supported by The Netherlands Scientific Organization (NWO 480‐05‐003) along with a supplement from the Dutch Brain Foundation and the VU University Amsterdam.
Supplementary Material
Acknowledgements
We thank Peter Smartt, Jake Carroll, Irek Porebski and the Queensland Brain Institute IT team for technical support.
Author contributions: N.R.W. was responsible for overall direction of the study, with analyses conducted by S.HL. E.M.B. conducted a literature review and wrote the first draft of the introduction generating hypotheses tested. N.R.W., S.H.L., S Raychaudhuri, E.M.B., B.J.M. and A.P. contributed to decisions about analyses conducted. N.R.W. wrote the first draft of the manuscript with substantive contributions including suggestions for follow-up analyses from S.H.L., S Raychaudhuri, P.F.S, A.P., M.O.D. and Y.O. S Ripke was responsible for initial QC of the GWAS data sets. A.A.E., O.A.A., T.F., A.G., X.H., V.M., J.J.McG., D.M., E.A.S., P.S. and Q.Z. contributed to secondary analyses.
Conflict of interest: Thomas Werge has acted as lecturer and consultant to the pharmaceutical company Lundbeck A/S (Denmark). Jeffrey D Greenberg is an employee of, and has stock in, Corrona, LLC, and is consultant for AstraZeneca, Celgene, Novartis and Pfizer. Paul Tak is now an employee of GSK and holds stock in GSK after completion of this study; GSK was not involved in this study.
References
- 1.Oken RJ, Schulzer M. At issue: schizophrenia and rheumatoid arthritis: the negative association revisited. Schizophr Bull 1999;25:625–38. [DOI] [PubMed] [Google Scholar]
- 2.Sellgren C, Frisell T, Lichtenstein P, Landen M, Askling J. The association between schizophrenia and rheumatoid arthritis: a nationwide population-based Swedish study on intraindividual and familial risks. Schizophr Bull 2014;40:1552–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Leon J, Diaz FJ. A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophr Res 2005;76:135–57. [DOI] [PubMed] [Google Scholar]
- 4.Bang SY, Lee KH, Cho SK, Lee HS, Lee KW, Bae SC. Smoking increases rheumatoid arthritis susceptibility in individuals carrying the HLA-DRB1 shared epitope, regardless of rheumatoid factor or anti-cyclic citrullinated peptide antibody status. Arthritis Rheum 2010;62:369–77. [DOI] [PubMed] [Google Scholar]
- 5.Torrey EF, Yolken RH. The schizophrenia-rheumatoid arthritis connection: infectious, immune, or both? Brain Behav Immun 2001;15:401–10. [DOI] [PubMed] [Google Scholar]
- 6.Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med 2005;2:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benros ME, Pedersen MG, Rasmussen H, Eaton WW, Nordentoft M, Mortensen PB. A nationwide study on the risk of autoimmune diseases in individuals with a personal or a family history of schizophrenia and related psychosis. Am J Psychiatry 2014;171:218–26. [DOI] [PubMed] [Google Scholar]
- 8.Lichtenstein P, Yip BH, Bjork C, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 2009;373:234–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frisell T, Holmqvist M, Kallberg H, Klareskog L, Alfredsson L, Askling J. Familial risks and heritability of rheumatoid arthritis: role of rheumatoid factor/anti-citrullinated protein antibody status, number and type of affected relatives, sex, and age. Arthritis Rheum 2013;65:2773–82. [DOI] [PubMed] [Google Scholar]
- 10.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry 2003;60:1187–92. [DOI] [PubMed] [Google Scholar]
- 11.MacGregor AJ, Snieder H, Rigby AS, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 2000;43:30–37. [DOI] [PubMed] [Google Scholar]
- 12.de Vries RR, van der Woude D, Houwing JJ, Toes RE. Genetics of ACPA-positive rheumatoid arthritis: the beginning of the end? Ann Rheum Dis 2011;70(Suppl 1):i51–54. [DOI] [PubMed] [Google Scholar]
- 13.Han B, Diogo D, Eyre S, et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am J Hum Genet 2014;94:522–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eaton WW, Hayward C, Ram R. Schizophrenia and rheumatoid arthritis: a review. Schizophr Res 1992;6:181–92. [DOI] [PubMed] [Google Scholar]
- 15.Taylor WM. Schizophrenia, rheumatoid arthritis and trytophan metabolism. J Clin Psychiatry 1978;39:499–503. [PubMed] [Google Scholar]
- 16.Horrobin DF. Schizophrenia as a prostaglandin deficiency disease. Lancet 1977;1:936–37. [DOI] [PubMed] [Google Scholar]
- 17.Horrobin DF, Ally AI, Karmali RA, Karmazyn M, Manku MS, Morgan RO. Prostaglandins and schizophrenia: further discussion of the evidence. Psychol Med 1978;8:43–48. [DOI] [PubMed] [Google Scholar]
- 18.Horrobin DF. Prostaglandins and schizophrenia. Lancet 1979;1:1031–32. [DOI] [PubMed] [Google Scholar]
- 19.Trevathan RD, Tatum JC. Rarity of concurrence of psychosis and rheumatoid arthritis in individual patients; report of a case. J Nerv Ment Dis 1954;120:83–84. [PubMed] [Google Scholar]
- 20.Blomstrom A, Karlsson H, Svensson A, et al. Hospital admission with infection during childhood and risk for psychotic illness - a population-based cohort study. Schizophr Bull 2014;40:1518–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol 2012;72:1272–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hultman CM, Sparen P, Takei N, Murray RM, Cnattingius S. Prenatal and perinatal risk factors for schizophrenia, affective psychosis, and reactive psychosis of early onset: case-control study. BMJ 1999;318:421–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strous RD, Shoenfeld Y. Schizophrenia, autoimmunity and immune system dysregulation: a comprehensive model updated and revisited. J Autoimmun 2006;27:71–80. [DOI] [PubMed] [Google Scholar]
- 24.Jones AL, Mowry BJ, Pender MP, Greer JM. Immune dysregulation and self-reactivity in schizophrenia: do some cases of schizophrenia have an autoimmune basis? Immunol Cell Biol 2005;83:9–17. [DOI] [PubMed] [Google Scholar]
- 25.Eaton WW, Byrne M, Ewald H, et al. Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am J Psychiatry 2006;163:521–28. [DOI] [PubMed] [Google Scholar]
- 26.Chen SJ, Chao YL, Chen CY, et al. Prevalence of autoimmune diseases in in-patients with schizophrenia: nationwide population-based study. Br J Psychiatry 2012;200:374–80. [DOI] [PubMed] [Google Scholar]
- 27.Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry 2011;168:1303–10. [DOI] [PubMed] [Google Scholar]
- 28.Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet 2008;4:e1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stastny P. Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N Engl J Med 1978;298:869–71. [DOI] [PubMed] [Google Scholar]
- 30.Newton JL, Harney SM, Wordsworth BP, Brown MA. A review of the MHC genetics of rheumatoid arthritis. Genes Immun 2004;5:151–57. [DOI] [PubMed] [Google Scholar]
- 31.Wright P, Nimgaonkar VL, Donaldson PT, Murray RM. Schizophrenia and HLA: a review. Schizophr Res 2001;47:1–12. [DOI] [PubMed] [Google Scholar]
- 32.Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;460:748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature 2009;460:744–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi J, Levinson DF, Duan J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009;460:753–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schizophrenia Working Group of the Psychiatric Genomics Consortium, Ripke S, Neale BM, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014;511:421–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Bakker PI, Raychaudhuri S. Interrogating the major histocompatibility complex with high-throughput genomics. Hum Mol Genet 2012;21:R29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet 2011;88:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stahl EA, Wegmann D, Trynka G, et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet 2012;44:483–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SH, DeCandia TR, Ripke S, et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet 2012;44:247–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gusev A, Bhatia G, Zaitlen N, et al. Quantifying missing heritability at known GWAS loci. PLoS Genet 2013;9:e1003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de la Fontaine L, Schwarz MJ, Riedel M, et al. Investigating disease susceptibility and the negative correlation of schizophrenia and rheumatoid arthritis focusing on MIF and CD14 gene polymorphisms. Psychiatr Res 2006;144:39–47. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe Y, Nunokawa A, Kaneko N, et al. Two-stage case-control association study of polymorphisms in rheumatoid arthritis susceptibility genes with schizophrenia. J Hum Genet 2009;54:62–65. [DOI] [PubMed] [Google Scholar]
- 44.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics 2012;28:2540–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SH, Ripke S, Neale BM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013;45:984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stahl EA, Raychaudhuri S, Remmers EF, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 2010;42:508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ripke S, Sanders AR, Kendler KS, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011;43:969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ripke S, O'Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 2013;45:1150–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Manolio TA, Pasquale LR, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet 2011;43:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lynch M, Walsh B. Genetics and Analysis of Quantitative Traits. Sunerland, MA: Sinauer Associates, 1998. [Google Scholar]
- 52.Visscher PM, Hemani G, Vinkhuyzen AAE, et al. Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet 2014;10:e1004269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falconer D. The inheritance of liability to certain diseases, estimates from the incidence among relatives. Ann Hum Genet 1965;29:51–76. [Google Scholar]
- 54.Reich T, Morris CA, James JW. Use of multiple thresholds in determining mode of transmission of semi-continuous traits. Ann Hum Genet 1972;36:163–84. [DOI] [PubMed] [Google Scholar]
- 55.Speed D, Hemani G, Johnson MR, Balding DJ. Improved heritability estimation from genome-wide SNPs. Am J Hum Genet 2012;91:1011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee SH, Yang J, Chen GB, et al. Estimation of SNP heritability from dense genotype data. Am J Hum Genet 2013;93:1151–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gusev A, Lee SH, Trynka G, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 2014;95:535–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu X, Kim H, Stahl E, Plenge R, Daly M, Raychaudhuri S. Integrating autoimmune risk loci with gene-expression data identifies specific pathogenic immune cell subsets. Am J Hum Genet 2011;89:496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheverud JM. A comparison of genetic and phenotypic correlations. Evolution 1988;42:958–68. [DOI] [PubMed] [Google Scholar]
- 60.Visscher PM, Goddard ME, Derks EM, Wray NR. Evidence-based psychiatric genetics, AKA the false dichotomy between common and rare variant hypotheses. Mol Psychiatr 2012;17:474–85. [DOI] [PubMed] [Google Scholar]
- 61.Torrey EF, Miller J, Rawlings R, Yolken RH. Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res 1997;28:1–38. [DOI] [PubMed] [Google Scholar]
- 62.Davies G, Welham J, Chant D, Torrey EF, McGrath J. A systematic review and meta-analysis of Northern Hemisphere season of birth studies in schizophrenia. Schizophr Bull 2003;29:587–93. [DOI] [PubMed] [Google Scholar]
- 63.Disanto G, Chaplin G, Morahan JM, et al. Month of birth, vitamin D and risk of immune-mediated disease: a case control study. BMC Med 2012;10:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chun RF, Liu PT, Modlin RL, Adams JS, Hewison M. Impact of vitamin D on immune function: lessons learned from genome-wide analysis. Front Physiol 2014;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang CY, Leung PS, Adamopoulos IE, Gershwin ME. The implication of vitamin D and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol 2013;45:217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol 2008;8:685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Euesden J, Breen G, Farmer A, McGuffin P, Lewis CM. The relationship between schizophrenia and rheumatoid arthritis revisited: genetic and epidemiological analyses. Am J Med Genet B Neuropsychiatr Genet 2015;168:8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bulik-Sullivan B, Finucane H, Antilla V, et al. An atlas of genetic correlations across human diseases and traits. BioRXiv. http://dx.doi.org/10.1101/014498. [DOI] [PMC free article] [PubMed]
- 69.Jiang L, Yin J, Ye L, et al. Novel risk loci for rheumatoid arthritis in Han Chinese and congruence with risk variants in Europeans. Arthritis Rheum 2014;66:1121–32. [DOI] [PubMed] [Google Scholar]
- 70.Wray NR, Lee SH, Kendler KS. Impact of diagnostic misclassification on estimation of genetic correlations using genome-wide genotypes. Eur J Hum Genet 2012;20:668–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Witte JS, Visscher PM, Wray NR. The contribution of genetic variants to disease depends on the ruler. Nat Rev Genet 2014;15:765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.