

Abstract
Purine biosynthetic enzymes organize into dynamic cellular bodies called purinosomes. Little is known about the spatiotemporal control of these structures. Using super-resolution microscopy, we demonstrated that purinosomes colocalized with mitochondria, and these results were supported by isolation of purinosome enzymes with mitochondria. Moreover, the number of purinosome containing cells responded to dysregulation of mitochondrial function and metabolism. To explore the role of intracellular signaling, we performed a kinome screen using a label-free assay and identified that mTOR influenced purinosome assembly. mTOR inhibition disrupted purinosome-mitochondria colocalization and suppressed purinosome formation stimulated by mitochondria dysregulation. Collectively, our data suggests an mTOR-mediated link between purinosomes and mitochondria and suggests a general means by which mTOR regulates nucleotide metabolism by spatiotemporal control over protein association.
Purine levels in mammalian cells are maintained by the coordinated action of complementary salvage and de novo biosynthetic pathways. While the salvage pathway maintains purine nucleotide levels under normal physiological conditions, the de novo pathway is upregulated during growth (1, 2) and altered in neoplastic cells (3, 4). Purinosomes are mesoscale assemblies formed to protect unstable intermediates and increase metabolic flux through the de novo pathway (5–9). These structures are dynamic and form reversibly in response to purine depletion and act to increase de novo purine biosynthesis (5, 6, 10). Their formation is cell cycle dependent and can be regulated by GPCR agonists and casein kinase 2 (11–14). Increased number of purinosome containing cells correlates positively with the degree of purine salvage deficiency in Lesch-Nyhan disease (15). Cellular conditions resulting in disruption of purinosome formation led to enhanced sensitivity to cancer chemotherapeutics (16). An analogous cellular phenotype was also recently reported for a multi-functional protein involved in pyrimidine biosynthesis (17). These structures are examples of an increasing number of reported higher order organizations involving metabolic proteins (7, 18, 19).
Considering that de novo purine biosynthesis not only provides the nucleotide precursors necessary for mitochondrial ATP production but also conversely demands ATP for its operation, we hypothesized that a synergistic relationship between purinosomes and mitochondria might exist. This synergy would be even more critical in cells that preferentially use oxidative phosphorylation for ATP production, such as several cervical cancer, breast carcinoma, hepatoma, pancreatic cancer and glioma cell lines (20, 21). The relationship would also supply one-carbon units generated by the mitochondrial conversion of serine to formate for incorporation into the purine ring during de novo biosynthesis. In this work, we investigated the physical and functional relationship between purinosome and mitochondria using super-resolution imaging, a dynamic mass redistribution assay, and other biochemical measures.
Conventional fluorescence microscopy images initially suggested spatial proximity between purinosomes and mitochondria, but the high density of the mitochondrial network precluded clear demonstration and quantitative characterization of the colocalization between these two structures at diffraction-limited image resolution (Fig. S1). To further investigate the spatial distribution of purinosomes, we used three-dimensional stochastic optical reconstruction microscopy (3D STORM), a super-resolution fluorescence imaging method (22–24), to image HeLa cells under conditions that promote the formation of purinosomes. Purinosomes were imaged via transient transfection of photoactivatable fluorescent protein (mEos2) (25) tagged formylglycinamidine ribonucleotide synthase (FGAMS or PFAS), a core purinosome component (5, 26). Given the residual dimerization tendency of mEos2, we also tagged FGAMS to recently developed monomeric photoactivatable fluorescent protein, mMaple3 (27). The number and size distributions of purinosomes were independent of the tagging method (Fig. S2).
To investigate purinosome-mitochondria colocalization, we induced purinosome formation by purine starvation, fixed the cells and immunostained for a mitochondrial outer membrane translocase (TOM20) using a photoswitchable fluorescent dye (Alexa Fluor 647). Two-color 3D STORM images of cells that exhibited purinosomes revealed a highly correlated spatial distribution of purinosomes and mitochondria (Fig. 1). Purinosomes were found colocalized with the mitochondria (Fig. 1B–F). Under the conditions tested, a substantially larger fraction of purinosomes were colocalized with mitochondria than what would be expected if the purinosomes were randomly distributed throughout the cytoplasm (Fig. 1G, S3).
Fig. 1.

Super-resolution imaging of purinosomes and mitochondria. (A) 2D projection of a 3D STORM image showing purinosomes labeled with mEos2 fused to a purinosome core protein FGAMS (magenta) and mitochondria immunolabeled against outer membrane protein TOM20 (green) in a HeLa cell grown under purine-depleted conditions. (B) Zoom in of the boxed region in panel (A) showing the close proximity between the two structures. (C) An 100 nm thick xy-cross section of the region in (B). (D, E) Comparison of the conventional fluorescence image (D) and corresponding 2D projection STORM image (E) of the boxed region in (B). (F) xz-cross section along the dotted line in (E) showing a purinosome and two neighboring mitochondria. (G) The percentage of purinosomes (mean ± standard deviation colocalized with mitochondria observed using STORM (65.1 ± 11.5%) is significantly higher than the expected value for a randomized purinosome distribution (33.7 ± 7.3%). N = 26 images, student’s t-test, p≪0.001 as denoted by ***. Scale bars: 250 nm in (E–F).
To provide further support for the potential physical interactions between purinosomes and mitochondria, we isolated mitochondria from cells after chemical cross-linking and compared the proteins present in these mitochondrial extracts to cytosolic fractions. Four different cross-linkers of varying length and reactivity were employed to minimize method bias, and the proteins that co-purified with the mitochondria were identified by mass spectrometry (Table S1). In addition to mitochondrial proteins such as ATP synthase, voltage-dependent anion channel, and malate dehydrogenase, one of the 174 proteins identified was adenylosuccinate lyase (ASL or ADSL), a known purinosome protein. ASL catalyzes the eighth step in de novo purine biosynthesis and was observed using three out of the four cross-linkers (Table S1). To validate ASL colocalization with mitochondria, mitochondria were purified from cells under purinosome forming conditions without chemical cross-linking and purinosome enzymes that co-purified with mitochondria were detected using Western blot. In addition to ASL, FGAMS, a core protein of the purinosome structure, also co-precipitated with isolated mitochondria (Fig. 2A). While these data demonstrate a physical link between purinosomes and mitochondria, further experiments are required to characterize the molecular details of this interaction and identify any structural intermediaries.
Fig. 2.
Physical and functional links between purinosomes and mitochondria. (A) Western blot of purified mitochondria showing that purinosome proteins FGAMS and ASL co-isolate with mitochondria in a rapamycin-dependent manner. Mitochondria were isolated from HeLa cells grown under purine-depleted conditions that transiently expressed FGAMS-3×FLAG after treatment with 1 μM rapamycin (+) or vehicle control (−) for 1 h. Inhibition of mTOR was verified by observing a decrease in the phosphorylated form (pT389) of the mTOR target S6 kinase (p70-S6K). VDAC1 was used as a mitochondria loading control and p70-S6Kserved as a cytoplasmic loading control. (B) The percentage of cells with visible purinosomes (determined from at least 100 total cells) as a function of modulators of mitochondrial metabolism and glycolysis at their specified concentrations for 1 h. Values reflect mean ± standard deviation, N=3. (C) Intracellular malate (gray) and lactate (black) levels were determined by colorimetric assay after various drug treatments for 1 h (2 h for MKT-077). Values reflect mean ± standard deviation, N=3.
To investigate the functional relationship of purinosomes with mitochondria, we first examined the effect of mitochondrial poisons on purinosome content of cells. Inhibition of electron transport (using antimycin A or rotenone) or oxidative phosphorylation (using oligomycin) increased the number of purinosome-positive cells by more than 2-fold (Fig. 2B). Inhibition of glycolysis (using 2-deoxyglucose), which also lowers cellular ATP concentrations (28), had no effect on purinosome levels (Fig. 2B). The latter result, combined with previous observations of the effect of exogenous ATP treatments (14), suggest that although mitochondria dysregulation induced a stimulation of purinosome formation in cells, the purinosome assembly is not governed by ATP concentration. Next, we examined the effect of purinosome levels on mitochondrial metabolism. As an approximate measure of glycolytic (cytosolic) and oxidative phosphorylation (mitochondria) activities, we assayed cellular lactate and malate concentrations, respectively (29, 30). Compounds known to disrupt or inhibit purinosome formation (17-AAG, MKT-077 and TBB) (11, 16) led to decreases in malate levels, while an increase in purinosome content induced by DMAT (11) significantly increased malate production (Fig. 2C). Lactate levels were not changed by any of the purinosome effectors. These results indicate that there is also a functional link between purinosomes and mitochondria.
Previously, we reported that the assembly of purinosomes was stimulated by agonist binding to the α2A-adenergic receptor and subsequent activation of the Gαi-mediated signaling pathway (14). To identify the intracellular signaling pathway employed in the control of the relationship between purinosomes and mitochondria, we conducted an shRNA screen of the human kinome using a two-step dynamic mass redistribution (DMR) assay (Fig. 3A). DMR is a label-free method that employs a resonant waveguide grating biosensor system to monitor, in real time, refractive index alterations resulting from stimulus-induced biomass changes near the surface of a sensor (Fig. S4, S5) (14, 31). Epinephrine (EPI) is known to induce purinosome assembly, which contributed to a DMR signal increase; TBB is known to cause purinosome disassembly, which contributed to a DMR signal decrease (14). Here we used the EPI-induced DMR signal as a purinosome assembly indicator and the EPI-stimulated TBB response as a purinosome disassembly indicator to identify kinases that influence purinosomes. Analyses of the robust z-score (32) for each shRNA treatment (Fig. 3A, 3B, S6; Table S2) and Gene Ontology (GO) enrichment (Table S3) suggest that some of the identified kinases are indeed associated with regulation of purine nucleotide metabolism. Using the STRING9.1 database that provides known and predicted protein-protein associations (33), networks were generated for the identified kinases that connect them to known components of endogenous α2A-receptor signaling and purinosome assembly, including casein kinase 2 and the six enzymes of the de novo purine biosynthetic pathway (Fig. 3C, S7, S8). This analysis identifies a putative kinase network involved in directly translating chemical signals into a purinosome response.
Fig. 3.

Human kinome screen identified kinases involved in α2A-adrenergic receptor (α2A-AR) activation-mediated purinosome formation. (A) Characteristic DMR of HeLa cells in response to sequential stimulation steps (S1 and S2). Red line: buffer (S1) – buffer (S2); black line: buffer – TBB; purple line: EPI – buffer; blue line: EPI – TBB. The DMR of assay buffer stimulation was used as the negative control. Buffer by itself triggered little DMR, and did not alter DMR induced by 100 nM epinephrine (EPI). EPI (100 nM) triggered a positive DMR. Conversely, TBB led to a negative DMR in the buffer pretreated cells, but a much greater negative DMR in EPI pretreated cells. (B) The robust z-score of EPI-induced DMR (green dots) or TBB-induced DMR (red dots) as a function of shRNA clones. Robust z-scores (a z-score not adversely affected by outliers) were calculated using [(experimental data − median)/median absolute deviation (MAD)] where the normalization set the median to 0 and the MAD to 1. (C) Network analysis of the α2A-AR activation mediated purinosome formation. This analysis combines all hits common to the EPI and TBB DMR responses identified using the current kinome screen with known signaling components of endogenous α2A-AR in HeLa cells, casein kinase 2 (CSNK2B, CSNK2A1, CSNK2A2) and six enzymes (PPAT, GART, PFAS, PAICS, ADSL, ATIC) involved in purine biosynthesis. Hits were selected when at least two shRNA clones for a kinase within the library gave a robust z-score of ≥ 3 or ≤ −3 (Table S2). The network was generated using STRING 9.1. Connecting lines are color coded by the type of evidence used to build the network (details in http://string-db.org/). Unconnected hits are listed at the bottom. (D) Top panel: The real-time DMR of EPI in the absence (red) or presence of everolimus (green). Bottom panel: the real-time DMR of TBB after EPI pre-stimulation in the absence (red) or presence of everolimus (green). The dose was 16 μM, 100 nM, or 20 μM for everolimus, EPI, or TBB, respectively. For (A, D), data represents mean + standard deviation, N=4. The standard deviation is shown in gray.
Among the kinases identified in this screen were several known to be master regulators of cellular metabolism. Interestingly, one of these kinases was the mechanistic target of rapamycin (mTOR). mTOR, which actively associates with mitochondria-associated ER membranes and modulates mitochondrial physiology, is also involved in regulating nucleotide metabolism (17, 30, 34, 35). Its role in modulating purine biosynthesis, however, is still unclear (36).
We examined the effect of mTOR inhibition on purinosome formation using the described DMR assay. Inhibition of mTOR with everolimus alone did not trigger a DMR response (Fig. S9), but partially inhibited the EPI-induced DMR signal in a dose-dependent manner (Fig. 3D). The EPI-induced DMR signal contains contributions from the Gαs and Gαi pathways, the latter of which is related to purinosome formation (6, 14). Moreover, mTOR inhibition also suppressed the EPI-potentiated TBB-induced DMR signal (Fig. 3D). Taken together, these results suggest a model wherein the inhibition of mTOR impairs α2A receptor activation stimulated purinosome formation.
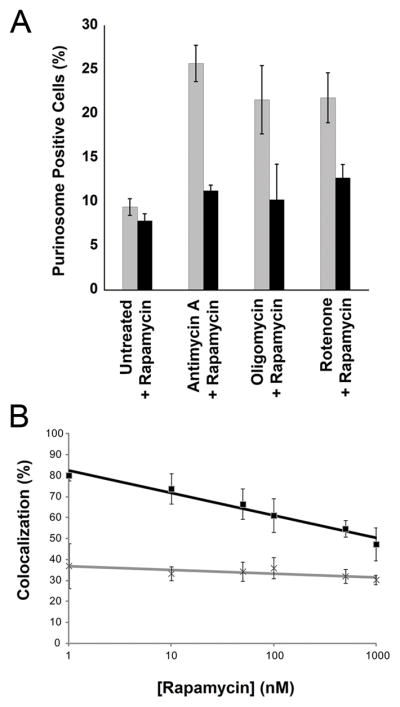
To test whether mTOR plays a role in mediating the link between mitochondria and purinosomes, we monitored the observed stimulation of the cellular purinosome level in response to mitochondrial dysregulation. While a large increase in the percentage of cells containing purinosomes was observed when mitochondria function was disrupted by antimycin A, oligomycin or rotenone, this response was abrogated by treatment with the mTOR inhibitor, rapamycin (Fig. 4A) similar to the response observed with everolimus treatment of EPI prestimulated cells (Fig. 3D). Rapamycin treatment alone had no effect on purinosome levels. Note that these observations were made without stimulation where such an effect would be difficult to detect. We then examined the colocalization between mitochondria and purinosomes in the presence of rapamycin using two-color STORM. Indeed, fractional colocalization between purinosomes and mitochondria decreased in a dose-dependent manner with increasing concentration of rapamycin (Fig. 4B), whereas both the number and size of purinosomes and the cellular distributions of mitochondria were unchanged up to concentrations of 500 nM (Fig. S10). Finally, to further support that mTOR plays a role in mediating a physical link between purinosomes and mitochondria, we probed for the presence of FGAMS in isolated mitochondria from cells treated with rapamycin (Fig. 2A). While these purinosome markers were observed in the mitochondrial fraction in the absence of rapamycin, it was either not observed or observed at a substantially reduced level in rapamycin treated cells. Taken together, these data suggest that mTOR plays a role in the link between purinosomes and mitochondria.
Fig. 4.

mTOR affects colocalization and functional links between purinosomes and mitochondria. (A) The percent of purinosome containing cells (determined from at least 100 cells) as a function of mitochondrial metabolism modulators in the absence (gray) and presence (black) of 100 nM rapamycin. Values reflect the mean ± standard deviation, N=3. (B) The percentage of purinosomes colocalized with mitochondria (black squares) as a function of increasing rapamycin concentration (10–1000 nM, 1 h). The results after randomization of the purinosome distribution are shown as gray crosses. The colocalization percentage is represented as the mean ± standard deviation, N=5 per condition.
Two recent reports detailed the mTOR-mediated stimulation of pyrimidine synthesis (17, 34). The mechanism of control exerted by mTOR on pyrimidine metabolism, the change in oligomerization and localization of the enzyme CAD, mirrors the observed effects reported herein. Pyrimidine biosynthesis also employs a mitochondrial enzyme, dihydroorotate dehydrogenase, further evidence for the relationship between nucleotide metabolism and the mitochondria. mTOR nucleates into two distinct multi-protein complexes (mTORC1 and mTORC2) and is known to regulate protein associations to control other cellular processes, such as autophagy (37–39).
The maintenance of nucleotide pools and the rapid response to changing levels of these critical building blocks are vital cellular processes. Management of metabolite levels in a dynamic microenvironment necessitates highly regulated post-translational control over metabolic flux. This study suggests a spatial mechanism of control. The mTOR-mediated link between purinosomes and mitochondria creates a functional synergy and highlights the interdependence of the relationship between mitochondrial function and nucleotide metabolism, which could provide a controllable response to changes in metabolic needs. This type of regulation is only beginning to be understood but is likely to emerge as a common mechanism by which cells exploit spatial and temporal control of enzymes and enzyme complexes to increase metabolic efficiency, protect unstable intermediates, and minimize off-target effects.
Supplementary Material
Acknowledgments
The authors thank Tatania Laremore at the proteomics and mass spectrometry core facility of the Huck Institutes of the Life Sciences at The Pennsylvania State University for assistance with data collection and analyses. The Orbitrap mass spectrometer was funded by a grant from the Pennsylvania Department of Health Tobacco Settlement Funds. JBF acknowledges the Canadian Institutes of Health Research for fellowship support. This work was funded by the National Institutes of Health grants NIH GM024129 (SJB) and 1R33EB019785-01 (TJH, SJB) as well as the Howard Hughes Medical Institute (XZ). JBF, SAJ, YF, XZ, and SJB designed the experiments; JBF, SAJ, HD, HH, AMP, CYC, DK, RJP, HZ, and YZ performed the experiments and analyzed the data; JBF, SAJ, AMP, RJP, and YF prepared the manuscript; JBF, YF, XZ, and SJB directed the research; all authors have reviewed and edited the manuscript. The authors declare that they have no competing interests. Additional data reported in this manuscript are available in the Supplementary Material.
Footnotes
References and Notes
- 1.Yamaoka T, et al. Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. The Journal of biological chemistry. 1997;272:17719–17725. doi: 10.1074/jbc.272.28.17719. [DOI] [PubMed] [Google Scholar]
- 2.Yamaoka T, et al. Feedback inhibition of amidophosphoribosyltransferase regulates the rate of cell growth via purine nucleotide, DNA, and protein syntheses. The Journal of biological chemistry. 2001;276:21285–21291. doi: 10.1074/jbc.M011103200. [DOI] [PubMed] [Google Scholar]
- 3.Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nature reviews Cancer. 2013;13:842–857. doi: 10.1038/nrc3613. [DOI] [PubMed] [Google Scholar]
- 4.Natsumeda Y, Prajda N, Donohue JP, Glover JL, Weber G. Enzymic capacities of purine de Novo and salvage pathways for nucleotide synthesis in normal and neoplastic tissues. Cancer research. 1984;44:2475–2479. [PubMed] [Google Scholar]
- 5.An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, French JB, Fang Y, Benkovic SJ. The purinosome, a multi-protein complex involved in the de novo biosynthesis of purines in humans. Chemical communications. 2013;49:4444–4452. doi: 10.1039/c3cc41437j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Havugimana PC, et al. A census of human soluble protein complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schendel FJ, Cheng YS, Otvos JD, Wehrli S, Stubbe J. Characterization and chemical properties of phosphoribosylamine, an unstable intermediate in the de novo purine biosynthetic pathway. Biochemistry. 1988;27:2614–2623. doi: 10.1021/bi00407a052. [DOI] [PubMed] [Google Scholar]
- 9.Zhang QC, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao H, et al. Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. The Journal of biological chemistry. 2015;290:6705–6713. doi: 10.1074/jbc.M114.628701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An S, Kyoung M, Allen JJ, Shokat KM, Benkovic SJ. Dynamic regulation of a metabolic multi-enzyme complex by protein kinase CK2. The Journal of biological chemistry. 2010;285:11093–11099. doi: 10.1074/jbc.M110.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan CY, et al. Purinosome formation as a function of the cell cycle. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1368–1373. doi: 10.1073/pnas.1423009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang Y, French J, Zhao H, Benkovic S. G-protein-coupled receptor regulation of de novo purine biosynthesis: a novel druggable mechanism. Biotechnology & genetic engineering reviews. 2013;29:31–48. doi: 10.1080/02648725.2013.801237. [DOI] [PubMed] [Google Scholar]
- 14.Verrier F, et al. GPCRs regulate the assembly of a multienzyme complex for purine biosynthesis. Nature chemical biology. 2011;7:909–915. doi: 10.1038/nchembio.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu R, et al. Clinical severity in Lesch-Nyhan disease: the role of residual enzyme and compensatory pathways. Mol Genet Metab. 2015;114:55–61. doi: 10.1016/j.ymgme.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.French JB, et al. Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2528–2533. doi: 10.1073/pnas.1300173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robitaille AM, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339:1320–1323. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- 18.Meyer FM, et al. Physical interactions between tricarboxylic acid cycle enzymes in Bacillus subtilis: evidence for a metabolon. Metabolic engineering. 2011;13:18–27. doi: 10.1016/j.ymben.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Straight PD, Fischbach MA, Walsh CT, Rudner DZ, Kolter R. A singular enzymatic megacomplex from Bacillus subtilis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:305–310. doi: 10.1073/pnas.0609073103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochimica et biophysica acta. 2011;1807:552–561. doi: 10.1016/j.bbabio.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. The Journal of biological chemistry. 1979;254:2669–2676. [PubMed] [Google Scholar]
- 22.Huang B, Jones SA, Brandenburg B, Zhuang X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nature methods. 2008;5:1047–1052. doi: 10.1038/nmeth.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nature methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nature methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng Y, et al. Mapping protein-protein proximity in the purinosome. The Journal of biological chemistry. 2012;287:36201–36207. doi: 10.1074/jbc.M112.407056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S, Moffitt JR, Dempsey GT, Xie XS, Zhuang X. Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:8452–8457. doi: 10.1073/pnas.1406593111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 29.Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer & metabolism. 2014;2:3. doi: 10.1186/2049-3002-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramanathan A, Schreiber SL. Direct control of mitochondrial function by mTOR. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:22229–22232. doi: 10.1073/pnas.0912074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang Y. Label-free drug discovery. Frontiers in Pharmacology. 2014;5:52. doi: 10.3389/fphar.2014.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birmingham A, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nature methods. 2009;6:569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franceschini A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic acids research. 2013;41:D808–815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nature reviews Molecular cell biology. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- 37.Ganley IG, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. The Journal of biological chemistry. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular biology of the cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of cell science. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.