

Abstract
Most malignant peripheral nerve sheath tumors (MPNSTs) exhibit combined inactivation of NF1, CDKN2A, and polycomb repressive complex 2 component genes (Embryonic Ectoderm Development [EED] and Suppressor of Zeste 12 [SUZ12]). Mutations in EED and SUZ12 induce loss of trimethylation at lysine 27 of histone 3 (H3K27me3), with subsequent aberrant transcriptional activation of polycomb repressive complex 2–repressed homeobox master regulators. These findings prompted us to investigate the performance of an anti-H3K27me3 monoclonal antibody clone C36B11 as an immunohistochemical marker for MPNSTs. We assessed the C36B11 reactivity pattern in a pathologically and genetically well-characterized cohort of 68 MPNSTs, spanning various clinical presentations, such as type 1 neurofibromatosis (NF1), radiotherapy, and sporadic MPNSTs. We found that 69% (n=47) of all MPNSTs demonstrated loss of H3K27me3 expression, with 42 (61%) showing complete loss and 5 (7%) showing partial loss, whereas 31% (n = 21) retained H3K27me3 expression. Among the NF1-related high-grade MPNSTs, 60% demonstrated loss of expression. In contrast, the majority of both sporadic (95%) and radiotherapy-related (91%) MPNSTs showed loss of H3K27me3 expression. Two of the 3 low-grade MPNSTs and all neurofibromas showed retained expression. Furthermore, all 5 epithelioid MPNSTs retained H3K27me3 labeling. The specificity of H3K27me3 loss as a marker for MPNSTs was studied by testing a large spectrum of lesions included in MPNST differential diagnosis, such as spindle/desmoplastic melanomas, synovial sarcomas, myoepithelial tumors, and other mesenchymal neoplasms, all of which retained expression of H3K27me3. We conclude that immunohistochemical analysis of H3K27me3 has good sensitivity and robust specificity for the diagnosis of MPNST, particularly outside of NF1 clinical history, which represents the most challenging diagnostic setting.
Keywords: malignant peripheral nerve sheath tumor, neurofibromatosis 1, polycomb repressor complex 2, EED, SUZ12, H3K27me3
Malignant peripheral nerve sheath tumors (MPNSTs) are rare neoplasms occurring mostly in adults and representing approximately 4% of all sarcomas.1 About 50% of MPNSTs occur in patients afflicted by type 1 neurofibromatosis (NF1), whereas about 10% of cases arise secondary to prior radiotherapy (RT).2,3 The remaining 40% of cases occur without known predisposition and include both conventional/spindle and epithelioid cell variants. A 2-tiered grading system with low-grade (LG) and high-grade (HG) MPNSTs is used, with the LG end being characterized by neurofibroma (NF)-like tumors composed of a cellular proliferation of bland, back-to-back spindle cells that appear to be in a continuum with the so-called “atypical/cellular neurofibroma.”4,5 Histologically, HG MPNSTs are characterized by an undifferentiated phenotype with limited, if any, evidence of schwannian differentiation at immunohistochemical (IHC) or ultrastructural levels. The majority of MPNSTs demonstrate a characteristic pattern of intersecting fascicles of relatively monotonous spindle cells with hyperchromatic nuclei, very high mitotic counts, and geographic areas of necrosis. This monomorphic spindle cell appearance is shared with other soft tissue sarcomas, chiefly monophasic synovial sarcomas (SSs) and adult-type fibrosarcomas among others.4,6 Rare morphologic variants include the pleomorphic type and MPNSTs with perineuriomatous differentiation.4,7 MPNSTs display limited expression of neural crest markers, such as S100 protein and Sox10, with only 30% to 39% and 49% to 67% of cases, respectively, showing positivity for these markers according to recent studies.8–10 In contrast, epithelioid MPNSTs consistently show diffuse S100 protein positivity and loss of INI1/SMARCB1 expression. Thus, accurate diagnosis of non-epithelioid MPNSTs outside the NF1 or radiation clinical history remains challenging because of lack of specific IHC and molecular markers.
Recently, our group and others demonstrated loss-of-function somatic alterations in different components of the polycomb repressive complex 2 (PRC2) in the majority of MPNST cases.11–13 These highly recurrent and specific inactivations of PRC2 components co-occurred with somatic alterations of CDKN2A and NF1. MPNSTs with PRC2 inactivation through EED or SUZ12 alterations showed consistent and complete loss of trimethylation at lysine 27 of histone H3 (H3K27me3) on IHC analysis. H3K27me3 represents an important intermediary of the PRC2 pathway of chromatin regulation.14,15 Although positive and negative H3K27me3 immunostaining was highly concordant with the genetic status of wild-type (WT) PRC2 components and homozygous loss of PRC2 components, respectively,11 heterozygous loss of PRC2 components was not predictive of H3K27me3 immunostaining. In fact, analysis of H3K27me3 expression at a protein level appeared to be more accurate than DNA sequencing, as reflected by RNA sequencing and transcriptional clustering. On the basis of these findings, we sought to investigate the specificity and sensitivity of a new monoclonal H3K27me3 antibody as an ancillary diagnostic marker in MPNSTs.
Materials and Methods
The diagnosis of MPNST was reviewed and confirmed on the basis of a constellation of findings including typical morphologic patterns, focal/patchy S100 protein/Sox10 reactivity (defined as <20% of tumor cells) or nonspecific immunoprofile, presence of a preexistent benign peripheral nerve sheath tumor, and clinical history of NF1 or prior radiation. The typical morphologic picture defining a conventional HG MPNST is represented by a fascicular, monomorphic spindle cell proliferation with a “marbled” low-power appearance and areas of geographic necrosis. Tumors with these characteristics were classified as having “classic” morphology (Figs. 1A, B). The diagnosis of LG MPNST was defined by the above-described morphologic features. Finally, examples exhibiting histomorphologic features that deviated from these classic LG and HG patterns were classified under MPNST with variant morphology, which mainly included tumors exhibiting pleomorphism (focal or diffuse) and a rare tumor showing small cell features, all these phenotypes being previously described in the literature.5,7 To accept a diagnosis of MPNST in the setting of nuclear pleomorphism other strict criteria were required, such as NF1 history, origin from a nerve or benign peripheral nerve sheath tumor, and/or focal S100 protein positivity (<20%) in the absence of other markers. The epithelioid MPNST cases displayed distinctive morphologic features with solid or nested growth of purely epithelioid cells, showing diffuse and strong S100 protein positivity and INI1/SMARCB1 loss. An effort was made to include cases with a complete IHC workup available for review, as well as cases with additional material for IHC analysis. The study was approved by the Institutional Review Board of both institutions.
Figure 1.
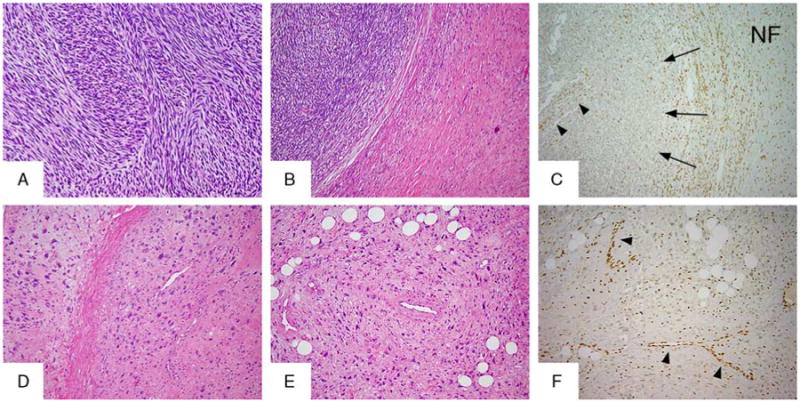
H3K27me3 immunostaining in morphologic variants of MPNSTs. Classic HG MPNST features with intersecting fascicles of monomorphic spindle cells shown here in an NF1-related tumor (A, B; MPNST03), with an adjacent plexiform NF (B, MPNST03, right side). H3K27me3 immunolabeling showed retained expression in NF (C), whereas the adjacent MPNST exhibited complete loss of expression (C, arrows). Note the internal positive controls: vessels (C, arrowheads), and inflammatory cells retaining staining. An RT-associated HG MPNST (MPNST48.1) with pleomorphic features (D, E) showing loss of H3K27me3 immunostaining (F, note internal control in vessels, arrowheads).
MPNST Patients and Tumor Samples
Our cohort consisted of a total of 68 samples from 58 MPNST patients, with 44 patients being previously included in the study by Lee et al.11 The selection of MPNST cases in that study was based on typical morphology, matching immunoprofile, and clinical findings, double-blinded from the molecular results. We centered our investigation on the prior MPNST study group to correlate the IHC findings of a new anti-H3K27me3 monoclonal antibody with the specific genetic alterations of PRC2 components and investigate the concordance with a previous IHC analysis using a H3K27me3 polyclonal antibody11 (see antibody characteristics). The study was further expanded with additional MPNST cases, as well as other entities frequently included in the differential diagnosis. There were 3 groups of samples included: from patients with NF1, from patients with a history of RT to the tumor site, and from patients without any prior history (sporadic). An effort was made to include matched samples from the same patient exhibiting different morphologic features and/or from different time points. Thus, when available we included NF-MPNST pairs from the same resection, including examples of so-called atypical NFs (when possible in near proximity), areas of different morphology (classic MPNST, pleomorphic MPNST, and tumors with divergent differentiation component), matched primary-metastases, as well as multiple primary MPNSTs arising in the same patient.
There were 32 male (55%) and 26 female (45%) patients, with an overall mean age at diagnosis of 42 years (range, 2 to 76y). The average age at diagnosis varied depending on the clinical setting of MPNST. The NF1-related MPNST patients presented at a younger age, with a mean of 37 years (range, 2 to 59y), whereas the patients with sporadic MPNST presented at a mean age of 48 years (range, 28 to 76y). In RT-related MPNST patients, tumors presented at an older age, with a mean of 51 years (range, 32 to 75y). Five epithelioid MPNST patients were included, with a mean age of 37 years (range, 26 to 59y). There were 27 NF1 MPNST patients (47%), 15 patients with sporadic MPNST (26%) and 11 patients with RT-related MPNSTs (19%). The 5 patients with epithelioid MPNSTs (9%) were grouped separately from those with conventional spindle cell sporadic MPNSTs.
Control Groups of Tumors Included in the Differential Diagnosis of MPNST
To test the specificity of this marker, we analyzed different entities that are often considered in the differential diagnosis of MPNST, as well as miscellaneous tumors as controls. This process was carried out by applying a monoclonal anti-H3K27me3 antibody to tissue microarray (TMA) slides as well as to whole-tissue sections (WTS) with examples of such entities. The majority of sarcoma tumor samples were obtained from the Memorial Sloan Kettering Cancer Center Pathology Department, whereas the melanoma samples were derived from Moffitt Cancer Center Pathology Department. A TMA containing a total of 53 cutaneous melanomas from the same number of patients was tested, including 51 primaries/recurrent lesions and 2 metastatic tumors to the lung. The melanoma cases were subdivided into 37 pure desmoplastic melanoma (DM) variants, 11 mixed DM with spindle and epithelioid components, and 5 spindle cell melanomas (SCM). All but 8 (15%) melanoma tumors (1 SCM, 2 pure DM, and 5 mixed DM/SCM) showed evidence of a melanoma in situ component. Immunoprofile data were available in all cases with the exception of 4 pure DM cases (7%), all of which showed melanoma in situ as well as classic pure DM features, both histopathologically (hypocellular proliferation of bland spindle cells in a sclerotic/myxoid stroma) and clinically (lack of lymph node metastases). All 48 tumors with available IHC exhibited diffuse and strong S100 positivity, and 24 of these tumors also showed the classic loss of Melan A and HMB45 staining. In addition, a TMA containing a large number (113) of SSs, including monophasic, biphasic, and poorly differentiated samples, was also studied. Also included were 6 soft tissue myoepithelial tumors (WTSs), 5 of them carrying the characteristic EWSR1 or FUS gene rearrangements. We investigated a TMA of 123 gastrointestinal stromal tumors (GISTs) from 113 patients, as well as WTS from KIT-mutant adult GIST, SDHB-deficient WT pediatric and adult GIST (13 cases), and KIT-negative de-differentiated GIST (1 case). Finally, TMAs containing 75 liposarcomas (31 well differentiated and 44 dedifferentiated) and 63 myxofibrosarcomas (MFSs) (both LG and HG), as well as 6 WTS of ossifying fibromyxoid tumors (OFMTs), were tested.
H3K27me3 IHC
IHC for H3K27me3 was performed utilizing rabbit monoclonal antibody, clone C36B11 (1:200 dilution; Cell Signaling Technology, Danvers, MA). All immunostaining studies were performed on a Leica Bond-RX and Bond-3 automated stainer platform (Leica, Buffalo Grove, IL) employing a polymeric secondary detection system (Refine; Leica), using diaminobenzidine as a chromogen. The immunostaining protocol was tested and optimized using samples from our pervious MPNST study using a rabbit polyclonal anti-HeK27me3 antibody (07-449; EMD Millipore, Billerica, MA).11 C36B11 was tested on TMA and WTS slides. Evaluation of the C36B11 IHC signal was recorded as positive/partially negative/negative according to the status of the nuclear signal, and the proportion of lesional cells showing lack of nuclear staining with the marker was also semi-quantitatively recorded as complete (≥95%) or partial (< 95%) loss of staining. The presence of positive internal controls (endothelial cells, stromal cells, and/or inflammatory cells) was strictly required to assess the IHC signal; samples not reaching this standard were excluded. The unpaired t test was used to assess for correlation between the different morphologic variables and H3K27me3 status.
Results
Clinicopathologic Features
A total of 68 MPNST samples from 58 patients were included in the study. Most samples (84%) originated from primary or recurrent lesions, whereas in 16% the available material was obtained from the metastatic lesions. The primary tumors were located in the limbs in 22 (37%) cases (upper limbs, 10; lower limbs, 12), in the trunk in 15 (26%) cases (11 paravertebral and 4 chest/mediastinum), in the abdominal region in 15 (26%) cases (retroperitoneum, 8; pelvis, 5; abdominal wall, 2), and in the head and neck in 5 (11%) cases. The majority of metastatic samples were located in the lung (9 cases), with 1 each in the mediastinum and adrenal gland. The primary lesions from which these metastases originated occurred in the lower limb (5 tumors), upper limb (2 tumors), and in the retroperitoneum and sacrum (1 each). Eighteen samples (26%) were status post prior chemoradiation therapy. In addition, 11 NFs were included, 8 being away from and 3 adjacent to the MPNST (evaluated in the same tissue section).
Of the 63 conventional (nonepithelioid) MPNSTs, 60 tumors (95%) showed an HG MPNST morphology, with the majority (72%) exhibiting a classic phenotype (Figs. 1A, B, Fig. 3C). Three cases (5%) showed LG MPNST features; all of them were NF1 related, with 1 tumor being associated with an atypical NF (MPNST13.1, 13.2). In the HG MPNST group (n = 60) there was a subset of 17 tumors (28%) exhibiting morphologic deviation from the classic features: 6 MPNSTs with pleomorphic features (4 NF1, 1 sporadic, and 1 RT) (Figs. 1D, E, Fig. 3E), 11 mostly classic MPNST but with focal pleomorphism(4NF1,4sporadic, and3RT) (Fig. 3A), and 1 tumor with focal pleomorphism and small cell features (sporadic, also displaying heterologous elements, Fig. 4D) Among these MPNSTs with variable pleomorphism, 4 (2 NF1 patients, 1 sporadic, and 1 RT) showed considerable morphologic intertumoral and intratumoral heterogeneity, with a classic HG MPNST component being invariably present in all cases. A total of 9 cases (13%) exhibited features of divergent differentiation (Fig. 4), 3 of them being NF1 related, 2 being RT induced, and the remaining 4 being sporadic. The most common heterologous component was rhabdomyosarcomatous (5 cases) (Figs. 4A, B), followed by glandular (3 cases) (Fig. 4B), chondrosarcomatous (2 cases), osteosarcomatous (1 case), and leiomyosarcomatous (1 case) (Fig. 4E). In 4 cases, the divergent differentiation consisted of 2 heterologous components (Figs. 4A, B). All 5 epithelioid MPNSTs exhibited the classic morphologic features of solid or nests of medium-sized to large-sized epithelioid cells bearing eosinophilic to amphophilic cytoplasm and vesicular nuclei with prominent nucleoli, in a background of a fibrotic and focally myxoid stroma (Figs. 2D, E).
Figure 3.
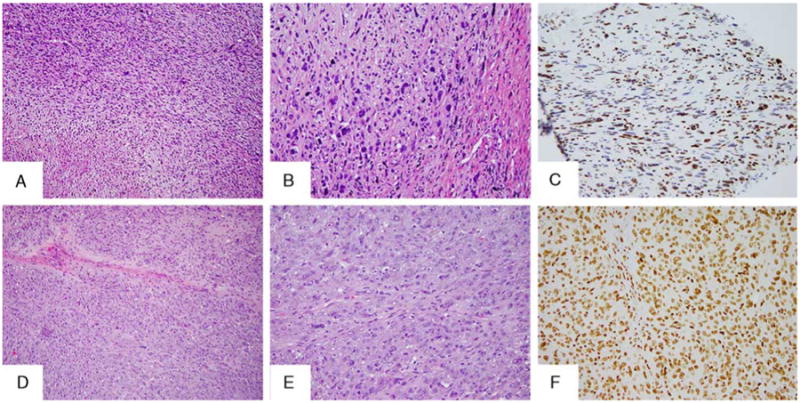
Morphologic and H3K27me3 IHC intertumoral heterogeneity in NF1-related MPNST. This patient (MPNST15) developed 3 different MPNST primaries, which showed variable morphology: HG with focal pleomorphism (MPNST15.1) (A), HG classic (C) arising with an adjacent plexiform NF (MPNST15.3), and HG with pleomorphic features (MPNST15.4) (E). Only the MPNST with pleomorphic features showed retained H3K27me3 (F), whereas the others lost expression (B, D).
Figure 4.
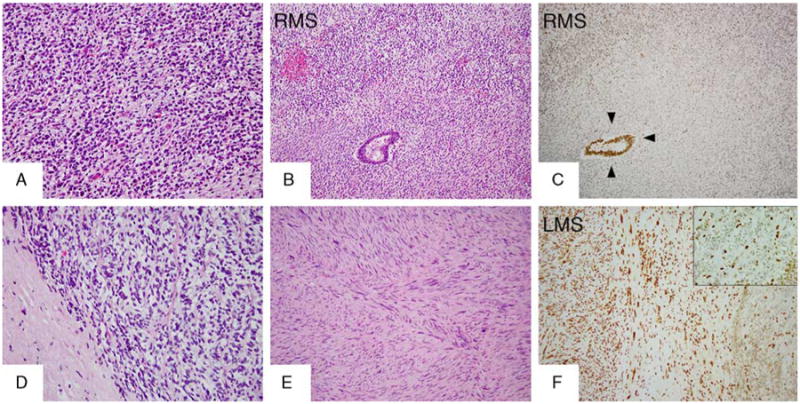
H3K27me3 staining in MPNSTs with heterologous components. A sporadic case (MPNST35) with discordant H3K27me3 expression between the 2 types of divergent differentiation components, rhabdomyosarcomatous (A, B, RMS) and glandular (B): lost expression in the homologous and RMS element (C, RMS) and retained expression in the glandular component (C, arrowheads). H3K27me3 differential expression in another sporadic MPNST (MPNST30) with 2 types of divergent differentiation components: loss of expression in the homologous (small cell) component (D, F right side and inset) and retained expression in both heterologous elements, leiomyosarcoma (E, F left side LMS) and glandular (not shown). LMS indicates leiomyosarcoma; RMS, rhabdomyosarcoma.
Figure 2.
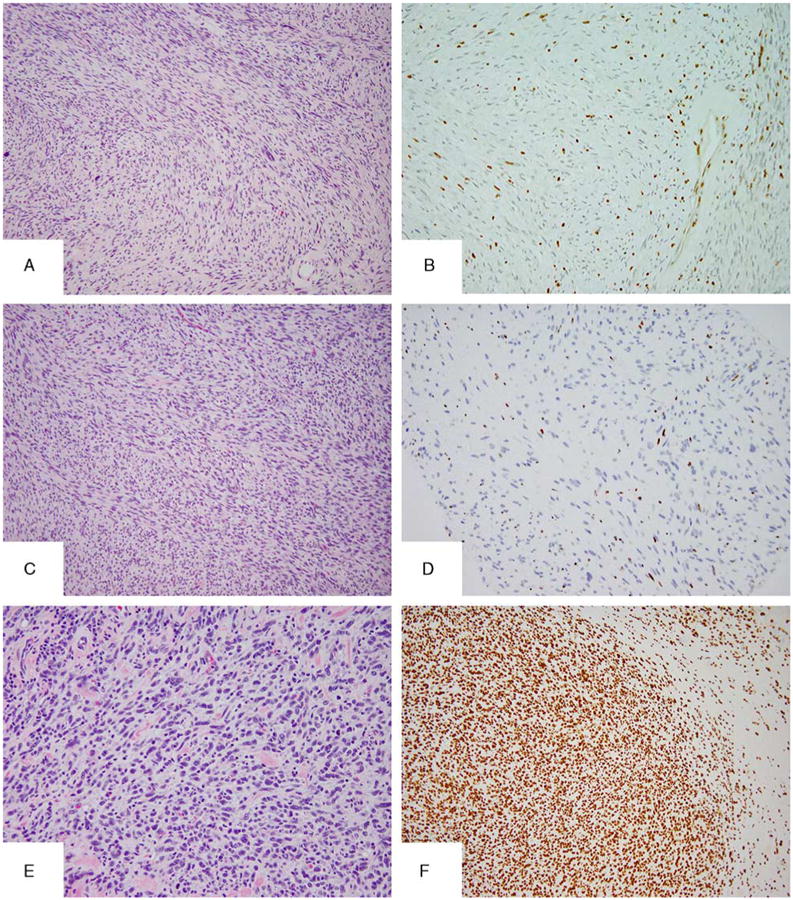
Additional patterns of H3K27me3 immunolabeling in MPNST. Partial loss or mosaic pattern of staining seen in 1 NF1-related tumor (A–C, MPNST12). Note the focal pleomorphism in MPNST12 (A, B). An epithelioid MPNST composed of solid nests of large polygonal cells with eosinophilic cytoplasm and vesicular nuclei with macronucleoli (D, E), showing diffuse immunolabeling (F).
The Majority of Sporadic and RT-related MPNSTs Showed Loss of H3K27me3 Expression
IHC evaluation with the anti-H3K27me3 monoclonal antibody revealed that 69% (n= 47) of MPNST cases demonstrated either complete (n=42, 61%) (Figs. 1C, F, Fig. 3B, D) or partial (n = 5, 7%) (Fig. 2C) loss of staining, whereas 31% (n = 21) of cases retained diffuse immunoreactivity (Fig. 3F). However, considerable differences were observed in terms of C36B11 immunostaining status among the different MPNST groups. When considering only the NF1-related HG MPNSTs (n=30), 18 cases (60%) demonstrated loss of H3K27me3 expression (14 tumors showing complete loss and 4 lesions showing partial loss), whereas H3K27me3 expression was retained in the remaining 12 (40%) NF1-related HG MPNSTs. Among the 3 LG MPNSTs only 1 tumor showed loss of H3K27me3 expression (MPNST02). All 11 NFs, either distant or adjacent to the MPNSTs, retained C36B11 nuclear labeling (Fig. 1C, right aspect of picture). In contrast, the majority of both sporadic and RT-related MPNSTs showed loss of H3K27me3 expression, in 95% (17/18) and 91% (10/11) of cases, respectively. Only 1 of the sporadic MPNST showed partial loss of H3K27me3 (MPNST41). All 5 epithelioid MPNSTs retained H3K27me3 expression (Fig. 2F). See Table 1 for a summary of results.
Table 1. Summary of the Results of H3K27me3 IHC of the Different MPNST Groups.
| H3K27me3 IHC Loss (n [%]) | |||
|---|---|---|---|
|
|
|||
| MPNST Groups | Complete | Partial | H3K27me3 IHC Retained |
| NF1-associated MPNSTs (N=33) | 15 (45) | 4 (12) | 14 (42) |
| Sporadic MPNSTs (N=18) | 16 (90) | 1 (5) | 1 (5) |
| RT-associated MPNSTs (N=12) | 11 (91) | — | 1 (9) |
| Epithelioid MPNSTs (N=5) | — | — | 5 (100) |
| Total MPNST cases (N=68) | 47 (69) | 21 (31) | |
There was no correlation (P = 0.25) between the presence of pleomorphic histology (n = 17) and the anti-H3K27me3 IHC status, with 12 (70%) tumors showing loss of H3K27me3 (11 total and 1 partial) and 5 retaining expression. When considering the different MPNST subgroups, differences can be noted with half of these cases occurring in the NF1 setting (n = 4) showing loss of staining (3 complete loss and 1 partial loss), whereas 5/6 sporadic and all 3 RT tumors exhibited complete loss of H3K27me3 expression. Among the 9 MPNSTs with divergent differentiation all except 1 (MPNST27, NF1-related) showed loss of H3K27me3 expression in the homologous component. H3K27me3 expression in the heterologous component was concordant with the homologous component in 2 of 4 cases evaluated, 2 showing loss, whereas 2 retained H3K27me3 expression (glandular and leiomyosarcomatous) (Fig. 4C, arrowheads and Fig. 4F, leiomyosarcoma “LMS”), suggesting intratumoral heterogeneity of H3K27me3 staining and perhaps of the mutational status of PRC2 gene components.
More than half (58%) of our conventional HG MPNST cohort exhibited at least focal positivity for S100 protein, whereas the remaining (42%) was completely S100 protein negative. No relationship was detected (P = 0.33) between the presence/absence of S100 immunostaining and the H3K27me3 IHC/PRC2 mutational status. The 2 retroperitoneal sporadic tumors (MPNST30 and 31) were negative for MDM2 and CDK4 immunoexpression. All 5 epithelioid MPNSTs were diffusely S100 protein positive, and 2 of the tumors tested with INI1/SMARCB1 (MPNST54, 58) also showed loss of expression of this marker.
Loss of H3K27me3 Expression Correlates With Genetic Alterations of PRC2 Components
The great majority of the MPNST cases with available molecular data (93%, 50/54) showed a good correlation between the H3K27me3 expression pattern (C36B11 immunostaining) and the PRC2 component genotype (EED and/or SUZ12). Cases with complete loss of C36B11 immunoreactivity correlated with either homozygous gene losses or point, frameshift, or splice mutations and structural variations, occurring alone or in combination with heterozygous losses. The “mosaic” pattern of partial loss of H3K27me3 expression detected in 4 NF1-related MPNSTs was associated with a SUZ12 heterozygous deletion and a WT EED in 3/4 cases, whereas a WT SUZ12 and EED heterozygous loss was found in the remaining case. No molecular data were available for the sporadic MPNST case showing partial loss of staining. Although H3K27me3-retained expression was seen in the majority of the WT/WT EED/SUZ12 genotype, there were 4 NF1-related tumors (MPNST16.1, 16.2, 17, and 18.1) in which discordant IHC/molecular results were found. Whereas the first 2 (16.1 and 16.2) showed lost H3K27me3 expression with a WT/WT EED/SUZ12 status, the other 2 exhibited diffuse H3K27me3 immunoreactivity in the presence of a SUZ12 heterozygous gene loss. A concordant pattern of WT/WT EED/SUZ12 profile and retained H3K27me3 expression was seen in all NF examples (n = 8) tested. For a complete list of the clinicopathologic findings and the mutational status of EED and SUZ12 please see Supplementary Tables 1 to 4 (Supplemental Digital Content 1, http://links.lww.com/PAS/A321).
When comparing immunoreactivity of the monoclonal antibody C36B11 with that of the polyclonal antibody used in our previous study, only 2/52 (4%) MPNST tumors and NFs were found to demonstrate discordant immunoreactivity11 (immunopositive with polyclonal antibody, immunonegative with C36B11 monoclonal antibody). Although there was an excellent correlation of the immunostaining of the 2 antibodies, the signal to noise ratio was superior with the monoclonal H3K27me3 antibody, which showed a crisper appearance with less background staining. This feature appears particularly important in the context of evaluating an IHC stain for loss of signal.
Intertumoral Heterogeneity of PRC2 Component Aberrations and H3K27me3 Expression Was Observed in Rare NF1-related MPNST Cases
Two examples in this category require a more detailed discussion. The first NF1-related MPNST exhibited different H3K27me3 expression patterns as well as different PRC2 component mutation profiles in the primary tumor (MPNST11.1) compared with the metastasis (MPNST11.2). Whereas the primary tumor exhibited complete loss of H3K27me3 expression due to an EED frameshift mutation and a SUZ12 heterozygous deletion, the metastatic deposit showed a WT EED and SUZ12 heterozygous deletion associated with partial loss of H3K27me3 expression.
The second NF1-related MPNST (MPNST15) developed 3 distinct soft tissue HG MPNSTs, with tissue available for IHC analysis of all lesions and molecular data available for analysis of 2/3 lesions. Morphologically, 1 tumor showed a classic HG phenotype with focal pleomorphism (MPNST15.1, retroperitoneum) (Fig. 3A), the second exhibited a classic HG phenotype (MPNST15.3, abdominal wall) (Fig. 3D), and the third primary was a pleomorphic MPNST (MPNST15.4, thigh) (Fig. 3E). Both the MPNST with focal pleomorphism (MPNST15.1) and the classic HG MPNST (MPNST15.3) showed complete loss of H3K27me3 (Figs. 3B, D), with MPNST15.1 harboring an SUZ12 frameshift mutation and heterozygous loss. The predominantly pleomorphic MPNST (MPNST15.4) revealed retained H3K27me3 expression (Fig. 3F), correlated with a WT/WT EED/SUZ12.
H3K27me3-retained Expression on IHC Study in All Look-alike Spindle Cell Neoplasms Tested and Other Miscellaneous Tumors
All tumors included as controls (see the Materials and methods section) exhibited positivity with the monoclonal antibody C36B11 to H3K27me3 (Fig. 5). A peculiar nucleolar pattern of signal accentuation was observed in some cases, particularly of SS (Fig. 5C) of uncertain significance. For a summarized display of these data, please refer Table 2.
Figure 5.
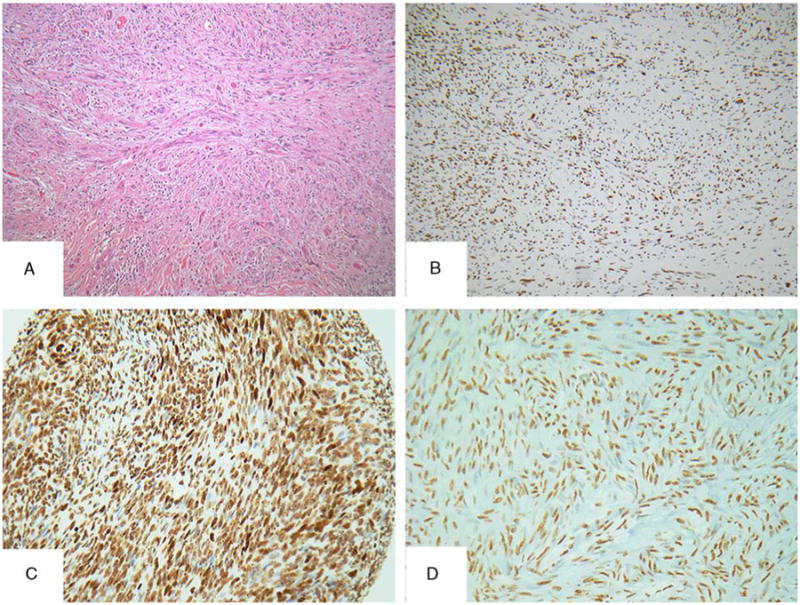
Morphologic mimics of MPNST showed retained H3K27me3 expression. An SCM with long intersecting fascicles of bland monomorphous spindle cells (A) showing diffuse and strong immunoreactivity for H3K27me3 (B). Selected cases of other entities included in the control group with H3K27me3-retained expression: monophasic SS (C) and GIST (D).
Table 2. H3K27me3 Monoclonal Antibody IHC Results of the Different Entities Included in the MPNST Differential Diagnosis and Miscellaneous Tumors.
| Diagnosis | H3K27me3 IHC Loss/Total Cases |
|---|---|
| Cutaneous melanoma | |
| Pure desmoplastic melanoma | 0/37 |
| Mixed desmoplastic melanoma | 0/11 |
| Spindle cell melanoma | 0/5 |
| Synovial sarcoma (MF, BF, and PD) | 0/113 |
| GIST | |
| KIT/PDGFRA mutant | 0/109 |
| SDHB-deficient WT pediatric and adult | 0/13 |
| WT dedifferentiated GIST | 0/1 |
| Liposarcoma | |
| Well differentiated | 0/31 |
| Dedifferentiated | 0/44 |
| Ossifying fibromyxoid tumor | 0/6 |
| Soft tissue myoepithelial carcinomas | 0/6 |
| MFS | 0/63 |
In the case of GIST, KIT and PDGFRA were WT.
BF indicates biphasic; MF, monophasic; PD, poorly differentiated.
Discussion
The diagnosis of MPNSTs remains challenging and somewhat arbitrary because of its nonspecific morphologic features and lack of pathognomonic ancillary IHC and molecular diagnostic markers. As a consequence, a high interobserver variability exists, even among experts, particularly when MPNST is encountered outside the known predisposing conditions (NF1, RT). Historically, the finding of either focal S100 positivity in 50% to 90% of cases16–19 or frequent allelic losses (20% to 50%) of NF1 in a background of complex karyotypes20 was considered consistent but relatively nonspecific features in these tumors. A more definitive diagnosis of MPNST was traditionally rendered in the context of particular clinical settings, such as in individuals affected by NF1 and tumors arising in a previously radiated area. When confronted with cases outside this clinical background (ie, in sporadic cases), the diagnosis of MPNST was often made on the basis of matching morphologic features, patchy S100 protein immunostaining, possible involvement of a nerve structure, and quite frequently as a diagnosis of exclusion from other spindle cell neoplasms.
A recent study from our group11 characterized novel molecular aberrations in MPNSTs that affected the EED (11q14.2-q22.3) and SUZ12 (17q11.2, formerly known as JJAZ1) genes, both of which code for homonymous proteins that form part of the PRC2. These aberrations were present in the majority (80%) of MPNSTs arising in a variety of clinical settings, such as NF1, RT, and sporadic scenarios, being in most cases mutually exclusive. These results were also independently confirmed and replicated by others.13,21 The EED and SUZ12 gene products, along with EZH1 and EZH2, constitute the core of the PRC2 complex and are chiefly responsible for the dimethylation and trimethylation of lysine 27 of histone H3 (H3K27me2 and H3K27me3), important epigenetic mediators of PRC2 function.14,15 PRC2 is heavily involved in chromatin compaction, heterochromatin formation, chromosome X inactivation, and polycomb-mediated gene silencing.22,23 Interestingly, SUZ12 somatic recombinations have been described in patients with NF1 mosaicism,24,25 which in part may explain the mosaicism encountered in our NF1-related cohort.
Our initial study showed that PRC2 inactivation through mutations in EED and SUZ12 induced loss of H3K27me3 IHC expression using a polyclonal anti-body.11 On the basis of these findings, we sought to investigate the performance of a new monoclonal H3K27me3 antibody as an ancillary diagnostic marker in MPNST. Thus, we expanded the initial MPNST cohort as well as investigated a large spectrum of lesions typically included in the differential diagnosis of MPNST. Our results showed loss of H3K27me3 expression in 69% of all MPNSTs (NF1, RT related, sporadic, and epithelioid). In isolation, this result appears to have an equivalent diagnostic value to S100 protein or Sox10 IHC staining. However, when considering the different MPNST clinical subsets, loss of H3K27me3 expression emerges as a highly sensitive marker, being seen in most RT-related and sporadic MPNSTs. This finding reinforces the pathologic and molecular identity of MPNSTs even in less-defined diagnostic circumstances, such as in non-NF1 patients. Although it remains uncertain why NF1-related HG MPNSTs showed the lowest incidence of molecular PRC2 complex abnormalities and exhibited loss of H3K27me3 expression in only 60% of cases, from a practical diagnostic viewpoint this particular clinical subset is rarely in need of a more definitive ancillary marker, as the majority of HG spindle cell sarcomas occurring in an NF1 patient are considered de facto MPNSTs. In addition, in this context most MPNSTs occur nearby or in association with an intraneural or plexiform NF. This scenario is obviously very different in the absence of NF1 clinical history, where the high performance of anti-H3K27me3 IHC could indeed validate the diagnosis of MPNST, otherwise being labeled by default as an unclassified spindle cell sarcoma. Fortunately, the finding of loss of H3K27me3 expression had the highest sensitivity in this particular group with 94% of sporadic and 91% of RT-related MPNSTs. All 5 epithelioid MPNSTs exhibited retention of H3K27me3 expression, further reinforcing that most of these tumors have distinct morphologic, IHC, and molecular features, compared with classic nonepithelioid MPNSTs.
In 50 of 54 (93%) MPNST cases with available IHC and molecular data the H3K27me3 expression status correlated with the EED and/or SUZ12 genetic aberrations. The 4 cases harboring discordant molecular/IHC results were NF1-related MPNSTs. In addition, 1 NF1 patient developing multiple MPNST primaries showed intertumoral heterogeneity at morphologic, H3K27me3 expression and molecular levels. Our study also attempted to investigate the impact of the histologic spectrum of MPNSTs, mostly represented by various degrees of pleomorphism, which typically falls outside the rigidly defined histologic phenotype of MPNST. When focusing on the 17 cases with pleomorphic features, although there was no correlation with the mutational status of the PCR2 components, 70% of these cases showed H3K27me3 loss of expression, particularly the tumors arising in the sporadic (5/6) and RT-related (3/3) clinical settings. Thus, the lack of anti-H3K27me3 staining may help in the diagnosis of MPNSTs with variant morphology, particularly in non-NF1 cases. Most MPNSTs with divergent differentiation demonstrated loss of H3K27me3 expression in the homologous component, with concurrent loss in the divergent component in a subset of cases. However, in 2 MPNSTs with 2 divergent components, whereas the “homologous” portion demonstrated mutations of EED and SUZ12 and concurrent loss of H3K27me3 expression, H3K27me3 positivity was retained in one of the heterologous elements, reinforcing the intratumoral genetic heterogeneity observed in some cases.
To determine the specificity of the H3K27me3 loss of expression in the context of the differential diagnosis of MPNST, we have investigated the pattern of expression of this marker in a large cohort of look-alike lesions. SS and MPNSTs share similar morphologic features, being composed of monomorphic, undifferentiated spindle cells, tightly packed in intersecting long fascicles. As this morphologic overlap can be quite significant, ancillary methods are often required for a definitive diagnosis. IHC analysis for S100 protein has limited utility in this context, as SS might show patchy reactivity ranging from 15% to 38%.8–10,26 Although recently 2 additional reliable IHC markers were added in this differential diagnosis—namely, Sox10 and TLE1—their low sensitivity and specificity have perpetuated the diagnostic challenges. Sox10 is positive in only 67% of MPNSTs, while also being present in 7% of SS cases, particularly in intraneural lesions.8,27 Similarly, the TLE1 antibody, initially hailed as a sensitive and specific marker for SS,28–30 experienced a subsequent waning of its specificity in later studies,31 including focal positivity in up to 30% of MPNSTs.29,31 Interestingly, MPNST can potentially express epithelial markers such as focal AE1:AE3 and low–molecular weight keratins,32 and the MPNST with perineurial differentiation will consistently express epithelial membrane antigen.33,34 The most reliable test in this differential diagnosis has been detecting the t(x;18) translocation in SS, either by reverse transcription polymerase chain reaction of the SS18-SSX1 or SS18-SSX2 gene fusions or by fluorescence in situ hybridization assay for SYT gene rearrangement.35,36 Nevertheless, molecular testing is not always available, and a prior controversial study has illustrated some of the pitfalls interpreting molecular results, which erroneously suggested that the t(x;18) translocation is not specific for SS but can also occur in MPNST or NFs.37 Our results suggest that H3K27me3 IHC might serve as an additional useful discriminating marker outside the NF1 setting, as all of the SS cases tested (n = 113), including monophasic, biphasic, and poorly differentiated examples, retained expression of this marker. A recent study showed that neurofibromin C-terminus (NFC) antibody, against the protein product of the NF1 gene, is a reliable ancillary marker that differentiates MPNST from other spindle cell neoplasms.38 As the majority of both NF1 and sporadic MPNSTs show NF1 gene mutations, NFC expression is lost in 88% and 43% of cases, respectively. In contrast, all SS cases included in the study (0/22), among several other spindle cell proliferations, showed retention of NFC staining.
The differential diagnosis of MPNST from SCM/DM is also very challenging, especially as these melanoma variants, particularly pure DMs, show no or little expression of melanocytic markers, such as MiTF, Melan A, and HMB45. Although most DM and SCM cases are immunoreactive with S100 protein and Sox10,8,10,39–41 loss of expression of these markers can occasionally be found,41,42 particularly in metastatic cases and when the rare phenomenon of dedifferentiation ensues.43,44 Altered PRC2 components may also play a role in melanoma progression, and they appear to be overwhelmingly composed of activating EZH2 mutations.45 Consequentially, a recent study46 demonstrated an over-expression of H3K27me3, particularly at the tumor front region, in superficial spreading, as well as in nodular, acral, and lentigo maligna melanoma tumors, with no cases of DM/SCM being included in that publication. In this study, all of the DM/SCM melanoma samples tested with H3K27me3 IHC analysis demonstrated retained expression.
Another potential problem is distinguishing deep-seated MPNSTs arising in the trunk or retroperitoneum from a dedifferentiated liposarcoma (DDL), particularly in the 10% to 15% of cases of DDL that will only show an HG spindle cell component. Similarities between DDL and MPNST include a nonspecific immunoprofile, a similar monomorphic spindle cell tumor morphologic picture, and the presence of occasional DDL cases (5% to 10%) with divergent differentiation, including rhabdomyosarcomatous elements.47,48 Nevertheless, although the diagnosis of DDL can be reliably confirmed in most cases by demonstrating either the IHC overexpression or gene amplification of MDM2 and CDK4,49 we believe that anti-H3K27me3 IHC could also be helpful in this context. The entirety of liposarcoma samples tested in this study, including 44 examples of dedifferentiated tumors, retained H3K27me3 expression.
Occasionally, GIST might be considered in the differential diagnosis of MPNST, particularly in the NF1 setting, where both GIST and MPNST can occur. Of note, most GISTs in the NF1 setting lack KIT/PDGFRA mutations, but they typically express diffuse and strong CD117 reactivity.50,51 Rare documented examples of KIT-independent imatinib-resistant GISTs may be seen to lose CD117 expression and show divergent differentiation toward a rhabdomyosarcomatous component.52 Our results reveal that none of the GIST samples tested (n = 123), spanning a large clinical and molecular spectrum, showed loss of H3K27me3 expression, adding a potential extra tool when confronted with this differential diagnosis.
Additional spindle cell lesions included in our study were MFS and OFMTs. All of our control cases of MFS and OFMT were immunoreactive with anti-H3K27me3.
In summary, our work advocates the utility of IHC analysis of H3K27me3 as a new and robust diagnostic tool in the armamentarium of soft tissue surgical pathology, in particular in the differential diagnosis of MPNSTs. In certain clinical contexts, such as RT-related or sporadic MPNSTs, loss of H3K27me3 expression emerges as a reliable ancillary marker, with excellent sensitivity and specificity. A lower level of sensitivity of this marker transpires in NF1-related MPNSTs, which is linked to a lower incidence of PRC2 inactivation. None of the entities included in the control groups, which are the most frequent considerations included in the differential diagnosis of MPNSTs, showed H3K27me3 loss of expression. Furthermore, our study indicates that monoclonal antibody clone C36B11 is an appropriate reagent for the analysis of H3K27me3 expression, demonstrating a good correlation between IHC expression and the EED/SUZ12 mutation status in the vast majority (93%) of cases.
Supplementary Material
Acknowledgments
Supported in part by the Tissue Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292); P50 CA140146-01 (C.R.A., P.C.); K08CA151660 (P.C.), DP2CA174499 (P.C.), Department of Defense, W81XWH-15-1-0124 (P.C.), Geoffrey Beene Cancer Research Center Fund, MSKCC (P.C.).
Footnotes
Conflicts of Interest and Source of Funding: The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website, www.ajsp.com.
References
- 1.Toro JR, Travis LB, Wu HJ, et al. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: an analysis of 26,758 cases. Int J Cancer. 2006;119:2922–2930. doi: 10.1002/ijc.22239. [DOI] [PubMed] [Google Scholar]
- 2.Ducatman BS, Scheithauer BW. Postirradiation neurofibrosarcoma. Cancer. 1983;51:1028–1033. doi: 10.1002/1097-0142(19830315)51:6<1028::aid-cncr2820510610>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 3.Foley KM, Woodruff JM, Ellis FT, et al. Radiation-induced malignant and atypical peripheral nerve sheath tumors. Ann Neurol. 1980;7:311–318. doi: 10.1002/ana.410070405. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295–319. doi: 10.1007/s00401-012-0954-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheithauer BW, Woodruff JM, Erlandson RA, et al. Tumors of the Peripheral Nervous System. Washington, DC: Armed Forces Institute of Pathology; 1999. [Google Scholar]
- 6.Bahrami A, Folpe AL. Adult-type fibrosarcoma: a reevaluation of 163 putative cases diagnosed at a single institution over a 48-year period. Am J Surg Pathol. 2010;34:1504–1513. doi: 10.1097/PAS.0b013e3181ef70b6. [DOI] [PubMed] [Google Scholar]
- 7.Thway K, Fisher C. Malignant peripheral nerve sheath tumor: pathology and genetics. Ann Diagn Pathol. 2014;18:109–116. doi: 10.1016/j.anndiagpath.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Kang Y, Pekmezci M, Folpe AL, et al. Diagnostic utility of SOX10 to distinguish malignant peripheral nerve sheath tumor from synovial sarcoma, including intraneural synovial sarcoma. Mod Pathol. 2014;27:55–61. doi: 10.1038/modpathol.2013.115. [DOI] [PubMed] [Google Scholar]
- 9.Karamchandani JR, Nielsen TO, van de Rijn M, et al. Sox10 and S100 in the diagnosis of soft-tissue neoplasms. Appl Immunohistochem Mol Morphol. 2012;20:445–450. doi: 10.1097/PAI.0b013e318244ff4b. [DOI] [PubMed] [Google Scholar]
- 10.Nonaka D, Chiriboga L, Rubin BP. Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol. 2008;32:1291–1298. doi: 10.1097/PAS.0b013e3181658c14. [DOI] [PubMed] [Google Scholar]
- 11.Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–251. doi: 10.1038/nature13561. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M, Wang Y, Jones S, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1170–1172. doi: 10.1038/ng.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 15.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 16.Matsunou H, Shimoda T, Kakimoto S, et al. Histopathologic and immunohistochemical study of malignant tumors of peripheral nerve sheath (malignant schwannoma) Cancer. 1985;56:2269–2279. doi: 10.1002/1097-0142(19851101)56:9<2269::aid-cncr2820560922>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Nakajima T, Watanabe S, Sato Y, et al. An immunoperoxidase study of S-100 protein distribution in normal and neoplastic tissues. Am J Surg Pathol. 1982;6:715–727. doi: 10.1097/00000478-198212000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Weiss SW, Langloss JM, Enzinger FM. Value of S-100 protein in the diagnosis of soft tissue tumors with particular reference to benign and malignant Schwann cell tumors. Lab Invest. 1983;49:299–308. [PubMed] [Google Scholar]
- 19.Wick MR, Swanson PE, Scheithauer BW, et al. Malignant peripheral nerve sheath tumor. An immunohistochemical study of 62 cases. Am J Clin Pathol. 1987;87:425–433. doi: 10.1093/ajcp/87.4.425. [DOI] [PubMed] [Google Scholar]
- 20.Mertens F, Dal Cin P, De Wever I, et al. Cytogenetic characterization of peripheral nerve sheath tumours: a report of the CHAMP study group. J Pathol. 2000;190:31–38. doi: 10.1002/(SICI)1096-9896(200001)190:1<31::AID-PATH505>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 21.Luscan A, Vidaud D, Ortonne N, et al. PRC2 alterations in NF1-associated malignant peripheral nerve sheath tumors: schwann cells with no complex. Med Sci (Paris) 2014;30:733–735. doi: 10.1051/medsci/20143008006. [DOI] [PubMed] [Google Scholar]
- 22.Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan W, Wu T, Fu H, et al. Dense chromatin activates polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science. 2012;337:971–975. doi: 10.1126/science.1225237. [DOI] [PubMed] [Google Scholar]
- 24.Kehrer-Sawatzki H, Kluwe L, Sandig C, et al. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet. 2004;75:410–423. doi: 10.1086/423624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petek E, Jenne DE, Smolle J, et al. Mitotic recombination mediated by the JJAZF1 (KIAA0160) gene causing somatic mosaicism and a new type of constitutional NF1 microdeletion in two children of a mosaic female with only few manifestations. J Med Genet. 2003;40:520–525. doi: 10.1136/jmg.40.7.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pelmus M, Guillou L, Hostein I, et al. Monophasic fibrous and poorly differentiated synovial sarcoma: immunohistochemical reassessment of 60 t(X;18)(SYT-SSX)-positive cases. Am J Surg Pathol. 2002;26:1434–1440. doi: 10.1097/00000478-200211000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Scheithauer BW, Amrami KK, Folpe AL, et al. Synovial sarcoma of nerve. Hum Pathol. 2011;42:568–577. doi: 10.1016/j.humpath.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 28.Jagdis A, Rubin BP, Tubbs RR, et al. Prospective evaluation of TLE1 as a diagnostic immunohistochemical marker in synovial sarcoma. Am J Surg Pathol. 2009;33:1743–1751. doi: 10.1097/PAS.0b013e3181b7ed36. [DOI] [PubMed] [Google Scholar]
- 29.Knosel T, Heretsch S, Altendorf-Hofmann A, et al. TLE1 is a robust diagnostic biomarker for synovial sarcomas and correlates with t(X;18): analysis of 319 cases. Eur J Cancer. 2010;46:1170–1176. doi: 10.1016/j.ejca.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 30.Terry J, Saito T, Subramanian S, et al. TLE1 as a diagnostic immunohistochemical marker for synovial sarcoma emerging from gene expression profiling studies. Am J Surg Pathol. 2007;31:240–246. doi: 10.1097/01.pas.0000213330.71745.39. [DOI] [PubMed] [Google Scholar]
- 31.Kosemehmetoglu K, Vrana JA, Folpe AL. TLE1 expression is not specific for synovial sarcoma: a whole section study of 163 soft tissue and bone neoplasms. Mod Pathol. 2009;22:872–878. doi: 10.1038/modpathol.2009.47. [DOI] [PubMed] [Google Scholar]
- 32.Smith TA, Machen SK, Fisher C, et al. Usefulness of cytokeratin subsets for distinguishing monophasic synovial sarcoma from malignant peripheral nerve sheath tumor. Am J Clin Pathol. 1999;112:641–648. doi: 10.1093/ajcp/112.5.641. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell A, Scheithauer BW, Doyon J, et al. Malignant perineurioma (malignant peripheral nerve sheath tumor with perineural differentiation) Clin Neuropathol. 2012;31:424–429. doi: 10.5414/NP300423. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg AS, Langee CL, Stevens GL, et al. Malignant peripheral nerve sheath tumor with perineurial differentiation: “malignant perineurioma”. J Cutan Pathol. 2002;29:362–367. doi: 10.1034/j.1600-0560.2001.290607.x. [DOI] [PubMed] [Google Scholar]
- 35.Coindre JM, Hostein I, Benhattar J, et al. Malignant peripheral nerve sheath tumors are t(X;18)-negative sarcomas. Molecular analysis of 25 cases occurring in neurofibromatosis type 1 patients, using two different RT-PCR-based methods of detection. Mod Pathol. 2002;15:589–592. doi: 10.1038/modpathol.3880570. [DOI] [PubMed] [Google Scholar]
- 36.Guillou L, Coindre J, Gallagher G, et al. Detection of the synovial sarcoma translocation t(X;18) (SYT;SSX) in paraffin-embedded tissues using reverse transcriptase-polymerase chain reaction: a reliable and powerful diagnostic tool for pathologists. A molecular analysis of 221 mesenchymal tumors fixed in different fixatives. Hum Pathol. 2001;32:105–112. doi: 10.1053/hupa.2001.21130. [DOI] [PubMed] [Google Scholar]
- 37.O'Sullivan MJ, Kyriakos M, Zhu X, et al. Malignant peripheral nerve sheath tumors with t(X;18). A pathologic and molecular genetic study. Mod Pathol. 2000;13:1253–1263. doi: 10.1038/modpathol.3880230. [DOI] [PubMed] [Google Scholar]
- 38.Reuss DE, Habel A, Hagenlocher C, et al. Neurofibromin specific antibody differentiates malignant peripheral nerve sheath tumors (MPNST) from other spindle cell neoplasms. Acta Neuropathol. 2014;127:565–572. doi: 10.1007/s00401-014-1246-6. [DOI] [PubMed] [Google Scholar]
- 39.Mohamed A, Gonzalez RS, Lawson D, et al. SOX10 expression in malignant melanoma, carcinoma, and normal tissues. Appl Immunohistochem Mol Morphol. 2013;21:506–510. doi: 10.1097/PAI.0b013e318279bc0a. [DOI] [PubMed] [Google Scholar]
- 40.Palla B, Su A, Binder S, et al. SOX10 expression distinguishes desmoplastic melanoma from its histologic mimics. Am J Dermatopathol. 2013;35:576–581. doi: 10.1097/DAD.0b013e31827a0b98. [DOI] [PubMed] [Google Scholar]
- 41.Nonaka D, Chiriboga L, Rubin BP. Differential expression of S100 protein subtypes in malignant melanoma, and benign and malignant peripheral nerve sheath tumors. J Cutan Pathol. 2008;35:1014–1019. doi: 10.1111/j.1600-0560.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- 42.Miller DD, Emley A, Yang S, et al. Mixed versus pure variants of desmoplastic melanoma: a genetic and immunohistochemical appraisal. Mod Pathol. 2012;25:505–515. doi: 10.1038/modpathol.2011.196. [DOI] [PubMed] [Google Scholar]
- 43.Kacerovska D, Michal M, Kutzner H, et al. Metastatic desmoplastic malignant melanoma associated with low-grade myofibroblastic sarcoma. Am J Dermatopathol. 2009;31:490–494. doi: 10.1097/DAD.0b013e31819afdaa. [DOI] [PubMed] [Google Scholar]
- 44.Kiuru M, McDermott G, Berger M, et al. Desmoplastic melanoma with sarcomatoid dedifferentiation. Am J Surg Pathol. 2014;38:864–870. doi: 10.1097/PAS.0000000000000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zingg D, Debbache J, Schaefer SM, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6:6051, 1, 17. doi: 10.1038/ncomms7051. [DOI] [PubMed] [Google Scholar]
- 46.Kampilafkos P, Melachrinou M, Kefalopoulou Z, et al. Epigenetic modifications in cutaneous malignant melanoma: EZH2, H3K4me2, and H3K27me3 immunohistochemical expression is enhanced at the invasion front of the tumor. Am J Dermatopathol. 2015;37:138–144. doi: 10.1097/DAD.0b013e31828a2d54. [DOI] [PubMed] [Google Scholar]
- 47.Pytel P, Taxy JB, Krausz T. Divergent differentiation in malignant soft tissue neoplasms: the paradigm of liposarcoma and malignant peripheral nerve sheath tumor. Int J Surg Pathol. 2005;13:19–28. doi: 10.1177/106689690501300103. [DOI] [PubMed] [Google Scholar]
- 48.Binh MB, Guillou L, Hostein I, et al. Dedifferentiated liposarcomas with divergent myosarcomatous differentiation developed in the internal trunk: a study of 27 cases and comparison to conventional dedifferentiated liposarcomas and leiomyosarcomas. Am J Surg Pathol. 2007;31:1557–1566. doi: 10.1097/PAS.0b013e31804b4109. [DOI] [PubMed] [Google Scholar]
- 49.Sirvent N, Coindre JM, Maire G, et al. Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. Am J Surg Pathol. 2007;31:1476–1489. doi: 10.1097/PAS.0b013e3180581fff. [DOI] [PubMed] [Google Scholar]
- 50.Andersson J, Sihto H, Meis-Kindblom JM, et al. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol. 2005;29:1170–1176. doi: 10.1097/01.pas.0000159775.77912.15. [DOI] [PubMed] [Google Scholar]
- 51.Kinoshita K, Hirota S, Isozaki K, et al. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol. 2004;202:80–85. doi: 10.1002/path.1487. [DOI] [PubMed] [Google Scholar]
- 52.Liegl B, Hornick JL, Antonescu CR, et al. Rhabdomyosarcomatous differentiation in gastrointestinal stromal tumors after tyrosine kinase inhibitor therapy: a novel form of tumor progression. Am J Surg Pathol. 2009;33:218–226. doi: 10.1097/PAS.0b013e31817ec2e6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.