

SUMMARY
NF-κB is considered a major contributor to tumor development, but how this factor functions in the initial stages of oncogenesis is not clear. In a model of Ras-induced transformation, we probed NF-κB function as preneoplastic cells formed tumors in mice. As previously shown, the p65 subunit of NF-κB acts as a tumor suppressor in normal cells by sustaining senescence following DNA damage. Our current data reveal that, following immortalization, p65 switches to an oncogene by counteracting the surveillance properties of immune cells. NF-κB exerts this effect by protecting transformed cells against macrophage-derived proapoptotic factors, tumor necrosis factor, and nitric oxide. Additionally, NF-κB acts through transforming growth factor beta (TGF-β) to mitigate T cell cytotoxicity and other factors to expand myeloid-derived suppressor cells. Together, these data suggest that NF-κB functions in the early stages of transformation by suppressing immune surveillance of both innate and adaptive immune cells, information that may be useful for targeted immunotherapies.
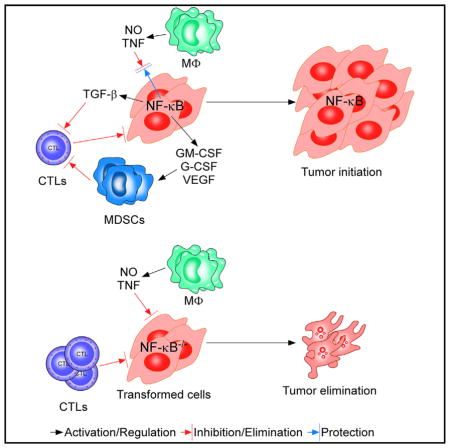
Graphical Abstract
INTRODUCTION
Cancer is a complex genetic disease involving multiple steps of activated proto-oncogenes and inactivation of tumor suppressor genes (Hanahan and Weinberg, 2000). In humans, these processes take decades, going through stages of tumor initiation, promotion, and progression. This likely explains why the majority of cancers occur in older adults (Hanahan and Weinberg, 2000). In mammals, normal cells experience constant genotoxic stresses, which lead to DNA damage and genomic instability. In response to those stresses, tumor suppressors are activated to mediate proliferation arrest, DNA repair, cellular senescence, or cell death, which function as intrinsic barriers against further genetic mutations and tumor initiation (Campisi, 2013; Lowe et al., 2004). In the case of cellular senescence, studies indicate that activated oncogenes trigger a senescence program rather than directly inducing transformation. Further loss of tumor suppressors allows cells to escape senescence and transition to an immortalized state that in the presence of additional genetic alterations can progress to cancer (Campisi, 2013; Collado and Serrano, 2010).
However, once cells become transformed, they still need to overcome an extrinsic tumor suppressive mechanism coordinated by innate and adaptive immune cells. These cells function to detect tumor antigens, derived from mutated or aberrantly expressed gene products during tumor initiation and progression. Antigen recognition activates the immune system, leading to the elimination of tumor cells, a process referred to as immune surveillance (Pardoll, 2003; Schreiber et al., 2011). Because cancerous cells are genetically unstable, a rare subset may survive the elimination phase whose expansion is kept in equilibrium with the continued presence of the immune system. Over time, additional genetic changes allow selected cells to acquire the ability to circumvent the immune barrier and develop into a fully cancerous state (Khong and Restifo, 2002; Schreiber et al., 2011). The capacity for early stage tumor cells to escape immune surveillance is in fact now considered as a principle hallmark of cancer (Hanahan and Weinberg, 2011).
As a ubiquitously expressed transcription factor, NF-κB is widely considered to play a major role in tumor development by promoting cell survival, proliferation, angiogenesis, and metastasis (Chaturvedi et al., 2011; Karin et al., 2002). Such activities are mediated through homo- or heterodimerizations of NF-κB subunits RelA/p65, c-Rel, RelB, p50, and p52, in which RelA/p65 and p50 are the most abundant subunits in vertebrates. These subunits share a Rel homology domain for DNA binding, protein interaction, and nuclear localization, but only RelA/p65 (here on referred to as p65), c-Rel, and RelB contain additional transactivation domains (Hayden and Ghosh, 2008).
In malignant cells, NF-κB is activated by the oncogenes Ras, HER2/neu, BCR-ABL, CARD11, and Bcl-10 (Chaturvedi et al., 2011; Mayo et al., 1997; Reuther et al., 1998; Staudt, 2010). Gene loci encoding c-rel, NFKBIA, B cell receptor, and A20 also undergo amplifications, mutations, or deletions, which further accounts for constitutive NF-κB activity (Chaturvedi et al., 2011; Staudt, 2010). NF-κB antagonizes p53 (Tergaonkar et al., 2002) and studies support the requirement of NF-κB in breast cancer and in inflammation-induced oncogenesis (Karin, 2009; Sovak et al., 1997). In spite of this overwhelming evidence that NF-κB acts as a tumor promoter, a growing number of reports indicate that NF-κB also possesses tumor suppressor activity. NF-κB is capable of inducing the proapoptotic gene CD95/Fas (Chan et al., 1999), and, in certain tissues such as keratinocytes, NF-κB promotes growth arrest through regulation of the p21-cyclin-dependent kinase inhibitor (Dajee et al., 2003; Seitz et al., 1998). In addition, conditional deletion of the NF-κB-activating kinase complex subunits IKKβ or NEMO in mice facilitates the development of heptocellular carcinoma (Luedde et al., 2007; Maeda et al., 2005).
How NF-κB possesses these seemingly opposite activities during tumor development remains elusive. One possibility is that either its tumor suppressor or tumor promoter activity is restricted to one cell type, as seen in keratinocytes (Dajee et al., 2003) or colon cells, respectively (Schwitalla et al., 2013; Shaked et al., 2012). Alternatively, NF-κB may function in a cell autologous manner, acting both as a tumor promoter and suppressor. To test these possibilities, we utilized a genetic model, which allowed us to study NF-κB function throughout the early stages of tumorigenesis. Our findings reveal that precancerous cells lacking p65 escape senescence and immortalize at a faster rate compared to wild-type cells. However, during a transition from immortalization to transformation mediated by Ras, NF-κB switches from a tumor suppressor to a tumor promoter. This switch allows Ras-expressing cells to overcome elimination by the innate and adaptive immune systems. Thus, in addition to the well-studied antiapoptotic activity of NF-κB (Finco et al., 1997; Hanson et al., 2004; Mayo et al., 1997), our current work highlights that during tumor initiation NF-κB protects transformed cells against a suppressive mechanism mediated by immune surveillance.
RESULTS
p65 Acts as a Tumor Suppressor in Precancerous Mouse Embryonic Fibroblasts
Previous results showed that p65 acts as a tumor suppressor in cytogenetically normal mouse and human fibroblasts by regulating genomic stability through DNA repair (Rovillain et al., 2011; Wang et al., 2009). Consistent with these results, we observed that primary p65−/− mouse embryonic fibroblasts (MEFs) escaped senescence and transitioned into a precancerous, immortalized state considerably faster than p65+/+ littermate cells (Figure 1A). This phenotype associated with defects in DNA repair, as measured by a delay in γ-H2AX recovery following a sublethal dose of irradiation (4–8 Gy) in p65−/− compared to p65+/+ cells (Figure S1A). In addition, DNA damage as assessed by comet analysis exhibited a persistent tail in p65−/− cells (Figure S1B). This equated to 20% DNA strand break repair in null cells compared to 70% repair in p65+/+ cells (p < 0.0001) (Figure S1C). Such data reaffirm our original findings (Wang et al., 2009) and support that NF-κB functions as a tumor suppressor by maintaining the genomic integrity of primary cells.
Figure 1. NF-κB Is Able to Switch from a Tumor Suppressor to a Tumor Promoter.

(A) Primary p65+/+ and p65−/− cells were subcultured every 3 days until immortalization. Graph depicts cumulative cell numbers at each passage (n = 3).
(B) Immortalized p65+/+ and p65−/− MEFs were inoculated and counted at indicated time points. Data are represented as mean ± SD from five out of seven pairs of immortalized lines.
(C) Same cells in (B) were grown in soft agar and colonies (>40 cells/cluster) were subsequently scored. Data are represented as mean ± SD from three independent experiments.
(D) Immortalized p65+/+ orp65−/− cells (1 × 107) were subcutaneously injected into SCID mice. Mice were photographed after 10 months, and data are representative of a minimum of ten mice per group that were injected with either p65+/+ or p65−/− cells.
(E) Western blotting was performed with a pan Ras antibody on cell extracts from immortalized p65+/+ and p65−/− cells infected with pBabe-Hygro or pBabe-H-RasG12V retroviruses.
(F) p65+/+Ras or p65−/−Ras cells were inoculated, and at indicated time points cells were counted. Data are representative of mean ± SD from three out of four pairs of Ras-expressing lines.
(G and H) To compare tumor growth, p65+/+Ras and p65−/−Ras cells were injected in SCID mice on left and right flanks, respectively. Photographs were taken after 4 weeks (G) or tumor sizes were measured at indicated time points and tumor diameter was plotted (H). Data are representative of at least 16 mice injected in three independent experiments (p = 0.004).
NF-κB Switches to a Tumor Promoter in Ras-Expressing MEFs
Next, we asked if loss of tumor suppressor activity in immortalized p65−/− cells was sufficient to promote transformation. Proliferation assays revealed that five out of seven p65−/− lines grew at an accelerated rate compared to p65+/+ littermate pairs (Figure 1B). In addition, p65−/− lines readily formed colonies in soft agar, whereas no colonies were observed in wild-type cells (Figure 1C). However, in spite of these tumorigenic features, none of the seven p65−/− lines that we established formed tumors in severe combined immunodeficiency (SCID) mice, even after 10 months of observation (Figure 1D). Thus, although p65 maintains tumor suppressor activity in preneoplastic cells, loss of this NF-κB subunit is not sufficient to promote tumorigenesis in mice.
To further understand the role of NF-κB in the early stages of tumor development, we transformed MEFs with stable expression of mutant Ha-RasG12V (referred to as p65+/+Ras and p65−/−Ras) (Figure 1E). Similar to immortalized cells, p65−/−Ras lines maintained a faster doubling time (Figure 1F). However, in contrast to p65+/+Ras cells that formed tumors as early as 5–7 days postinjection in 16/16 SCID mice with as little as 1 × 105 injected cells, tumor onset from p65−/−Ras cells was substantially delayed, appearing only after 15–25 days in 15/16 mice (p < 0.004) (Figures 1G and 1H). Similar results were observed in C57BL/6 nude mice (Figure S1D). Thus, as previously demonstrated, oncogenic Ras requires NF-κB to facilitate tumorigenesis (Finco et al., 1997; Meylan et al., 2009). Taken together, these results suggest that p65 is capable of switching activities from a tumor suppressor in normal cells to a tumor promoter in transformed cells.
NF-κB Protects Transformed Cells from Macrophage-Induced Cell Death
Mechanistically, how Ras utilizes NF-κB to facilitate tumorigenesis remains unclear. To gain insight, we histologically analyzed p65−/−Ras tumors that had eventually formed in SCID mice. Consistent with the notion that cancer development is accompanied by active changes in the stroma (Mueller and Fusenig, 2004), host cellular infiltrates were observed that stained positive for p65 (Figure 2A). Similarly, cells positive for the macrophage marker, F4/80, were also observed in these tumor sections (Figure 2B). This indicated that tumor development is accompanied by active recruitment of innate immune cells, which still occurs in SCID and nude mice lacking adaptive immunity. Given that tumor development from p65−/−Ras cells was delayed in SCID and nude mice, we considered that this lag derived from infiltrating macrophages (MΦs) that functioned in surveillance to eliminate Ras-transformed cells lacking NF-κB. To test this notion, we probed for the presence of MΦs following peritoneal injections of p65+/+Ras or p65−/−Ras cells in SCID mice. After only 5 days, infiltrating immune cells were clearly detected, of which >90% were MΦs (Figures S2A and 2C). Next, SCID mice were treated with liposomes containing the chemical, clodronate, which depletes MΦ (Figure S2B). This treatment caused adverse effects in SCID mice (likely due to a highly compromised immune system; Movie S1). However, from the mice that survived, we observed that p65−/−Ras tumors formed faster than in control mice treated with PBS/liposomes (Figure 2D; p < 0.0001). In contrast, p65+/+Ras cells showed a significantly delayed tumor growth (Figure S2C; p = 0.0338). These data highlighted a function of NF-κB in an early stage of tumor development that provides transformed cells resistance against innate immunity.
Figure 2. p65−/− Ras Tumors Show Host Immune Cell Infiltration.

(A and B) Tumors arising in SCID mice from p65+/+Ras and p65−/−Ras cells were sectioned and subsequently stained immunohistochemically for p65 (A) or F4/80 (green) counterstained with DAPI (blue) (B).
(C) p65+/+Ras and p65−/−Ras cells were injected peritoneally into SCID mice and infiltrating cells were harvested after 5 days and quantitated for F4/80 staining by FACS. Data were plotted as mean ± SD from two independent experiments with at least four mice in each group. Both total cell and MΦ numbers were compared to the PBS group: *p < 0.01; **p < 0.0005.
(D) Once per week, clodronate/liposome or its carrier, PBS/liposome were injected intravenously via tail vein. Two days after first injections, p65−/−Ras cells were injected subcutaneously into SCID mice. At indicated time points, tumor size was measured and plotted by tumor diameter. Data are representative of three independent experiments (p < 0.0001).
We then determined how NF-κB was required for this resistance. We designed an experiment where an equivalent number of p65+/+Ras and p65−/−Ras cells were “painted” with fluorescent tracker dyes, CMRA (orange) and CFSE (green), respectively, and mixed together in culture. This mixed population was subsequently cocultured with increasing ratios of activated MΦ. Following an overnight incubation, these cells were fixed and analyzed by fluorescence microscopy to score for viability based on how many CMRA+ and CFSE+ cells remained on the plate. Compared to p65+/+Ras cells, which remained viable with increasing ratios of MΦ, p65−/−Ras viability was pronouncedly reduced (Figures 3A and S3A), showing that Ras-transformed cells lacking NF-κB are sensitive to MΦ-induced killing.
Figure 3. Ras Cells Lacking p65 Are Sensitive to Innate Immunity-Induced Cell Death.

(A) p65+/+Ras and p65−/−Ras cells were respectively stained with fluorescence dyes, CMRA and CFSE, and then cocultured with activated macrophages (MΦs). Viability was scored by calculating the percentage of CMRA and CFSE-positive cells that remained on the culture dish. Data are representative of three independent experiments. *p < 0.001.
(B) p65+/+Ras or p65−/−Ras cells were incubated with MΦ, either wild-type or null for TNF, and viability was scored by a trypan blue exclusion assay. Cell survival was normalized to untreated cells, set to 100%. Data are representative of at least three independent experiments, each from two independent pairs of Ras-transformed cells. *p < 0.05, **p < 0.04.
(C) p65+/+Ras and p65−/−Ras cells were incubated with the NO donor SNP at indicated concentrations, and viability was scored with an MTS assay.
(D) Similar as (B) except that MΦs were used that were either wild-type or null for iNOS. *p < 0.005, **p < 0.002.
(E) Similar as (B) except that MΦs were used that were either wild-type or double null for TNF and iNOS. *p < 0.001, **p < 0.0002.
(F) p65+/+Ras and p65−/−Ras cells were cocultured with NO plus TNF or with increasing ratios of MΦs overnight. Cells were then stained for Annexin V, 7-AAD, and CD11b. CD11b− cells positive for Annexin V or positive for 7-AAD and negative for Annexin V were graphed.
(A–F) Data are represented as mean ± SD. *p < 0.02 is compared to p65+/+Ras cells; **p < 0.02 is compared to p65−/−Ras cells.
Because MΦs are a major source of tumor necrosis factor (TNF), which in the absence of NF-κB functions as a potent pro-apoptotic factor (Gapuzan et al., 2005), we investigated whether elimination of p65−/−Ras cells was dependent on this cytokine. Therefore, p65+/+Ras and p65−/−Ras cells were cocultured with increasing ratios of MΦ derived from TNF−/− mice. Compared to wild-type MΦ, those deficient in TNF were significantly less toxic to p65−/−Ras cells, although toxicity was still observed (Figure 3B). These data supported that Ras cells require p65 to survive the proapoptotic activity of TNF produced from MΦ.
We then employed an MTS strategy to screen for additional proapoptotic factors secreted from MΦ, including interleukin (IL)-12, IL-15, IFN-β, H2O2, and nitric oxide (NO). Regardless of dose, IL-12, IL-15, IFN-β, and H2O2 had little effect on the viability of p65+/+Ras or p65−/−Ras cells (Figures S3B–S3E). However, addition of the NO donor, sodium nitroprusside (SNP), was extremely potent in eliminating p65−/−Ras cells (Figure 3C). Given that NO is generated by inducible nitric oxide synthase-2 (iNOS), we addressed the role of iNOS in MΦ-mediated killing of p65−/−Ras cells by coculturing Ras-transformed cells with MΦ in the presence or absence of the iNOS inhibitor, aminoguanidine. Compared to untreated MΦ, those exposed to aminoguanidine were significantly less effective at eliminating p65−/−Ras cells (Figure S3F). Similarly, MΦs from iNOS−/− mice were equally less potent in eliminating p65−/−Ras cells (Figure 3D). Importantly, p65−/−Ras cells were completely resistant to MΦ lacking both iNOS and TNF (DKO) (Figure 3E). We further used fluorescence-activated cell sorting (FACS), and markers of apoptosis (Annexin V) and necrosis (7-AAD), to determine the mechanism of MΦ-mediated cell killing. Although some level of necrosis was observed, the majority of killing by TNF + NO or activated MΦ derived from apoptosis (Figure 3F). Moreover, measurement of NF-κB antiapoptotic genes revealed that c-IAP1/2, and to a lesser extent, BCL-XL and BCL-2, were reduced in p65−/−Ras compared to p65−/−Ras cells (Figure S3G). Together, these data suggest that in the initial stages of Ras transformation, infiltrating MΦs participate in tumor elimination by secreting cytotoxic factors TNF and NO, but this surveillance property can be circumvented by the antiapoptotic activity of NF-κB in tumor cells.
p65−/−Ras Cells Are Genetically Unstable, Leading to Tumor Formation
Having shown that Ras cells lacking NF-κB are sensitive to innate immune cells, and that this sensitivity leads to a delay in tumor onset, we next asked why transplanted p65−/−Ras cells were eventually able to overcome the surveillance property of MΦs and develop tumors as seen in Figures 1G and S1D. To address this point, we surgically removed tumors developed in SCID mice and reconstituted p65+/+Ras and p65−/−Ras cells in culture under selection to eliminate any host cell contaminants. Immunoblots confirmed that selected cells, now referred to as RasT (for tumor derived), maintained an appropriate p65 genotype (Figure 4A). Interestingly, unlike p65−/−Ras cells, p65−/−RasT cells were no longer sensitive to TNF killing (Figure S4A). Likewise, p65−/−RasT cells were completely refractory to increasing ratios of MΦ and were more resistant to NO-induced apoptosis (Figures 4B and S4B). This suggested that p65−/−RasT cells had developed a mechanism to escape the surveillance property of MΦs.
Figure 4. p65−/−Ras Cells Reconstituted from p65−/−Ras Tumors Develop Resistance to Innate Immune Cell-Mediated Cell Death.

(A) p65+/+Ras and p65−/−Ras tumors were surgically removed from SCID mice and reconstituted in culture with antibiotic selection. Lysates were prepared and westerns performed probing for p65.
(B) p65+/+RasT and p65−/−RasT cells were cocultured with activated MΦ and viability was scored by trypan blue exclusion. Data are represented as mean ± SD of three independent experiments from two pairs of cells.
(C) Frozen sections from p65+/+ and p65−/− tumors were stained with anti-γ-H2AX (green) and anti-pBP1 (red). Cell nuclei were counterstained with DAPI (blue).
(D) p65+/+Ras and p65−/−Ras cells were infected with a pBabe retrovirus expressing LacZ. Cells were stained for LacZ immediately after retrovirus infection or after 2 weeks of puromycin selection.
(E) Microarray, M-A gene expression plots of p65+/+RasT or p65−/−RasT cells compared to p65+/+Ras or p65−/−Ras cells. Red markings denote genes that have >2-fold expression changes in RasT cells compared to Ras cells.
(F) Histogram representing the number of genes exhibiting significant up or downregulated expression changes between p65+/+RasT and p65+/+Ras or p65−/−RasT and p65−/−Ras cells.
(G) p65+/+RasT and p65−/−RasT cells were injected subcutaneously into SCID mice, and, at indicated times, tumors were measured with a digital caliber and represented as mean ± SD; p < 0.0001.
Cells expressing oncogenes are genetically unstable and are under continuous immune selection in vivo (Khong and Restifo, 2002). Over time, a rare subset of these cells develop mechanisms to overcome elimination by immune cells, thus allowing for their expansion (Khong and Restifo, 2002; Schreiber et al., 2011). Because we showed that cells lacking p65 are compromised in DNA repair (Wang et al., 2009) (Figure S1), we hypothesized that similar cells in vivo would be susceptible to continuous genetic insults leading to greater genetic instability and, in turn, would acquire the ability to evade immune surveillance. To test this notion, frozen sections from p65+/+Ras and p65−/−Ras tumors were stained for γ-H2AX. Although both tumor types exhibited DNA damage, γ-H2AX foci were noticeably higher in tumors lacking p65 (Figure 4C), which correlated with a second DNA damage repair marker, p53BP1 (Noon et al., 2010). This indicated that in vivo, p65−/−Ras tumor cells undergo augmented genomic alterations. To confirm these results, we performed a LacZ genomic stability assay, which as shown previously (Wang et al., 2009), is an effective method to quantitatively measure genomic integrity. This assay relies on the stable integration of a retrovirus expressing the β-galactosidase (LacZ) reporter gene, which is lost as a result of DNA deletions, mutations, or epigenetic silencing. Results showed that compared to p65+/+Ras cells, LacZ expression was completely absent in p65−/−Ras cells (Figure 4D). Given that retrovirus integration in mammalian genomes require proper DNA repair (Skalka and Katz, 2005), our findings suggest that Ras-transformed cells lacking p65 exhibit higher defects in DNA repair, leading to increased genomic instability.
Because genetic alterations are commonly reflected by changes in gene expression, high-density gene expression profiling was performed between two pairs of populations, p65+/+Ras versus p65+/+RasT and p65−/−Ras versus p65−/−RasT cells. Consistent with genomic stability results, we found that only ~300 genes were differentially expressed following the transition from p65+/+Ras to p65+/+RasT cells (Figure 4E). In contrast, ~2,000 genes were significantly altered as p65−/−Ras progressed to p65−/−RasT cells, and the vast majority of these genes (>1,900) were upregulated (Figure 4F). Such results demonstrate that genetic alterations occur at a higher frequency during the transition from p65−/−Ras to p65−/−RasT cells. These data further indicate that tumor formation from p65−/−Ras cells in immune-compromised mice results from inefficient DNA repair and genomic instability, which, in turn, allows selected cells to escape the cytotoxic effects of TNF and NO from innate immune cells.
From these results above, we reasoned that p65−/−RasT cells should be competent to form tumors in SCID mice because they had already undergone selection in vivo, making them capable of overcoming MΦ-induced cell death. Indeed, not only did p65−/−RasT cells overcome the initial delay period in tumor formation seen with p65−/−Ras cells, but, impressively, tumors also developed with faster kinetics than those from p65+/+RasT cells (Figure 4G). Although we showed that p65−/−RasT cells were resistant to TNF and NO, we asked whether their selection in vivo also made them resistant to other proapoptotic activities mediated by chemotherapeutic compounds. Unlike TNF and NO, in vitro treatment with, doxorubicin was similarly effective in killing p65−/−Ras and p65−/−RasT cells, as compared to p65+/+ cells (Figure S4C). In addition, although doxorubicin administration in mice had no effect on the growth rate of p65+/+RasT tumors, those same doses caused significant reduced growth of p65−/−RasT tumors (Figures S4D and S4E). Similar results were observed with etoposide (data not shown). These findings indicate that mutations that arose during the development of p65−/−Ras tumors were likely selected for their resistance against the innate immune system, but not other pro-apoptotic pathways elicited by chemotherapy.
Ras Cells Lacking p65 Are Completely Inhibited from Forming Tumors in Immune-Competent Mice
Because our results supported that NF-κB acts in the initiating stages of oncogenesis to protect transformed cells from MΦ, we next asked whether such protection could be extended to adaptive immune cells. Therefore, we subcutaneously injected p65+/+Ras and p65−/−Ras cells into syngeneic C57BL/6 mice, which unlike SCID or nude mice contain a fully competent immune system. In striking contrast to wild-type cells, which readily formed tumors after only 10 days postimplantation, Ras cells lacking p65 completely failed to form tumors in 100% (30/30) of injected mice, even after 10 months of observation (Figure 5A; data not shown). This dramatic result underscores the pivotal role that NF-κB plays in oncogenic Ras-expressing cells in evading the surveillance mediated by both innate and adaptive immune cells. To further test this point, we took p65−/−RasT cells, which resist MΦ-induced cell death and injected them into immune-competent C57BL/6 mice. Although p65−/−RasT cells were capable of initially forming tumor nodules in the first 10 days postinjection, these nodules completely regressed between 16 and 20 days, and no further tumor formation was observed in 97% (29/30) of injected mice, as compared to tumors that rapidly formed from p65+/+RasT cells (Figure 5B and inset; Figure S5). We suspected that this initial growth of p65−/−RasT tumors at 10 days postinjection reflected cells that were resistant to activated MΦ but were eventually eliminated by adaptive immune cells. To confirm this notion, C57BL/6 mice were preimmunized by injection of p65−/−Ras cells, and after 2 weeks further injected with p65−/−RasT cells. Compared to nonimmunized mice where injection of p65−/−RasT cells were observed to again induce tumor nodules that subsequently regressed, those preimmunized were completely unable to form tumors (Figure 5C). These results strongly suggest that NF-κB functions in transformation as a regulator of immunoevasion from both innate and adaptive immune systems.
Figure 5. p65 Mediates Immune Tolerance.

(A) p65+/+Ras and p65−/−Ras cells were injected subcutaneously into C57BL/6 mice and tumors were measured at indicated time points.
(B) p65+/+RasT and p65−/−RasT cells were injected subcutaneously into C57BL/6 mice and similar to (A) tumors were measured with a digital caliber.
(C) PBS (No preimmunization) or p65−/−Ras cells (Preimmunized) were injected in the right flank of C57BL/6 mice. Two weeks later, p65−/−RasT cells were injected in the left flank of the same mice. Tumor size was measured at the times as shown in (C).
(D) p65+/+Ras and p65−/−Ras cell-specific CTLs were obtained as described in Experimental Procedures and subsequently cocultured with p65+/+Ras or p65−/−Ras cells at indicated CTL:Target ratios. After 36–48 hr, cell viability was determined by trypan blue exclusion.
(E) Similar to (D) with the exception that HeLa and 293T cells were used and cocultured with primed CTLs.
In (C)–(E), data are represented as mean ± SD.
NF-κB Promotes Ras Tumor Initiation by Overcoming Adaptive Immunity
Because tumor cells are known to express antigens that are recognized by immune cells to mediate tumor surveillance (Schreiber et al., 2011), we considered the possibility that NF-κB functioned to evade adaptive immunity by circumventing the expression of surface antigens recognized by cytotoxic T lymphocytes (CTLs). To test this prediction, we first irradiated p65+/+Ras and p65−/−Ras cells with X-ray (50 Gy) to suppress their growth, and then syngeneic C57BL/6 mice were immunized by injecting these cells. Two weeks later, splenocytes were isolated and antigen specific CTLs were expanded by coculturing with growth arrested p65+/+Ras or p65−/−Ras cells as immunogen. Results showed that these enriched CTLs exhibited extremely strong killing activity that did not discriminate between p65+/+ and p65−/− status (Figure 5D). This immune response was specific to p65 Ras cells because considerably less T cell cytotoxicity was observed with HeLa or 293T cells, used as controls (Figure 5E). These data indicate that Ras cells expressing p65 remain immunogenic, and thus susceptible to T cell-mediated killing.
To explore what other mechanisms are responsible for allowing p65+/+Ras cells to develop tumors in the presence of an adaptive immunity, we reanalyzed our high-density profiling data and noticed that several known immune suppressor genes were downregulated in p65−/−Ras cells, including transforming growth factor beta (TGF-β), IL-10, GM-CSF, G-CSF, and VEGF. This regulation was validated by qualitative RT-PCR (Figure 6A), suggesting that these genes are regulated by NF-κB in Ras-transformed cells. This was confirmed by first showing that these genes were induced by an NF-κB-activating signal (Figure S6A), and second, by demonstrating that inhibition of NF-κB by stable expression of the IκBα-SR in p65+/+Ras cells led to a reduction in TGF-β, IL-10, GM-CSF, G-CSF, and VEGF (Figures S6B and S6C). TGF-β and IL-10 are considered immune suppressive cytokines that inhibit dendritic cell maturation and CTL function (Vesely et al., 2011), whereas GM-CSF, G-CSF, and VEGF exhibit immunosuppressive activity by stimulating myeloid derived suppressor cells (MDSCs) to diminish CTL-mediated immune surveillance (Vesely et al., 2011). Thus, NF-κB appears capable of coordinating a network of immune suppressor genes whose products are important for tumor cells to evade adaptive immunity.
Figure 6. p65-Regulated TGF-β Is Responsible for Tumor Immune Tolerance.

(A) Validation of immune suppressive genes from microarray analysis from Figure 4E was performed with real-time RT-PCR. Data are plotted as mean expression ± SD for each gene. All *p < 0.001.
(B) p65+/+Ras cells were injected subcutaneously into C57BL/6 mice and 2 days postinjection mice were administered a monoclonal antibody against TGF-β (α-TGF-β) or rat IgG (IgG) via tail vein once per week. Tumor growth was measured at indicated time points. Arrowheads indicate the times when anti-TGF-β antibody was injected. Data are representative of mean tumor sizes ± SD from two independent experiments. p < 0.05.
(C) Similar to (B), except that the survival of mice was recorded.
(D and E) p65+/+Ras cells were infected with a lentivirus expressing GFP (V) or shRNA against TGF-β (shTGF). Indicated cell lines with confirmed TGF-β knockdown were injected into C57BL/6 mice, and tumor size was measured compared to vector control lines, graphed separately (D) or as averaged values ±SD (E, shVector: p = 0.22; shTGF-β: p = 0.025).
We tested the contribution of one of these immune suppressor genes, TGF-β, by systemically treating mice with injected p65+/+Ras cells with a monoclonal antibody for this factor. Neutralization of TGF-β had little effect on the initiation of tumors during the first 10 days of treatment, which supports our earlier conclusion that adaptive immune cells are not fully active during this early phase of tumor development (Figure 6B). However, shortly afterward, tumor growth was significantly delayed, and survival considerably improved, in mice treated with anti-TGF-β compared to immunoglobulin (Ig) G control (Figures 6B and 6C). Next, we tested the direct contribution of TGF-β from p65+/+Ras cells by generating cell lines stably expressing a TGF-β-silencing shRNA. Two clones (p65+/+Ras-shTGF7 and p65+/+Ras-shTGF10) that contained efficient knockdown of TGF-β mRNA and protein (Figures S6D and S6E) were further examined for their effects on tumor initiation. Results showed that tumor development was delayed from both injected p65+/+Ras-shTGF7 and p65+/+Ras-shTGF10 clones compared to vector control lines, p65+/+Ras-V2 and p65+/+Ras-V4 (Figures 6D and 6E as averaged values). Together, these data support the notion that TGF-β represents at least one, among multiple, NF-κB-regulated genes that contributes to the development of Ras tumors.
NF-κB Activation in Transformed Cells Shows Enhanced MDSC Mobilization
MDSCs and T regulatory cells (Treg) represent two major cell types in the immunosuppressive network (Schreiber et al., 2011; Vesely et al., 2011). To investigate if the increase in GM-CSF, G-CSF, and VEGF in p65+/+Ras cells induce the production of immunosuppressive cells, splenocytes were isolated from C57BL/6 mice injected with p65+/+Ras and p65−/−Ras cells and subsequently stained with appropriate antibodies against markers of MDSC and Tregs. Results showed that as early as 9 days postinjection, tumor growth from p65+/+Ras cells was accompanied with a significant increase in MDSCs (CD11b+; Gr-1+) compared to injected saline control or p65−/−Ras cells (p < 0.005) (Figures 7A and 7B). In contrast, no differences in the number of Tregs (CD4+; CD25+; Foxp3+) were observed at this stage of tumor development (Figures S7A–S7C). To further examine the regulation of MDSCs by NF-κB, bone marrow (BM) cells were isolated from C57BL/6 mice and cocultured with p65+/+Ras or p65−/−Ras cells. After 6–7 days, a significantly higher number of MDSCs were observed from BM cells cocultured with p65+/+Ras compare to p65−/−Ras cells (Figures 7C and 7D). Similarly, MDSCs were also expanded from BM cells in the presence of conditioned media from p65+/+Ras (Figures 7E and S7D), but this expansion was reduced when conditioned media was incubated with antibodies against GM-CSF, G-CSF, or VEGF (Figure S7E). These data suggest that NF-κB regulates the production of MDSCs through a combination of target genes, GM-CSF, G-CSF, and VEGF. Because MDSCs participate in an immunosuppressive network by mitigating a T cell response, we next asked if the regulation of MDSCs by NF-κB-affected T cells. Therefore, T cells were isolated and stimulated by CD3/CD28 antibodies, and proliferation of CD4+ and CD8+ T cells was monitored in cocultures with MDSCs obtained from mice injected with p65+/+Ras cells. Compared to control conditions, cultures containing MDSCs caused a significant growth defect in both CD4+ and CD8+ T cells (Figures 7F and 7G), suggesting that MDSCs derived from p65+/+Ras-injected mice are capable of inhibiting T cell activation. To further evaluate this regulation in vivo, p65−/−RasT cells were injected subcutaneously into C57BL/6 mice (cohort 1). Two days postinjection, MDSCs, isolated from a separate cohort of mice preinoculated with p65+/+Ras cells for 14–16 days (Figure S7F), were administered intravenously once per week into cohort 1, and tumor initiation was subsequently monitored. Compared to mice without MDSC administration, where tumors completely regressed, mice injected with p65−/−RasT cells receiving weekly doses of MDSCs showed significantly higher tumor growth that was sustained even after 40 days of observation (Figure 7H). Such results support the conclusion that NF-κB-mediated transcription regulates MDSC production to promote tumor initiation by evading the surveillance of adaptive immune cells.
Figure 7. p65-Regulated Genes Are Responsible for Increased MDSC Mobilization.

(A) FACS results of splenic cells from C57BL/6 mice 7–10 days after injecting PBS, p65+/+Ras or p65−/−Ras cells. MDSCs are identified as CD11b/Gr-1 double-positive cells.
(B) From data in (A), percentages of MDSCs in mouse spleens were graphed as mean ± SD. Data are representative of three independent experiments with at least three mice injected (*, **p < 0.005).
(C) Bone marrow cells were cocultured with either p65+/+Ras or p65−/−Ras cells. After 6 days, cells were harvested and stained for CD11b and Gr-1 the same as describe in (A).
(D) From data in (C), the percentage of MDSCs was calculated. Data are representative of three independent experiments (mean ± SD). *p < 0.0001.
(E) Bone marrow cells were either incubated with conditioned media from p65+/+Ras cells (p65+/+Ras/CM) or p65−/−Ras cells (p65−/−Ras/CM), or incubated with 10 ng/ml GM-CSF as control for 6 days. The same as in (C), but cells were harvested and stained for MDSCs. Data are represented as mean ± SD from at least two independent experiments. *p < 0.001.
(F) CFSE-stained T cells isolated from C57BL/6 spleens were activated by culturing with anti-CD3/CD28 magnetic beads in the presence or absence of MDSCs isolated from p65+/+Ras-injected mice. Two to 3 days later, cultured cells were harvested and stained with anti-mouse CD8 and CD4 and analyzed by FACS.
(G) Percentages of CD8+ or CD4+ cells with lower fluorescence intensity than T cells cultured without CD3/CD28 beads were calculated as percentage of cell growth. * and **p < 0.0001. Data are represented as mean percentage of T cell growth ±SD.
(H) p65−/−RasT cells were injected subcutaneously into two groups of C57BL/6 mice. Two days after this injection, MDSCs were isolated from spleens of p65+/+Ras-injected mice (days 14–16). PBS (C) or isolated MDSCs (MDSC) were subsequently injected into p65−/−RasT cell-injected mice via tail veins (arrow heads). At indicated time points, tumor sizes were measured with a digital caliber.
DISCUSSION
Results in this study highlight several key points about how NF-κB functions in the initial stages of tumor development. One is that p65 is capable of switching from a tumor suppressor to a tumor promoter based on the staging of tumorigenesis. Previous studies have postulated that NF-κB can possess tumor suppressor activity (Chaturvedi et al., 2011; Perkins, 2004), but such function has not been shown to change over time based on the genetic background of the cell. In contrast, our results suggest that this regulation can occur in a cell autologous manner. Although Ras was used to drive oncogenesis in this study, the fact that NF-κB is activated by a variety of oncogenic signaling pathways (Chaturvedi et al., 2011; Mayo et al., 1997; Reuther et al., 1998; Staudt, 2010) suggests that the decision for NF-κB to switch from a tumor suppressor to a tumor promoter is likely to be regulated by more than Ras.
A significant feature of cancer development involves the interplay between tumor cells and surrounding stroma (Mueller and Fusenig, 2004). MΦs are immune cells that play multifaceted roles in the initiation and progression, as well as the immune tolerance of tumor cells (Allavena et al., 2008; Qian and Pollard, 2010). Previous studies indicated that NF-κB maintains an intrinsic cell survival property, which is important for Ras-mediated oncogenesis (Finco et al., 1997; Hanson et al., 2004; Mayo et al., 1997). In addition to these reports, our current findings show that NF-κB is required to protect transformed cells in their initial stage of tumor growth from MΦ-induced apoptosis, mediated through secretion of TNF and NO. Such results signify a key role that NF-κB plays between cancer cells and an inflammatory microenvironment.
Our results also showed that the link between NF-κB and tumorigenesis extends to adaptive immunity. Whereas transplantation of p65−/−Ras and p65−/−RasT cells into immune compromised mice develop tumors, these same lines, no matter how genetically unstable, were unable to form tumors when transplanted into mice with a fully competent immune system. This is in dramatic contrast to p65+/+Ras and p65+/+RasT where tumors readily formed in mice irrelevant of lymphocyte status. Cancer cells have long been considered to be immunogenic, and their survival is continuously threatened by immune surveillance (Pardoll, 2003; Schreiber et al., 2011), a point that was recently reaffirmed in a K-Ras mouse model of sarcoma, as well as through the identification of the tumor antigen, spectrin-β2 (DuPage et al., 2012; Matsushita et al., 2012). Studies also indicate that NF-κB activation promotes antigen presentation of advanced Lewis lung carcinoma cells (Hopewell et al., 2013). However, in our model, NF-κB did not compromise antigen expression because both p65+/+Ras and p65−/−Ras cells were equally sensitive to CTL-mediated killing. Rather, our results support that Ras requires NF-κB to mediate an immune-tolerant environment through the expression of multiple immunosuppression genes that separately or together counteract surveillance properties of adaptive immune cells by increasing the population of MDSCs. Thus, in addition to the previously well-described antiapoptotic activity of NF-κB in response to Ras (Hanson et al., 2004; Mayo et al., 1997), we propose that an added vital function of NF-κB in cancer is to provide developing tumors the ability to circumvent elimination from immune surveillance. Because these factors are regulated by NF-κB, and TNF is a potent activator of NF-κB, it is also possible that TNF secreted from infiltrating MΦs could play some role regulating the immunoevasive property of NF-κB against adaptive immunity. Importantly, our findings are distinct from the interplay previously described in prostate cancer, where in that scenario NF-κB was activated in epithelial cells by B lymphocytes to stimulate cell proliferation and maintain stem cell renewal necessary for tumor recurrence (Ammirante et al., 2010). Recently, investigators demonstrated that overexpression of IKKβ in lung epithelium promotes the accumulation of Tregs (Zaynagetdinov et al., 2012). Such findings, in combination with our current results, highlight the multiple signaling interactions that occur between tumor and surrounding immune cells.
Even after decades of intensive efforts, progress to develop effective tumor immunotherapies has been hampered by the ability of cancer cells to develop diverse strategies to overcome elimination by innate and adaptive immune cells (Alpizar et al., 2011; Zou, 2005). Our results reveal that NF-κB appears to play a pivotal role in tumor immune tolerance, because transformed cells lacking NF-κB are completely incapable of forming tumors in immune competent mice. Although certain factors have been identified as mediators of immune tolerance, the underlying mechanisms of immune surveillance evasion remains unclear. We believe our findings are significant for they point to NF-κB as a possible Achilles’ heel of cancer cells. If this is the case, current immunotherapy in combination with NF-κB inhibition may be considered an effective strategy to treat cancer.
EXPERIMENTAL PROCEDURES
Cell Survival Assays
For cell painting analysis, 106 Ras cells were washed and stained in PBS 0.05% BSA with CMRA (orange, for p65+/+Ras cells) or CFSE (green, for p65−/−Ras cells) fluorescence dyes according to the manufacturer (Invitrogen). Stained cells were then mixed in a 1:1 ratio and applied to 35 mm dishes containing coverslips. Two hours later, isolated MΦs were applied and incubated at 37°C with 5% CO2 for an additional 16–20 hr. Cells were fixed with 4% paraformaldehyde in PBS and mounted onto slides with DAPI-containing mounting solution (Electron Microscopy Sciences). Mounted cells were then observed with a fluorescence microscope and red to green cell ratios were calculated. For apoptosis analysis, cells were coincubated with harvested MΦs overnight. The next day, cells were harvested by trypsinization and stained with FITC-rat anti-mouse CD11b (BioLegend). Then cells were washed with Annexin V staining buffer and stained for Annexin V and 7-AAD with an phycoerythrin (PE) conjugate Annexin V staining kit (BD Pharmingen) as recommended by the manufacturer and analyzed by FACS. For CTL assays, p65+/+Ras or p65−/−Ras cell-specific CTLs were prepared (Supplemental Experimental Procedures) and cocultured with tumor cells in 96 well plates with 5–10 units/ml of IL-2. Thirty-six to 48 hours later, plates were trypsinized and viable cells were counted by a trypan blue exclusion assay.
T Cell Activation Analysis
T cells were isolated from spleens of C57BL/6 mice using a murine T cell negative isolation kit (Invitrogen). Isolated T cells were then stained with 5 μM of CFSE and subsequently activated by coculturing with CD3/CD28 magnetic beads (Invitrogen) with or without MDSCs. Forty-eight hours later, cells were harvested and fixed for 30 min on ice in 2% paraformaldehyde/PBS. Fixed cells were then stained with PE-conjugated rat anti-CD8 and APC/Cy7-conjugated rat anti-mouse CD4 monoclonal antibodies, which were then analyzed by FACS.
Macrophage Depletion, TGF-β Neutralization, and MDSC Treatment
Treatments were carried out by intravenous injections via tail vein with at a dose of 100 μl/10 g body weight of mice. For MΦ depletion, clodronate/liposome or PBS/liposome was vortexed vigorously before injection. Rat-anti-TGF-β (BioLegend) was diluted with PBS at 20–50 μg/200 μl before injection. For MDSC isolation, p65+/+Ras cells were injected subcutaneously into C57BL/6 mice. Ten to sixteen days after injections, MDSCs were separated from spleen and surrounding lymph nodes with an MDSC isolation kit (Miltenyi Biotec). MDSCs purity was confirmed by staining a small portion of isolated cells with fluorescent antibodies against CD11b and Gr-1, and verifying by FACS (Figure S7F). At the arranged time point, p65−/−RasT cells were also injected subcutaneously into C57BL/6 mice. Two days after tumor cell inoculation, isolated MDSCs were resuspended in PBS at 5–8 × 106 cells/200 μl and subsequently injected intravenously in mice once per week.
Statistical Analysis
For tumor growth analysis, an analysis of variance (ANOVA) approach was adopted to analyze differences between each group. Student’s t tests were used for all other analysis.
Supplementary Material
Highlights.
NF-κB is required for tumor initiation
Ras utilizes NF-κB to protect against macrophage-mediated killing
NF-κB is also responsible for tumor cells to evade adaptive immunity
NF-κB regulates TGF-β and MDSCs to protect tumor cells against immunosurveillance
Acknowledgments
We are grateful for A. Beg for generously providing p65 mice, G. Leone for helpful reagents, and members of Guttridge laboratory for engaging discussions throughout the course of this manuscript. Support for this work was provided by the Solid Tumor Biology Program at the Ohio State University Comprehensive Cancer Center and NIH funding P50CA140158 to J.C.B.
Footnotes
ACCESSION NUMBER
Microarray data were submitted to NCBI Gene Expression Omnibus with an accession number of GSE59545.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, one table, and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2014.08.049.
AUTHOR CONTRIBUTIONS
D.J.W. and D.C.G. designed and performed the experiments with assistance from N.M.R. J.C.B. provided expertise in immunology, and D.C.G. directed the overall study.
References
- Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–161. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- Alpizar YA, Chain B, Collins MK, Greenwood J, Katz D, Stauss HJ, Mitchison NA. Ten years of progress in vaccination against cancer: the need to counteract cancer evasion by dual targeting in future therapies. Cancer Immunol Immunother. 2011;60:1127–1135. doi: 10.1007/s00262-011-0985-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010;464:302–305. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H, Bartos DP, Owen-Schaub LB. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50-p65 recruitment. Mol Cell Biol. 1999;19:2098–2108. doi: 10.1128/mcb.19.3.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-κB addiction and its role in cancer: ‘one size does not fit all’. Oncogene. 2011;30:1615–1630. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finco TS, Westwick JK, Norris JL, Beg AA, Der CJ, Baldwin AS., Jr Oncogenic Ha-Ras-induced signaling activates NF-kappaB transcriptional activity, which is required for cellular transformation. J Biol Chem. 1997;272:24113–24116. doi: 10.1074/jbc.272.39.24113. [DOI] [PubMed] [Google Scholar]
- Gapuzan ME, Schmah O, Pollock AD, Hoffmann A, Gilmore TD. Immortalized fibroblasts from NF-kappaB RelA knockout mice show phenotypic heterogeneity and maintain increased sensitivity to tumor necrosis factor alpha after transformation by v-Ras. Oncogene. 2005;24:6574–6583. doi: 10.1038/sj.onc.1208809. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hanson JL, Hawke NA, Kashatus D, Baldwin AS. The nuclear factor kappaB subunits RelA/p65 and c-Rel potentiate but are not required for Ras-induced cellular transformation. Cancer Res. 2004;64:7248–7255. doi: 10.1158/0008-5472.CAN-03-3898. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hopewell EL, Zhao W, Fulp WJ, Bronk CC, Lopez AS, Massengill M, Antonia S, Celis E, Haura EB, Enkemann SA, et al. Lung tumor NF-κB signaling promotes T cell-mediated immune surveillance. J Clin Invest. 2013;123:2509–2522. doi: 10.1172/JCI67250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKK-gamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Wang CY, Cogswell PC, Rogers-Graham KS, Lowe SW, Der CJ, Baldwin AS., Jr Requirement of NF-kappaB activation to suppress p53-independent apoptosis induced by oncogenic Ras. Science. 1997;278:1812–1815. doi: 10.1126/science.278.5344.1812. [DOI] [PubMed] [Google Scholar]
- Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- Noon AT, Shibata A, Rief N, Löbrich M, Stewart GS, Jeggo PA, Goodarzi AA. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol. 2010;12:177–184. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–839. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 2004;14:64–69. doi: 10.1016/j.tcb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuther JY, Reuther GW, Cortez D, Pendergast AM, Baldwin AS., Jr A requirement for NF-kappaB activation in Bcr-Abl-mediated transformation. Genes Dev. 1998;12:968–981. doi: 10.1101/gad.12.7.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovillain E, Mansfield L, Caetano C, Alvarez-Fernandez M, Caballero OL, Medema RH, Hummerich H, Jat PS. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene. 2011;30:2356–2366. doi: 10.1038/onc.2010.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Seitz CS, Lin Q, Deng H, Khavari PA. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc Natl Acad Sci USA. 1998;95:2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked H, Hofseth LJ, Chumanevich A, Chumanevich AA, Wang J, Wang Y, Taniguchi K, Guma M, Shenouda S, Clevers H, et al. Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc Natl Acad Sci USA. 2012;109:14007–14012. doi: 10.1073/pnas.1211509109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalka AM, Katz RA. Retroviral DNA integration and the DNA damage response. Cell Death Differ. 2005;12(Suppl 1):971–978. doi: 10.1038/sj.cdd.4401573. [DOI] [PubMed] [Google Scholar]
- Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100:2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]
- Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- Wang J, Jacob NK, Ladner KJ, Beg A, Perko JD, Tanner SM, Liyanarachchi S, Fishel R, Guttridge DC. RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep. 2009;10:1272–1278. doi: 10.1038/embor.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaynagetdinov R, Stathopoulos GT, Sherrill TP, Cheng DS, McLoed AG, Ausborn JA, Polosukhin VV, Connelly L, Zhou W, Fingleton B, et al. Epithelial nuclear factor-κB signaling promotes lung carcinogenesis via recruitment of regulatory T lymphocytes. Oncogene. 2012;31:3164–3176. doi: 10.1038/onc.2011.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.