

Abstract
Permeability of the mitochondrial outer membrane is determined by the activity of voltage-dependent anion channels (VDAC) which are regulated by many factors and proteins. One of the main partner-regulator of VDAC is the 18 kDa translocator protein (TSPO), whose role in the regulation of membrane permeability is not completely understood. We show that TSPO ligands, 1 μM PPIX and PK11195 at concentrations of 50 μM, accelerate opening of permeability transition pores (mPTP) in Ca2+-overloaded rat brain mitochondria (RBM). By contrast, PK11195 at 100 nM and anti-TSPO antibodies suppressed pore opening. Participation of VDAC in these processes was demonstrated by blocking VDAC with G3139, an 18-mer phosphorothioate oligonucleotides, which sensitized mitochondria to Ca2+-induced mPTP opening. Despite the inhibitory effect of 100 nM PK11195 and anti-TSPO antibodies alone, their combination with G3139 considerably stimulated the mPTP opening. Thus, 100 nM PK11195 and anti-TSPO antibody can modify permeability of the VDAC channel and mPTP. When VDAC channels are closed and TSPO is blocked, permeability of the VDAC for calcium seems to be the highest, which leads to accelerated pore opening.
Keywords: VDAC, TSPO, Mitochondria, Permeability transition pore, PK11195, G3139
1. Introduction
Normal cell function is ensured by the coordinated exchange of metabolites between cells and the environment as well as between intracellular compartments. Mitochondria make a considerable contribution to cell metabolism regulation: they not only synthesize adenosine triphosphate (ATP) for cells, but they also participate in a number of vital processes such as Ca2+ signaling, cell cycle, differentiation, and programmed cell death [1,2].
Mitochondrial metabolism requires a continuous exchange of substrates between the cytoplasm and the mitochondrial matrix. For normal exchange, these metabolites must pass two (the outer and inner) mitochondrial membranes. Transport across the inner impermeable membrane for water-soluble metabolites is carried out by numerous specific protein transporters [3–5]. Water soluble metabolites penetrate through the outer membrane into the intermembrane space via voltage-dependent anion channels (VDAC), which plays a significant role in maintenance of the normal exchange of metabolites between the cytoplasm and the mitochondria [6–9]. Being major protein of the outer mitochondrial membrane, VDAC determines permeability of these membranes [6,7]. VDAC channels are usually opened, and the size of their pores is sufficient for the passage of all hydrophilic metabolites involved in oxidative phosphorylation, such as pyruvate, oxaloacetate, malate, succinate, ATP, ADP, and inorganic phosphate. In its closed state, VDAC becomes cation-selective, and its permeability actually increases for small cations, while the channel becomes essentially impermeable to large anions such as ATP [6,10].
VDAC closely interacts with other proteins (hexokinase, tubulin, adenine nucleotide translocase (ANT), and translocator protein (TSPO)), which in turn influence VDAC function. Binding of VDAC with hexokinase or tubulin results in closure (decreased conductance) of VDAC channels [11,12]. The interaction of VDAC with ANT and TSPO (previously known as the peripheral benzodiazepine receptor) results in the formation of a complex. Tight interaction between TSPO and VDAC occurs during VDAC purification; VDAC preparations are not free from TSPO [13]. TSPO is widely expressed in many tissues, and TSPO is of great physiological significance because of its involvement in a number of biological functions including steroid and neurosteroid synthesis, porphyrin transport in heme biosynthesis, cellular proliferation, and apoptosis [14,15]. As part of a complex with ANT and VDAC, which regulate the permeability of the outer and inner membranes, TSPO may be intimately involved in the regulation of mitochondrial membranes permeability, as well as in the initiation and regulation of apoptosis in various cells [16,17].
TSPO is the only established receptor in mitochondria. TSPO is a highly hydrophobic tryptophan rich protein of consisting of 169 amino acids. Topographic studies reveal that TSPO may form clusters of 4–6 18 kDa monomers associated with one VDAC subunit [18].
Different endogenous and synthetic TSPO ligands have been identified. Among the synthetic TSPO ligands, two families have been mainly characterized: benzodiazepines and isoquinoline carboxamides [15]. Benzodiazepine shows a large variability in TSPO affinity and specificity, with Ro5-4864 (7chloro-1,3dihydro-1-methyl-5-(p-chlorophenyl)-2H-1,4-benzodiazepine) having the highest affinity (nanomolar range) [19]. The isoquinoline carboxamides, which differ structurally from benzodiazepines, also have a high affinity and selectivity for TSPO. The most widely used member of this family is 1-(2 chlorophenyl)-N-methyl-(1-methypropyl)-3-isoquinoline carboxamide (PK11195). PK11195 has an affinity in the nanomolar range. TSPO was also named as an isoquinoline carboxamide-binding protein due to its high specificity for TSPO. Endogenous ligands for TSPO include cholesterol and the porphyrins (protoporphyrin IX, mesoporphyrin IX, deuteroporphyrin IX, hemin) [20,21]. Both cholesterol and porphyrins exhibit a very high (nanomolar) affinity for TSPO. Cholesterol binds better to TSPO monomers than to polymers, whereas some ligands (for example, PK11195) exhibit higher binding affinity to TSPO polymers [22]. It should be noted that VDAC binds also cholesterol [23,24].
TSPO ligands at high concentrations (above 100 μM) can modulate oxidative stress and mitochondrial permeability pore (mPTP) opening, which can initiate the mitochondrial pathway to apoptosis [25]. However, it is not clear how permeability of the outer membrane is linked to the permeability transition [26]. We suggested that PK11195 and PPIX, another TSPO ligand, might modify VDAC activity due to the tight interaction between VDAC and TSPO with a following modulation of the inner membrane permeability. Here, this hypothesis was examined in experiments investigating the effect of the combined application of PK11195 and PPIX on the induction of mPTP under conditions when VDAC channels were opened or blocked by G3139. G3139 (Oblimersen Genasense, Bcl-2 antisense), is a synthetic, 18-base, single-stranded phosphorothioate DNA oligonucleotide originally designed to downregulate bcl-2 mRNA expression and is widely used as an inhibitor of mitochondrial VDAC [27].
2. Materials and methods
2.1. Isolation of rat brain mitochondria (RBM)
Rat brains were rapidly removed (within 30 s) and placed in ice-cold solution, containing 0.32 M sucrose, 0.5 mM EDTA, 0.5 mM EGTA, 0.2% bovine serum albumin (BSA) (fraction V), and 10 mM Tris–HCl (pH 7.4). All solutions used were ice-cold, and all manipulations were carried out at +4 °C. The tissue was homogenized in a glass homogenizer with a ratio of brain tissue to isolation medium of 1:10 (w/v). The homogenate was centrifuged at 2000× g for 3 min. The mitochondrial pellet was obtained by centrifugation of the supernatant at 12,500× g for 10 min. At the next step in representative experiments, the mitochondria were purified on a Percoll gradient (10%–15%–24%) by centrifugation at 31,300× g for 10 min. RBM were suspended in ice-cold solution, containing 0.32 M sucrose and 10 mM Tris–HCl (pH 7.4) and they were additionally washed by centrifugation at 11,500× g for 10 min. Protein concentrations in the stock mitochondrial suspensions were 25–30 mg/mL. All animal procedures were approved by the ethics committee of the German federal state of Sachsen-Anhalt and they were conducted in accordance with the European Communities Council Directive (86/609/EEC).
2.2. Evaluation of mitochondrial functions
The mitochondrial membrane potential was measured as described earlier [28,29] by determining the distribution of tetra-phenylphosphonium ions (TPP+) in the incubation medium with a TPP+-selective electrode, and Ca2+ transport was determined with a Ca2+-sensitive electrode (Nico Analyt, Moscow, Russia) in the 1 mL chamber volume. Mitochondria (2.0 mg protein/mL) were incubated in the medium containing 125 mM KCl, 10 mM Tris–HCl, 0.4 mM KH2PO4, pH 7.4 at 25 °C. Succinate (5 mM potassium succinate) was used as mitochondrial respiratory substrate in the presence of 2 μM rotenone (inhibitor of complex I). In every mitochondrial preparation, threshold calcium concentration was determined before the beginning of the experiment. mPTP opening in RBM was induced by threshold Ca2+ loading by two pulses. All tested drugs were added into the chamber to the mitochondrial suspension before calcium.
G3139 was a generous gift from Dr. Robert Brown (Genta, Inc, Berkeley Heights, NJ, USA). Unless otherwise stated, all chemicals used were obtained from Sigma (St. Louis, MO, USA).
Mitochondrial parameters (Ca2+ influx rate (VCa2+in), lag time before Ca2+ release and Ca2+-capacity) were calculated as described previously [29]. Briefly, Ca2+ influx rate (VCa2+in) revealed the slope of the Ca2+-electrode trace in the direction of decrease in Ca2+ concentration in the incubation medium after the second addition of Ca2+ into mitochondrial suspension; lag time before Ca2+ release was calculated as time period between the loading of the second Ca2+ addition and subsequent Ca2+-release; Ca2+-capacity revealed maximal Ca2+ accumulation by mitochondria before PTP opening and respective Ca2+-release (See [29] for detailed graphical representation).
For statistical analysis, data were expressed as means ± standard deviations (SD) from at least 3–4 independent experiments. Significance was determined using Student’s t test. A value of P < 0.05 was considered to be significant.
3. Results
3.1. Combined effect of 100 nM PK11195 and G3139 on Ca2+-induced mPTP opening in purified RBM
Recently, we showed the presence of the TSPO in both pools of brain mitochondria (synaptic and nonsynaptic) obtained after their purification in Percoll gradient. Among the synthetic TSPO ligands, two families have been primarily characterized: benzodiazepines and isoquinoline carboxamides [15]. In the present study, we used PK11195 which is the most widely used member of isoquinoline carboxamide family. It has high affinity and selectivity for TSPO and recognized as a specific binding drug for TSPO. Earlier we reported that synthetic and natural TSPO ligands are able to modulate the permeability transition in the inner membrane of Ca2+-loaded mitochondria [29–31]. We showed that PK11195 taken at nanomolar concentration is able to prevent mPTP opening in calcium-overloaded mitochondria, while micromolar concentrations of PK11195 cause an acceleration of pore opening. The involvement of TSPO in regulation of mitochondrial membrane permeability remains incompletely understood, therefore we examined whether the combined application of PK11195 with G3139 is able to modify permeability transition in isolated purified nonsynaptic mitochondria.
Fig. 1 shows the combined effect of 100 nM PK11195 and G3139 on the function of RBM. The functional state of mitochondria was determined by simultaneous measurement of ΔΨm and [Ca2+] with TPP+- and Ca2+-selective electrodes. The mitochondria maintained a high ΔΨm level in the presence of succinate and rotenone. As shown in Fig. 1A, the first pulse of Ca2+ (added to RBM) induced a sharp decrease in ΔΨm (dashed line), as measured by the membrane potential indicator TPP+. Then, restoration of ΔΨm followed and at the same time, Ca2+ (solid line) rapidly accumulated into the mitochondrial matrix. However, the second calcium addition caused irreversible decrease in ΔΨm and then Ca2+ efflux from mitochondrial matrix within approximately 3 min after the second pulse of Ca2+, indicating the initiation of pore opening.
Fig. 1.

Combined effect of 100 nM PK11195 and G3139 on Ca2+-induced mPTP opening in purified RBM. Mitochondrial parameters were measured as described in Materials and methods. (A) Control RBM, (B) 100 nM PK11195-treated RBM, (C) 5 μM G3139-treated RBM, (D) 100 nM PK11195 and 5 μM G3139-treated RBM. The arrows indicate where CaCl2 (150 and 220 μM) was added to the mitochondrial suspension.
Addition of 5 μM G3139 alone stimulates induction of mPTP. The pore was also opened after the second calcium addition, but in this case, the calcium retention time (lag time period before pore opening) was diminished from 130 s (in control) to 70–80 s in the presence of G3139 (Fig. 1C). Thus, closing or blocking the VDAC channel may lead to increased Ca2+ flux and the acceleration of mPTP opening. In the presence of 100 nM PK11195 alone (Fig. 1B), suppression of mPTP opening was shown, which supports our earlier data [28]. Calcium retention is increased from 130 s in the control to 200 s in the presence of 100 nM PK11195. By contrast together with G3139, PK11195 displayed the opposite effect: it stimulated mPTP opening, causing calcium release immediately after the second calcium addition (panel D).
3.2. Effect of anti-TSPO antibody on G3139-induced activation of mPTP opening in purified RBM
Next, we examined whether TSPO itself can be involved in the observed effect of G3139. To assess that we used a specific anti-TSPO antibody generated against a peptide that corresponds to a certain fragment of TSPO, (antibodies against the site VGLTLVPSLGGFMGAYFVR [amino acids 9–27 of the TSPO sequence, placed in the first transmembrane helix and partly in the first loop, faced into cytosol]) and examined the effect of the anti-TSPO antibody on Ca2+-induced mPTP opening.
Fig. 2A demonstrates that in the control probe, the addition of the second calcium pulse leads to initiation of mPTP. In the absence any additions, opening of mPTP was found to be initiated within 150 s after the second addition of Ca2+. The effect of anti-TSPO antibody was checked by two ways: 1) RBM were pre-incubated with TSPO antibody and then G3139 was added; and 2) RBM were first pre-incubated with G3139, and the anti-TSPO antibodies were added subsequently. As shown in Fig. 2C, pre-incubation of RBM with anti-TSPO antibody with following addition of G3139 leads to the acceleration of mPTP opening (threshold calcium concentration decreased, pore opened immediately after the second addition, and lag time close to 50–70 s, which was similar to activation caused by PK11195 and G3139. When RBM were first pre-incubated with G3139 and then the anti-TSPO antibodies were added, the acceleration of mPTP opening was strengthened (Fig. 2D). Thus, the anti-TSPO antibody is likely able to alter the permeability of the VDAC channel and mPTP as well. When the VDAC channel is closed and TSPO is blocked, permeability of the VDAC for calcium seems to be highest, leading to the acceleration of pore opening.
Fig. 2.

Combined effect of anti-TSPO antibody and G3139 on Ca2+-induced mPTP opening in RBM. Mitochondrial parameters were measured as described in Materials and methods. (A) Control RBM, (B) anti-TSPO antibody (1:1500) treated RBM, (C) RBM pre-treated with anti-TSPO antibody and then incubated with 5 μM G3139, (D) RBM pre-treated with 5 μM G3139 and then incubated with anti-TSPO antibody. The arrows indicate where CaCl2 (150 and 220 μM) was added to the mitochondrial suspension.
3.3. Combined effect of 50 μM PK11195 and G3139 on Ca2+-induced mPTP opening in purified RBM
Earlier, we showed that PK11195 in a range of micromolar concentrations stimulates the opening of mPTP [28]. Here, we verified the combined effect of 50 μM PK11195 and G3139 on initiating the permeability transition. Fig. 3A shows that under control conditions, the non-selective mitochondrial pore is opened following the second calcium additions, as described earlier. The addition of 50 μM PK11195 (Fig. 3B) stimulated the opening of mPTP, while pre-incubation of RBM with G3139 (Fig. 3C) with subsequent addition of 50 μM PK11195 did not lead to additional activation of pore opening. Thus, it is probable that VDAC is a target of PK11195 at high concentrations of the drug.
Fig. 3.

Effect of 50 μM PK11195 in the presence of 5 μM G3139 on Ca2+-induced mPTP opening in RBM. Mitochondrial parameters were measured as described in Materials and methods. (A) Control RBM, (B) 50 μM PK11195 treated RBM, (C) 50 μM PK11195 and 5 μM G3139 treated RBM. The arrows indicate where CaCl2 (150 and 220 μM) was added to the mitochondrial suspension.
3.4. Combined effect of PPIX and G3139 on Ca2+-induced mPTP opening in purified RBM
It was recently shown that hemin and PPIX at 5 or 10 μM reduced the VDAC conductance. In VDAC-TSPO-ANT complex isolated from heart mitochondria and incorporated into the lipid bilayer, hemin (2.5–10 μM) incorporation was able to change channel state from open to closed [32]. Therefore, the effect of PPIX on mPTP opening in the presence of VDAC blocker has been examined.
Fig. 4 shows the effects of PPIX and G3139 on Ca2+ accumulation and Ca2+-induced mPTP opening in RBM. The functional state of mitochondria was determined as described above. As shown in Fig. 4, the second addition of Ca2+ caused the initiation of mPTP opening in the control probe and in G3139-or PPIX-treated RBM. The lag time period prior to pore opening lasted for 12–13 min in the control (Fig. 4A), but it was shorter in the presence of G3139 (which was 9–10 min, Fig. 4B) and in the presence of 1 μM PPIX (which was 7 min, Fig. 4C). Used together with G3139, PPIX was able to strengthen the acceleration of PTP opening, lowering lag time to 1 min (Fig. 4D).
Fig. 4.

Effect of 1 μM protoporphyrin IX and G3139 on Ca2+-induced mPTP opening in RBM. Mitochondrial parameters were measured as described in Materials and methods. (A) Control RBM, (B) 1 μM protoporphyrin IX treated RBM, (C) 5 μM G3139 treated RBM, (D) 1 μM protoporphyrin IX and 5 μM G3139 treated RBM. The arrows indicate where CaCl2 (150 and 220 μM) was added to the mitochondrial suspension.
3.5. Combined effect of G3139 with PK11195 and PPIX on calcium capacity in RBM
The combined effect of PK11195 and G3139 was tested on calcium capacity (Fig. 5), using serial calcium pulses added to the mitochondrial suspension to determine threshold calcium concentrations initiating Ca2+ release and opening of mPTP. In the control experiments, a total 7 additions of calcium (60 μM each and 420 μM in total) resulted in Ca2+ efflux (Fig. 5, trace a), whereas in the presence of G3139 6 additions of calcium (360 μM Ca2+) led to mPTP opening (Fig. 5, trace b). Only 5 calcium additions (300 μM Ca2+) caused pore opening in the presence of PPIX plus G3139 (Fig. 5, trace c), and 4 calcium additions (240 μM Ca2+) were enough to cause Ca2+ release in the presence of both 100 nM PK11195 and G3139 (Fig. 5, trace d).
Fig. 5.

Ca2+-capacity of RBM in the presence of TSPO ligands and G3139. Ca2+-capacity was measured with Ca2+-sensitive electrode as described in Materials and methods in control RBM (trace a) and in RBM incubated in the presence of 5 μM G3139 alone (trace b) and in its combination with 1 μM protoporphyrin IX (trace c) and 100 nM PK11195 (trace d). The arrows indicate where CaCl2 (60 μM) was added to the mitochondrial suspension.
The comparative summary data on the effect of PK11195, PPIX and G3139 on the parameters of mPTP opening – such as the Ca2+ uptake rate, lag time period and calcium capacity – are demonstrated in Fig. 6, where the characteristics of mPTP opening in control were taken as 100%. The diagram shows that the presence of G3139 alone causes a decrease in all parameters tested, which is evidence of stimulation of mPTP opening. Nanomolar concentrations of PK11195 retained calcium accumulation ability, increased threshold [Ca2+] and lag time, and lead to prevention of mPTP opening. On the contrary, PK11195 in combination with G3139 lost its inhibitory effect, decreasing calcium accumulation and retention, as well as amount of calcium required for mPTP induction. PPIX also decreased threshold [Ca2+] and lag time, causing an induction of mPTP. Taken together, PPIX and G3139 are able to strengthen one another’s effects, decreasing threshold [Ca2+] and retention of calcium in the matrix. However, the strongest effect was established for 100 nM PK11195 combined with G3139. The mechanism of action underlying mPTP induction remains unclear; however, the data allowed us to suggest that an interaction between TSPO and VDAC might considerably strengthen the action of TSPO ligands.
Fig. 6.
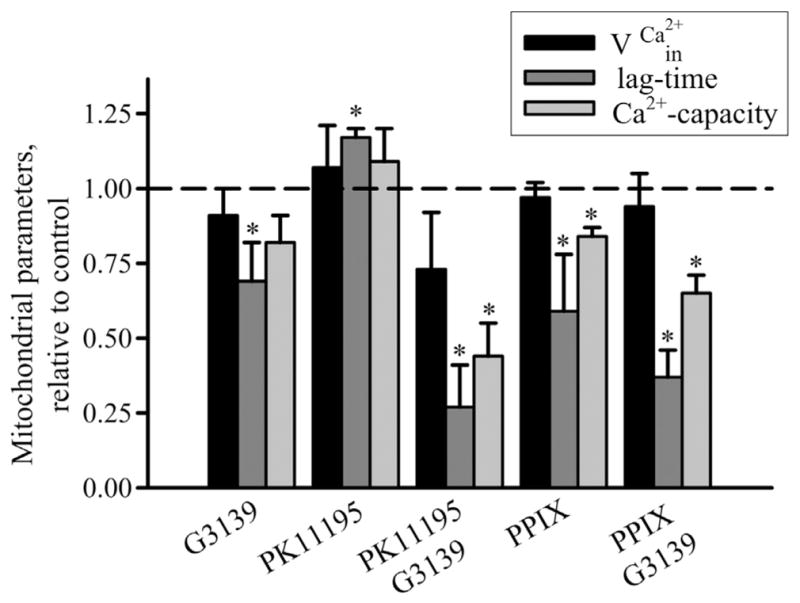
Mitochondrial parameters in the presence of 100 nM PK11195, 1 μM PPIX, 5 μM G3139 alone and in combination. Mitochondrial parameters (rate of Ca2+-influx (vCa2+in), lag time before Ca2+ release and Ca2+-capacity) were calculated as described in [29]. Column 1: mitochondrial parameters in the presence of 5 μM G3139, column 2: in the presence of 100 nM PK11195, column 3: in the presence of 5 μM G3139 and 100 nM PK11195, column 4: in the presence of 1 μM protoporphyrin IX and column 5: in the presence of 5 μM G3139 and 1 μM protoporphyrin IX. All parameters of RBM under experimental conditions were normalized to those of control RBM without any additions. Values represent means ± standard deviation from four independent experiments. *P < 0.05 versus control, as determined by Student’s t-test.
4. Discussion
The main properties of VDAC are associated with the permeability of the outer membrane in mitochondria. Structural studies reveal VDAC as a beta barrel composed of 19 β-strands [33–35]. A single 30–32 kDa polypeptide forms the channel [36]. In the open state, VDAC is anion selective and permeable to multivalent anionic metabolites [37,38]. In the closed state, the pore size diminishes, and the channel comes cation selective [39]. There is evidence suggesting that VDAC’s opening/closure is linked to apoptosis. VDAC closure blocks the ingress and egress of ATP from mitochondria [40]. The subsequent changes in cytosolic ATP levels could act as a signal. The voltage gating process of VDAC controls not only the flux of metabolites, but it also regulates the flux of calcium ions. Ca2+ flux into mitochondria through VDAC can lead to induction of mPTP opening and subsequent initiation of apoptosis. Although VDAC has some permeability to Ca2+ in its normal open state, after VDAC closure the permeability to Ca2+ can be increased by up to 10 times [41]. Thus, this gating suggests that Ca2+ normally permeates through the cation-selective closed VDAC channels in mitochondria. Therefore, regulation of the opening/closing of VDAC might serve as a control site through which to regulate the opening of mPTP and to initiate apoptosis. The permeability of VDAC is determined not only by membrane voltage, but also its conductance state determined, in part, by associated regulatory proteins such as G-actin [42], Bcl-XL [43], hexokinase and tubulin [11,12]. Additionally, TSPO, which resides in the outer mitochondrial membrane, is also able to interact tightly with VDAC, modulating VDAC conductance and mitochondrial function under a variety of physiologic and pathophysiologic conditions [25], including apoptosis [16,17] and stress adaptation [44]. It has been proposed that the major function of TSPO is to transport small molecules, including cholesterol and intermediates of heme biosynthesis, into or out of the mitochondria for different metabolic pathways [14,15,45]. In addition, TSPO has been found to be strongly expressed in areas of brain injury and inflammation [46], in aggressive cancers [25,47] as well as in the brains of Alzheimer and Huntington disease patients.
Earlier pharmacological and biochemical studies together with recent structural data support TSPO’s ability to bind numerous classes of drugs and endogenous ligands, including cholesterol and porphyrins [15,20,48–51]. While the pharmacology of TSPO is broadly accepted, the genetics of TSPO is more controversial. TSPO is an ancient protein highly conserved in all domains of life [52], suggesting an essential role.
TSPO ligands have potential diagnostic and therapeutic applications [16,25]. The best-characterized ligand of TSPO is PK11195, which binds to TSPO with nanomolar affinity in many species [13,53,54]. PK11195 is used as a biomarker in positron emission tomography to visualize brain inflammation in patients with neuronal damage [46,55,56].
The initiation of apoptosis is usually preceded by the loss of the mitochondrial membrane potential (ΔΨm), high-amplitude swelling of the mitochondria, and apoptotic factor release, which are found during the permeability transition pore opening. Until now, the exact composition of the pore complex has not been established, but it is clear that increased permeability of the inner membrane also depends on the permeability of the outer mitochondrial membrane.
Recently, we showed that TSPO synthetic ligands such as PK11195 and Ro5-486, as well as endogenous ligand PPIX were able to modulate the permeability of the inner membrane, mPTP [29]. The involvement of TSPO in the regulation of the inner mitochondrial membrane permeability is questionable and poorly understood. Therefore, we examined whether combined application of PK11195 with G3139 is able to modify the permeability transition in isolated purified nonsynaptic mitochondria, since G3139 (a phosphorothioate oligonucleotides) is the most effective specific inhibitor of VDAC yet identified. Normal VDAC closure results in partial conductance loss, with remaining VDAC conductance to small electrolytes at a level 40–60% of that found in the open state. G3139 induces rapid gating with long-lived closing events. In the presence of G3139, the extent of closure is much greater and represents a complete loss of VDAC conductance. G3139 acts directly on VDAC, preferentially from one side of the channel, showing one-to-one binding of G3139 to VDAC [27]. The functional state of mitochondria was determined via the simultaneous measurement of ΔΨm and Ca2+ fluxes with TPP+- and Ca2+-selective electrodes. Our experiments demonstrate that PK11195 at nanomolar concentration suppressed the initiation of mPTP in Ca2+-overloaded RBM (Fig. 1), increasing lag time before Ca2+-induced calcium release, while micromolar concentrations of PK11195 accelerated pore opening in Ca2+-overloaded RBM.
The addition of 5 μM G3139 alone stimulated the induction of mPTP in nonsynaptic brain mitochondria, diminishing its calcium retention time (the lag time before pore opening) almost by 2-fold. This is in agreement with finding that G3139 blocks VDAC when the channel is in its open state. Earlier, treatment of isolated rat liver mitochondria with 5 μM G3139 was also shown to cause the acceleration of mPTP opening [57].
Thus, closing or blocking the VDAC channel may lead to increased Ca2+ flux and the acceleration of mPTP opening. Calcium retention time increased from 130 s in control to 200 s in the presence of 100 nM PK11195 (Fig. 1B). Interestingly in spite of the preventive effect of 100 nM PK11195 alone, the combination with G3139, PK11195 led to the opposite effect and stimulated mPTP opening, lowered the threshold calcium concentration and decreased the lag time period almost by 5-fold. It might be important in cancer cells for their growth suppression using low specific concentrations of PK11195 in complex with G3139.
We also checked whether TSPO itself participates in PK11195-dependent effect. For that, anti-TSPO antibody was used which is able to prevent mPTP opening [29]. This effect is specific, because both fatty acid free BSA and an unrelated antibody has no effect on the Ca2+-induced mPTP opening. However, in complex with G3139, the anti-TSPO antibody exhibits pore stimulating effects similar to the combined effect of 100 nM PK11195 and G3139 (Fig. 2). PK11195 was shown to interact mainly on the first half of the loop 1 of TSPO [58] as well as anti TSPO antibody. That allowed us to suggest that loop 1 and transmembrane helix 1 are probably participate in the effect of PK11195 as well as TSPO antibody. It should be noted that the potency of VDAC blockage/closure in isolated mitochondria by G3139 is 50 fold higher than that found in experiments with pure VDAC incorporated into planar membranes, indicating possible existence of a VDAC-associated protein, which is able to increase the affinity of VDAC for phosphorothioate (G3139) [27].
A strong acceleration of mPTP in Ca2+-overloaded RBM occurred with high concentrations (50 μM) of PK11195 (Fig. 3). Earlier, we examined effect of different concentration of PK11195 on mPTP opening and found that 10 nM–1 μM PK11195 suppressed pore opening [31], while stimulation of pore opening was observed at 5–200 μM of PK11195 (unpublished data). The addition of G3139 to the mitochondrial suspension did not cause any alteration when compared with effect of 50 μM PK11195 alone. It should be noted that concentrations of PK11195 in range of 50–100 μM might have a nonspecific effect due to its binding with the membrane lipid bilayer. Indeed, it was recently shown that, the effect of high concentrations of PK11195 might be linked to lipid fluidity changes of the outer membrane following modulation of the function of other proteins [59].
The combined effect of G3139 with another endogenous TSPO ligand PPIX on the induction of mPTP opening was also studied. Porphyrins are tetrapyrrolic pigments formed in the biosynthesis pathway of heme, mitochondrial cytochrome, hemoglobin, and other tetrapyrrol-containing enzymes, such as catalase, peroxidase, superoxide dismutase, and calcineurin. Here, it was shown that PPIX (1 μM) suppressed the Ca2+ capacity by 40–55%, shortened the lag time by 40–60% (Fig. 4B, D and Fig. 6) and significantly stimulated Ca2+ efflux, supporting our previous data that PPIX promotes permeability transition in isolated RBM [29]. Interestingly, PPIX binds with loop 1 also, as PK11195 does, although at different sites [49]. Used together with G3139, PPIX (Fig. 4D) was further able to strengthen the acceleration of mPTP opening. It was recently shown that PPIX is able to reduce VDAC conductance. The VDAC-TSPO-ANT “putative” complex isolated from heart mitochondria and incorporated into lipid bilayers was found to change the channel state from open (in the absence of PPIX) to completely closed (in the presence of 2.5–10 μM PPIX) [32]. Moreover, PPIX was shown to bind specifically to ANT [60,61] and recently we showed that immunoprecipitates of TSPO in rat brain mitochondria contain ANT [29]. We suggest that the effect of PPIX on induction of mPTP is exerted through its binding to the trimeric complex of VDAC-TSPO-ANT.
Thus the addition of TSPO ligands to G3139-treated mitochondria under mPTP opening likely stabilized complex through conformational changes in the transient G3139–VDAC–PPIX/PK11195 complex, leading to rapid VDAC closure, an increase in Ca2+ permeation, and sensitization of the mitochondria to apoptotic signals. Such like events might underlie in the mechanism of complete conductance loss of VDAC.
All these effects indicate that VDAC might be a key point at which various signals are integrated into a single response: channel opening or closure. TSPO ligands and TSPO itself seems to modulate VDAC conductance.
Acknowledgments
This study was supported by grants from the Russian Foundation for Basic Research (13-04-00935 to T.A.), the Russian Federation Government (N14.Z50.0028 to J.L.), Deutsche Forschungs gemeinschaft (DFG grant Re563/22-1 to G.R.) the Canadian Institutes of Health Research (MOP 125983 to V.P.), and a Canada Research Chair in Biochemical Pharmacology (V.P.).
Abbreviations
- ANT
adenine nucleotide translocase
- PK11195
1-(2-Chlorophenyl-N-methylpropyl)-3-isoquinolinecarboxamide
- PPIX
protoporphyrin IX
- mPTP
mitochondrial permeability transition pore
- RBM
rat brain mitochondria
- ROS
reactive oxygen species
- TPP
tetraphenylphosphonium
- TSPO
translocator protein (18 kDa)
- VDAC
voltage-dependent anion channel
- ΔΨm
mitochondrial transmembrane potential
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Contributor Information
T. Azarashvili, Email: tamara.azarashvili@gmail.com.
O. Krestinina, Email: krestinina@rambler.ru.
Yu Baburina, Email: byul@rambler.ru.
I. Odinokova, Email: odinokova@rambler.ru.
D. Grachev, Email: grachew@rambler.ru.
V. Papadopoulos, Email: vassilios.papadopoulos@mcgill.ca.
V. Akatov, Email: akatov.vladimir@gmail.com.
J.J. Lemasters, Email: JJLemasters@musc.edu.
G. Reiser, Email: georg.reiser@med.ovgu.de.
References
- 1.Murgia M, Giorgi C, Pinton P, Rizzuto R. J Mol Cell Cardiol. 2009;46:781–788. doi: 10.1016/j.yjmcc.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh C, Barrow S, Voronina S, Chvanov M, Petersen OH, Tepikin A. Biochim Biophys Acta. 2009;1787:1374–1382. doi: 10.1016/j.bbabio.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Rasola A, Bernardi P. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- 4.Palmieri F, Bisaccia F, Capobianco L, Dolce V, Fiermonte G, Iacobazzi V, Indiveri C, Palmieri L. Biochim Biophys Acta. 1996;1275:127–132. doi: 10.1016/0005-2728(96)00062-x. [DOI] [PubMed] [Google Scholar]
- 5.Rossi CS, Carafoli E, Lehninger AL. Protoplasma. 1967;63:90–94. [PubMed] [Google Scholar]
- 6.Colombini M. Mol Cell Biochem. 2004;256–257:107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- 7.Benz R. Biochim Biophys Acta. 1994;1197:167–196. doi: 10.1016/0304-4157(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 8.Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. Curr Pharm Des. 2006;12:2249–2270. doi: 10.2174/138161206777585111. [DOI] [PubMed] [Google Scholar]
- 9.Lemasters JJ, Holmuhamedov E. Biochim Biophys Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Rostovtseva T, Colombini M. Biophys J. 1997;72:1954–1962. doi: 10.1016/S0006-3495(97)78841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurnev PA, Rostovtseva TK, Bezrukov SM. FEBS Lett. 2011;585:2363–2366. doi: 10.1016/j.febslet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ. Cancer Res. 2010;70:10192–10201. doi: 10.1158/0008-5472.CAN-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Proc Natl Acad Sci U S A. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casellas P, Galiegue S, Basile AS. Neurochem Int. 2002;40:475–486. doi: 10.1016/s0197-0186(01)00118-8. [DOI] [PubMed] [Google Scholar]
- 15.Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, Gavish M. Trends Pharmacol Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Jorda EG, Jimenez A, Verdaguer E, Canudas AM, Folch J, Sureda FX, Camins A, Pallas M. Apoptosis. 2005;10:91–104. doi: 10.1007/s10495-005-6064-9. [DOI] [PubMed] [Google Scholar]
- 17.Veenman L, Shandalov Y, Gavish M, Bioenerg J. Biomembr. 2008;40:199–205. doi: 10.1007/s10863-008-9142-1. [DOI] [PubMed] [Google Scholar]
- 18.Golani I, Weizman A, Leschiner S, Spanier I, Eckstein N, Limor R, Yanai J, Maaser K, Scherubl H, Weisinger G, Gavish M. Biochemistry. 2001;40:10213–10222. doi: 10.1021/bi010431+. [DOI] [PubMed] [Google Scholar]
- 19.Gavish M, Bachman I, Shoukrun R, Katz Y, Veenman L, Weisinger G, Weizman A. Pharmacol Rev. 1999;51:629–650. [PubMed] [Google Scholar]
- 20.Lacapere JJ, Papadopoulos V. Steroids. 2003;68:569–585. doi: 10.1016/s0039-128x(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 21.Snyder SH, Verma A, Trifiletti RR. FASEB J. 1987;1:282–288. doi: 10.1096/fasebj.1.4.2820823. [DOI] [PubMed] [Google Scholar]
- 22.Delavoie F, Li H, Hardwick M, Robert JC, Giatzakis C, Peranzi G, Yao ZX, Maccario J, Lacapere JJ, Papadopoulos V. Biochemistry. 2003;42:4506–4519. doi: 10.1021/bi0267487. [DOI] [PubMed] [Google Scholar]
- 23.Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR. Science. 2012;336:1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiser BP, Salari R, Eckenhoff RG, Brannigan G. J Phys Chem B. 2014;118:9852–9860. doi: 10.1021/jp504516a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Decaudin D, Castedo M, Nemati F, Beurdeley-Thomas A, De PG, Caron A, Pouillart P, Wijdenes J, Rouillard D, Kroemer G, Poupon MF. Cancer Res. 2002;62:1388–1393. [PubMed] [Google Scholar]
- 26.Ricchelli F, Sileikyte J, Bernardi P. Biochim Biophys Acta. 2011;1807:482–490. doi: 10.1016/j.bbabio.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Tan W, Loke YH, Stein CA, Miller P, Colombini M. Biophys J. 2007;93:1184–1191. doi: 10.1529/biophysj.107.105379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azarashvili T, Krestinina O, Yurkov I, Evtodienko Y, Reiser G. J Neurochem. 2005;94:1054–1062. doi: 10.1111/j.1471-4159.2005.03260.x. [DOI] [PubMed] [Google Scholar]
- 29.Azarashvili T, Grachev D, Krestinina O, Evtodienko Y, Yurkov I, Papadopoulos V, Reiser G. Cell Calcium. 2007;42:27–39. doi: 10.1016/j.ceca.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Azarashvili T, Stricker R, Reiser G. Biol Chem. 2010;391:619–629. doi: 10.1515/BC.2010.070. [DOI] [PubMed] [Google Scholar]
- 31.Krestinina OV, Grachev DE, Odinokova IV, Reiser G, Evtodienko YV, Azarashvili TS. Biochem Mosc. 2009;74:421–429. doi: 10.1134/s0006297909040105. [DOI] [PubMed] [Google Scholar]
- 32.Tamse CT, Lu X, Mortel EG, Cabrales E, Feng W, Schaefer S. J Clin Basic Cardiol. 2008;11:24–29. [Google Scholar]
- 33.Song J, Midson C, Blachly-Dyson E, Forte M, Colombini M. J Biol Chem. 1998;273:24406–24413. doi: 10.1074/jbc.273.38.24406. [DOI] [PubMed] [Google Scholar]
- 34.Blachly-Dyson E, Peng S, Colombini M, Forte M. Science. 1990;247:1233–1236. doi: 10.1126/science.1690454. [DOI] [PubMed] [Google Scholar]
- 35.Thomas L, Blachly-Dyson E, Colombini M, Forte M. Proc Natl Acad Sci U S A. 1993;90:5446–5449. doi: 10.1073/pnas.90.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colombini M, Blachly-Dyson E, Forte M. Ion Channels. 1996;4:169–202. doi: 10.1007/978-1-4899-1775-1_5. [DOI] [PubMed] [Google Scholar]
- 37.Lee AC, Xu X, Blachly-Dyson E, Forte M, Colombini M. J Membr Biol. 1998;161:173–181. doi: 10.1007/s002329900324. [DOI] [PubMed] [Google Scholar]
- 38.Xu X, Decker W, Sampson MJ, Craigen WJ, Colombini M. J Membr Biol. 1999;170:89–102. doi: 10.1007/s002329900540. [DOI] [PubMed] [Google Scholar]
- 39.Song J, Midson C, Blachly-Dyson E, Forte M, Colombini M. Biophys J. 1998;74:2926–2944. doi: 10.1016/S0006-3495(98)78000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rostovtseva T, Colombini M. J Biol Chem. 1996;271:28006–28008. doi: 10.1074/jbc.271.45.28006. [DOI] [PubMed] [Google Scholar]
- 41.Tan W, Colombini M. Biochim Biophys Acta. 2007;1768:2510–2515. doi: 10.1016/j.bbamem.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu X, Forbes JG, Colombini M. J Membr Biol. 2001;180:73–81. doi: 10.1007/s002320010060. [DOI] [PubMed] [Google Scholar]
- 43.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. J Biol Chem. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 44.Itzhak Y, Norenberg MD. Brain Res. 1994;660:346–348. doi: 10.1016/0006-8993(94)91311-0. [DOI] [PubMed] [Google Scholar]
- 45.Azuma M, Kabe Y, Kuramori C, Kondo M, Yamaguchi Y, Handa H. PLoS One. 2008;3:e3070. doi: 10.1371/journal.pone.0003070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banati RB, Myers R, Kreutzberg GW. J Neurocytol. 1997;26:77–82. doi: 10.1023/a:1018567510105. [DOI] [PubMed] [Google Scholar]
- 47.Rechichi M, Salvetti A, Chelli B, Costa B, Da PE, Spinetti F, Lena A, Evangelista M, Rainaldi G, Martini C, Gremigni V, Rossi L. Biochim Biophys Acta. 2008;1782:118–125. doi: 10.1016/j.bbadis.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Guo Y, Kalathur RC, Liu Q, Kloss B, Bruni R, Ginter C, Kloppmann E, Rost B, Hendrickson WA. Science. 2015;347:551–555. doi: 10.1126/science.aaa1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaremko L, Jaremko M, Giller K, Becker S, Zweckstetter M. Science. 2014;343:1363–1366. doi: 10.1126/science.1248725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li F, Liu J, Zheng Y, Garavito RM, Ferguson-Miller S. Science. 2015;347:555–558. doi: 10.1126/science.1260590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Nat Rev Drug Discov. 2010;9:971–988. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- 52.Fan J, Lindemann P, Feuilloley MG, Papadopoulos V. Curr Mol Med. 2012;12:369–386. doi: 10.2174/1566524011207040369. [DOI] [PubMed] [Google Scholar]
- 53.Guo Q, Owen DR, Rabiner EA, Turkheimer FE, Gunn RN. Neuroimage. 2012;60:902–910. doi: 10.1016/j.neuroimage.2011.12.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirsch T, Decaudin D, Susin SA, Marchetti P, Larochette N, Resche-Rigon M, Kroemer G. Exp Cell Res. 1998;241:426–434. doi: 10.1006/excr.1998.4084. [DOI] [PubMed] [Google Scholar]
- 55.Owen DR, Matthews PM. Int Rev Neurobiol. 2011;101:19–39. doi: 10.1016/B978-0-12-387718-5.00002-X. [DOI] [PubMed] [Google Scholar]
- 56.Rao VL, Bowen KK, Rao AM, Dempsey RJ. J Neurosci Res. 2001;64:493–500. doi: 10.1002/jnr.1101. [DOI] [PubMed] [Google Scholar]
- 57.Tikunov A, Johnson CB, Pediaditakis P, Markevich N, Macdonald JM, Lemasters JJ, Holmuhamedov E. Arch Biochem Biophys. 2010;495:174–181. doi: 10.1016/j.abb.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li F, Xia Y, Meiler J, Ferguson-Miller S. Biochemistry. 2013;52:5884–5899. doi: 10.1021/bi400431t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hatty CR, Le Brun AP, Lake V, Clifton LA, Liu GJ, James M, Banati RB. Biochim Biophys Acta. 2014;1838:1019–1030. doi: 10.1016/j.bbamem.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 60.Taketani S, Kohno H, Furukawa T, Tokunaga R. J Biochem. 1995;117:875–880. doi: 10.1093/oxfordjournals.jbchem.a124790. [DOI] [PubMed] [Google Scholar]
- 61.Wendler G, Lindemann P, Lacapere JJ, Papadopoulos V. Biochem Biophys Res Commun. 2003;311:847–852. doi: 10.1016/j.bbrc.2003.10.070. [DOI] [PubMed] [Google Scholar]