

Abstract
Bridged bicyclic metallacyclopentenes generated from the [4+2] cycloaddition of metallacyclopentadienes with alkenes have been proposed as reactive intermediates in the course of [2+2+2] annulation reactions. Recently a collection of alkoxide-directed Ti-mediated [2+2+2] annulation reactions have been discovered for the synthesis of densely functionalized hydrindanes, where the bridged bicyclic metallacyclopentenes from intramolecular [4+2] were treated as fleeting intermediates en route to cyclohexadiene products by formal cheletropic extrusion of Ti(Oi-Pr)2. In studies aimed at understanding the course of these organometallic cascade reactions it was later discovered that these bridged bicyclic intermediates can be trapped by various elimination processes. Here, we have realized metallacycle-mediated annulation reactions for the assembly of angularly substituted decalins – structural motifs that are ubiquitous in natural products and molecules of pharmaceutical relevance. In addition to defining the basic annulation reaction we have discovered a surprising stability associated with the complex organometallic intermediates generated in the course of this coupling process and document here the ability to control the fate of this species. Ligand-induced cheletropic extrusion of the titanium center delivers cyclohexadiene-containing products, while several distinct protonation events have been identified to realize polycyclic products that contain three new stereocenters (one of which is the angular quaternary center that is a hallmark of alkoxide-directed titanium-mediated [2+2+2] annulation reactions). Examples of this metallacycle-mediated annulation reaction are provided to demonstrate that a range of stereodefined fused bicyclo[4.4.0]-decanes are accessible, including those that contain aromatic and aliphatic substituents, and an empirical model is presented to accompany the observations made.
TOC Graphic
INTRODUCTION
Metal-centered [2+2+2] annulation is a powerful means to prepare complex carbocyclic skeletons found in numerous complex natural products and pharmaceutically relevant agents.1, 2 Early examples include Co-mediated intramolecular cyclization reactions of enediynes (and related polyunsaturated species) that deliver polycyclic diene-containing products, often with excellent levels of stereoselectivity.3 While much progress has been made in developing this area of chemical reactivity, there remain substantial challenges that limit the great potential utility of this mode of reactivity to afford pathways for the efficient synthesis of a range of carbocyclic targets. For example, while effective in wholly intramolecular settings, it is often the case that intermolecular variants are complicated by issues related to the control of regio- and stereoselectivity in various steps of the cascade process, thereby complicating the use of this reactivity for convergent synthesis. This situation is particularly relevant in the context of carbocyclic targets that contain angular substitution (a quaternary stereocenter at the ring fusion) and is therefore an issue that substantially limits the utility of [2+2+2] chemistry for the efficient assembly of terpene-derived targets.
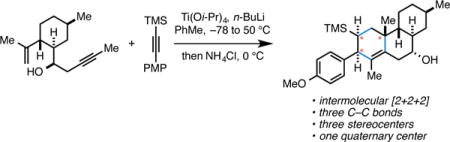
Recently we reported a titanium alkoxide-centered cascade reaction that defined a concise, convergent, and highly selective synthesis of angularly substituted hydrindanes from acyclic precursors (Figure 1; 1 + 2 → 4).4 This process was proposed to proceed by initial alkoxide-directed metallacycle-mediated alkyne–alkyne coupling, followed by intramolecular [4+2] cycloaddition to generate the presumed bridged bicyclic metallacyclopentene intermediate 3. This species was then envisioned to undergo a formal cheletropic extrusion of Ti(Oi-Pr)2 and produce the angularly substituted diene-containing hydrindane product 4. In later work, we realized a strategy to trap intermediate 3 by double elimination (Y = OMe, R1 = CH2OR5) and, in so doing, secured a pathway to stereodefined angularly substituted hydrindanes possessing a highly reactive cross-conjugated triene (5).5 In other efforts, installation of an appropriately situated allylic ether in 3 (i.e. R1 = CH2OPh or CH2OPMB) led to rapid elimination and the formation of a transient allylic titanium species that could be protonated with site- and stereoselectivity. Products of this latter annulation process are trans-fused hydroindanes 6.6
Figure 1.
Titanium-centered intermolecular [2+2+2] chemistry by way of a polycylic metallacyclopenetene intermediate (3).
While defining a means to accomplish intermolecular variants of [2+2+2] chemistry that furnish angularly substituted hydrindanes with high levels of stereoselectivity was viewed as a significant success, this reactivity was observed only for annulation reactions that delivered hydroindanes – i.e. with substrates that could, after alkoxide-directed alkyne–alkyne coupling,7 undergo intramolecular cycloaddition to form a five-membered ring. Also, while one could imagine a rich reactivity profile associated with the metallacyclopentene intermediate 3, we have been able to divert its reactivity away from standard cheletropic extrusion of Ti(Oi-Pr)2 only by one of two elimination pathways (en route to 5 or 6).5, 6 Here, we report a substantial advance in this general area of chemical reactivity, defining a means to realize intermolecular alkoxide-directed titanium-centered [2+2+2] annulation chemistry for the synthesis of angularly substituted decalins, and discovering that the resulting metallacyclopentene intermediates formed on cycloaddition (3, n = 1) can be coerced down distinct reaction pathways en rote to more stereochemically complex products. Our studies have resulted in convergent annulation reactions that can be terminated not only by ligand induced cheletropic extrusion (7) but also by site- and stereoselective protonation (8–10). These latter pathways define novel reaction cascades in the broader area of metal-centered [2+2+2] annulation and provide convenient access to densely functionalized and stereochemically rich decalin-containing polycycles.
RESULTS AND DISCUSSION
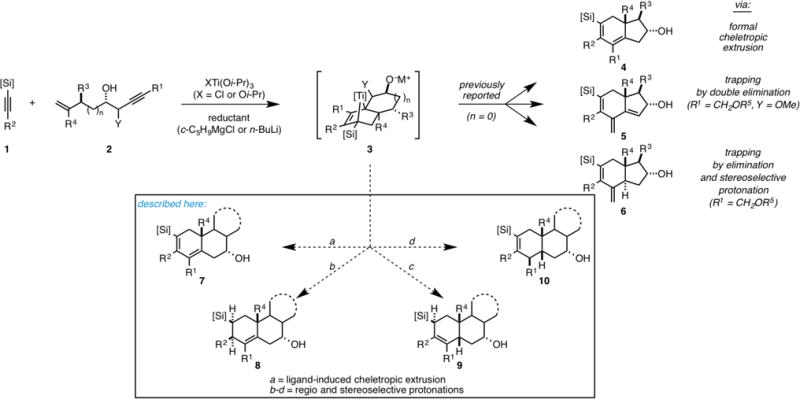
The present study began with an inquiry aimed at understanding the scope and limitations of titanium-centered alkoxide-directed metallacycle-mediated [2+2+2] annulation of TMS-alkynes with substituted enynes. With our goals focused on realizing a means to achieve annulation reactions for the synthesis of angularly substituted decalins, we began exploring the reactivity of enyne substrates in metallacycle-mediated coupling chemistry that have an increased tether length (the number of carbons between the two π-unsaturated units would be increased from three- to four). Perhaps not surprisingly, our initial attempt in this vein was met with failure (Table 1, entry 1). Here, Ti-mediated union of enyne 11 with TMS-alkyne 12 did not deliver the angularly substituted decalin 13. Rather, on protonation of the presumed intermediate organometallic species, the expected product of simple alkoxide-directed alkyne–alkyne coupling was observed (a trisubstituted 1,3-diene, not shown).7 This result is consistent with the conclusion that, while the alkyne–alkyne coupling remains effective for this substrate class, there exists a larger energetic barrier for intramolecular cycloaddition in comparison to that associated with the annulation processes previously achieved for the convergent synthesis of hydrindanes.4–6
Table 1.
|
Recognizing that our failure to achieve the desired reactivity profile likely derives from a greater entropic penalty associated with intramolecular cycloaddition, we contemplated the use of substrates that are more conformationally predisposed to cyclization. The first experiment in this vein is depicted in entry 2 of Table 1 and is based on employing a tether that incorporates an o-disubstituted aromatic. Here, treatment of alkyne 12 with the combination of Ti(Oi-Pr)4 and n-BuLi,8 followed by exposure of the resulting organometallic intermediate to the Li-alkoxide of 14, and quenching with benzaldehyde resulted in the formation of the tricyclic carbocycle 15 in 59% yield (dr ≥ 20:1, rs ≥ 20:1). Similarly, coupling of the methoxy-substituted aromatic 16 with TMS-alkyne 12 delivered 17, again with exquisite levels of regio and stereselectivity (entry 3). As depicted in entries 4–6, this annulation reaction is also effective with a conjugated alkyne possessing a functionalized silane (18), as well as propargylic cyclopropane (21). Here, tricyclic products 19, 20 and 22 were generated in 64, 48 and 52% yield.
With these early examples demonstrating the ability to accomplish alkoxide-directed and Ti-mediated convergent annulation for the synthesis of angularly substituted decalins that contain a fused aromatic, we directed our attention toward realizing this annulation reaction with enynes that contain fully aliphatic tethers. As illustrated in entries 7–13, the annulation reaction is quite effective in this series, delivering stereodefined carbocyclic products in 53–71% yield. Notably, the metallacycle-mediated annulation reaction is also effective for the generation of linearly fused tricycles. As illustrated in entry 14 of Table 1, union of enyne 36 with alkyne 12 proceeds to deliver the tricyclic product 37 in 57% isolated yield. In addition to nicely extending the scope of this annulation process to address the stereoselective synthesis of linearly fused tricycles, this is the first example of an alkoxide-directed annulation cascade that employs a tertiary alkoxide-containing enyne.
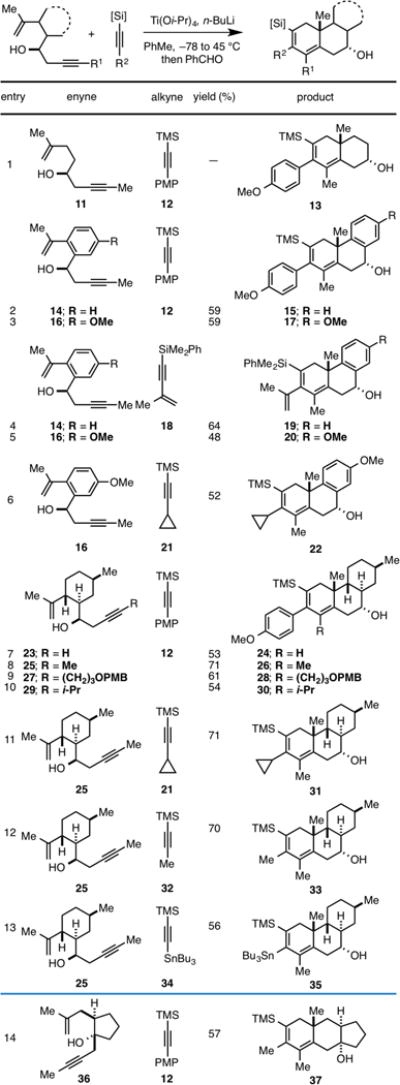
During the course of our studies we discovered that termination of the annulation reaction with a proton source results in the generation of a structurally unique product that harbors two additional stereocenters. For example, when reaction of enyne 14 with alkyne 12 is terminated with methanol at −78 °C (rather than quenching with benzaldehyde), the stereodefined reduced product of [2+2+2], 38 is generated in 54% yield (Table 2, entry 1). Here, stereoselectivity was quite high, positioning both the TMS- and PMP-group on the β-face of the carbocyclic product. When the reaction is instead quenched with CD3OD, as in entry 2, the dideuterated carbo-cycle 39 is generated. These observations are consistent with the conclusion that the organometallic intermediate generated after [4+2] cycloaddition has a lifetime that is sufficiently long to allow for manipulating the fate of this species down a reaction pathway that is distinct from our prior studies (cheletropic extrusion of titanium, or carefully orechestrated elimination reactions).
Table 2.
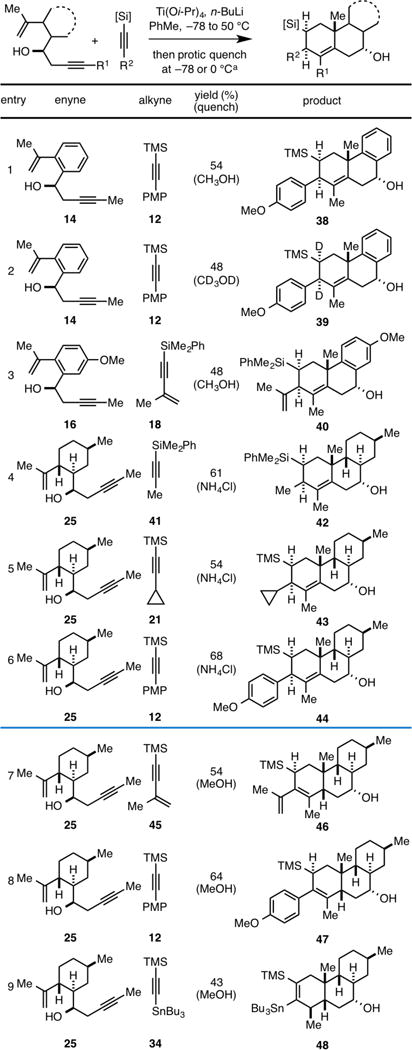
|
=Quenching with MeOH was conducted at −78°C, while quenching with NH4Cl (aq) was conducted at 0 °C
This reductive version of the alkoxide-directed Ti-mediated [2+2+2] annulation is also effective with a conjugated alkyne (18), in this case delivering the functionalized carbocycle 40 in 48% yield (entry 3). In efforts focused on exploring the scope of this process, we found that in some cases quenching with aqueous ammonium chloride was more effective than with methanol. As illustrated in entries 4–6, reductive [2+2+2] reactions between enyne 25 and alkynes 41, 21, and 12 are also effective, delivering the stereodefined products 42–44 in 61, 54 and 68% yield, respectively. Interestingly, as illustrated in entries 7–9, when the annulation reactions of enyne 25 with alkynes 45, 12, and 34 are quenched with methanol at −78 °C, structurally unique products of reductive annulation are generated. In the case of entries 7 and 8, reduction takes place in a net 1,4-manner to deliver the cis-fused carbocycles 46 and 47 in 54 and 64% yield, while in entry 9, the major product 48 is the formal product of a regioisomeric 1,2 reduction that generates an isomeric cis-fused carbocycle (the structure of 48 was confirmed by X-ray diffraction data collected for a simple derivative – see Supporting Information for details).
While these results document the feasibility of accomplishing metallacycle-mediated [2+2+2] for the convergent assembly of a variety of angularly substituted decalin-containing carbocycles, it is perhaps not surprising that some substrates were not compatible with the process. As described in Figure 2, enyne 49 could not be converted to carbocycle 50, and the oxygenated enyne 51 (either isomer) could not be converted to 52. In this latter case, each diastereomer of 51 was converted to the triene resulting from alkoxide-directed metallacycle-mediated alkyne–alkyne coupling7 without evidence being observed for progression of the reaction through the intramolecular [4+2] cycloaddition.
Figure 2.
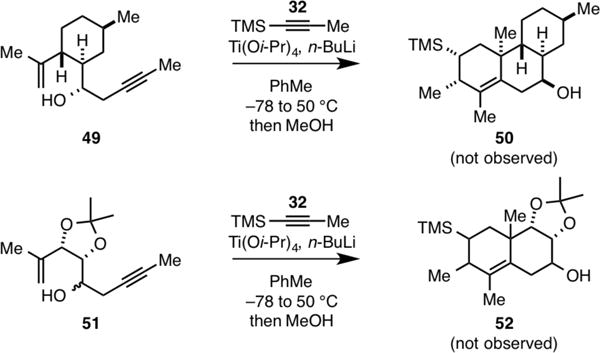
Success in this annulation is a function of substrate stereo-chemistry and substitution: Examples of substrates that failed in attempted annulation with TMS-propyne.
Our results have revealed that metallacycle-mediated intermolecular [2+2+2] annulation is a productive mode of reactivity for the convergent and stereoselective synthesis of angularly substituted decalin-containing carbocycles. Further, these studies have revealed an unexpected lifetime of the bridged polycyclic organometallic intermediate proposed to result from intramolecular cycloaddition. As depicted in Figure 3 exposure of this intermediate (generalized as structure I) to benzaldehyde results in cheletropic extrusion of the metal and production of a cyclohexadiene (I→III). Alternatively, quenching with methanol or ammonium chloride leads to one of three different reduced products. A simple model to visualize these stereoseletcive pathways is shown via initial regioselective syn SE’ protonation (I→A→B, or I→C→D), followed by a second protonation of the resulting tertiary organotitanium intermediate that proceeds with either inversion (B→II) or retention (D→IV). Alternately, we have observed products that appear to result from direct protonation of the bridged bicyclic metallacycle but may derive from a more complex and less clear sequence of steps (I→V). Surprisingly, the stereoselectivity associated with each pathway appears to be quite high, but distinguishing between and predicting which reaction path will predominate as a function of substrate structure or nature of the protic quench is less clear.
Figure 3.
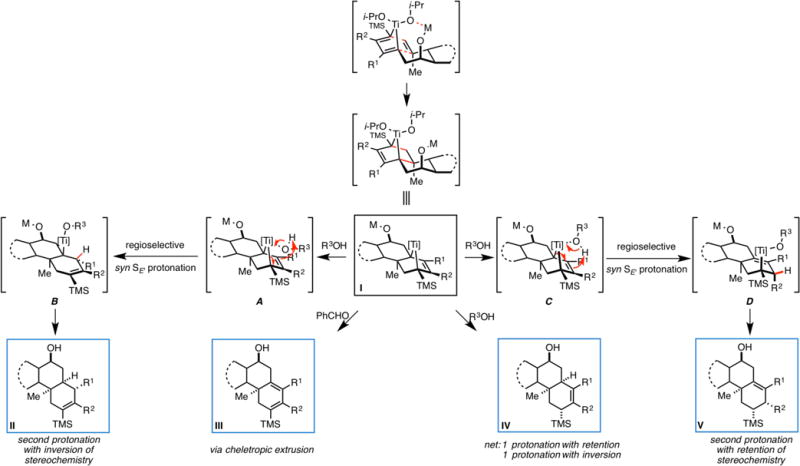
Summary of Ti-mediated annulation reactions en route to functionalized and angularly substituted decalins.
CONCLUSION
In summary, we have discovered a metal-centered reaction cascade for the convergent and stereoselective synthesis of angularly fused decalins. While defining the first convergent [2+2+2]-based strategy to accomplish such a feat, these investigations have also led to the elucidation of unique modes of reactivity associated with the presumed complex organometallic intermediates generated under the reaction conditions. We demonstrate cheletropic extrusion of the metal can be induced to proceed by addition of benzaldehyde, defining a highly stereoselective pathway to angularly substituted decalins that contain a highly substituted cyclohexadiene. Notably, we have also discovered that the complex organometallic intermediate in these annulations can be diverted away from this cheletropic reaction down one of several stereoselective pathways to reduced products of [2+2+2] annulation. While the nature of the proton source plays a role in path selectivity, so does the substrate structure – in combination defining a complex matrix that can be employed to gain access to decalin products through an annulation reaction that forges three carbon–carbon bonds and three new stereocenters, one of which is quaternary. We look forward to exploring the complexity of this reaction process and further investigating its utility in complex molecule synthesis.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work by the National Institutes of Health – NIGMS (GM80266) and postdoctoral fellowship support to HM from the Uehara Memorial Foundation.
Footnotes
SUPPORTING INFORMATION
Experimental procedures and tabulated spectroscopic data for new compounds (PDF) are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of metal-centered [2+2+2] annulation, see:; (a) Vollhardt KPC. Acc Chem Res. 1977;10:1–8. [Google Scholar]; (b) Aubert C, Buisine O, Petit M, Slowinski F, Malacria M. Pure Appl Chem. 1999;71:1463–1470. [Google Scholar]; (c) Rubin M, Sromek AW, Gevorgyan V. Synlett. 2003:2265–2291. [Google Scholar]; (d) Gandon V, Aubert C, Malacria M. Chem Commun. 2006:2209–2217. doi: 10.1039/b517696b. [DOI] [PubMed] [Google Scholar]; (e) Shibata T, Tsuchikama K. Org Biomol Chem. 2008;6:1317–1323. doi: 10.1039/b720031e. [DOI] [PubMed] [Google Scholar]; (f) Tanaka K. Chem Asian J. 2009;4:508–518. doi: 10.1002/asia.200800378. [DOI] [PubMed] [Google Scholar]; (g) Leboeuf D, Gandon V, Malacria M. Transition Metal-Mediated [2 + 2 + 2] Cycloaddtions. In: Ma S, editor. Handbook of Cyclization Reactions. Vol. 1. Wiley–VCH; Weinheim: 2009. p. 367. [Google Scholar]
- 2.(a) González MA. Nat Prod Rep. 2015;32 doi: 10.1039/C4NP00110A. [DOI] [PubMed] [Google Scholar]; (b) Heasley B. Chem Eur J. 2012;18:3092–3120. doi: 10.1002/chem.201103733. [DOI] [PubMed] [Google Scholar]; (c) Chapelon A-S, Moraléda D, Rodriguez R, Ollivier C, Santelli M. Tetrahedron. 2007;63:11511–11616. [Google Scholar]; (d) Godtfredsen WO, von Daehne W, Vangedal S, Marquet A, Arigoni D, Melera A. Tetrahedron. 1965;21:3505–3530. doi: 10.1016/s0040-4020(01)96970-4. [DOI] [PubMed] [Google Scholar]; (e) Zhao M, Gödecke T, Gunn J, Duan J-A, Che C-T. Molecules. 2013;18:4054–4080. doi: 10.3390/molecules18044054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Sternberg ED, Vollhardt KPC. J Am Chem Soc. 1980;102:4839–4841. [Google Scholar]; (b) Sternberg ED, Vollhardt KPC. J Org Chem. 1982;47:3447–3450. [Google Scholar]; (c) Clinet J-C, Duñach E, Vollhardt KPC. J Am Chem Soc. 1983;105:6710–6712. [Google Scholar]; (d) Butenschön H, Winkler M, Vollhardt KPC. J Chem Soc, Chem Commun. 1986:388–390. [Google Scholar]; (e) Grotjahn DB, Vollhardt KPC. J Am Chem Soc. 1986;108:2091–2093. [Google Scholar]; (f) Germanas J, Aubert C, Vollhardt KPC. J Am Chem Soc. 1991;113:4006–4008. [Google Scholar]; (g) Johnson EP, Vollhardt KPC. J Am Chem Soc. 1991;113:381–382. [Google Scholar]
- 4.(a) Greszler SN, Reichard HA, Micalizio GC. J Am Chem Soc. 2012;134:2766–2774. doi: 10.1021/ja2105043. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent review of alkoxide-directed metallacycle-mediated cross-coupling, see:; (b) Micalizio GC, Hale SB. Acc Chem Res. 2015;48:663–673. doi: 10.1021/ar500408e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng X, Micalizio GC. Org Lett. 2014;16:5144–5147. doi: 10.1021/ol502496d. [DOI] [PMC free article] [PubMed] [Google Scholar]; The cross-conjugated triene-containing hydrindanes produced by this coupling reaction served as substrates for intermolecular Diel-Alder cycloaddition reactions with a variety of highly activated dienophiles.
- 6.(a) Jeso V, Aquino C, Cheng X, Mizoguchi H, Nakashige M, Micalizio GC. J Am Chem Soc. 2014;136:8209–8212. doi: 10.1021/ja504374j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim WS, Aquino C, Mizoguchi H, Micalizio GC. Tetrahedron Lett. 2015;56 doi: 10.1016/j.tetlet.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan J, Micalizio GC. J Am Chem Soc. 2006;128:2764–2765. doi: 10.1021/ja057352w. [DOI] [PubMed] [Google Scholar]
- 8.(a) Obora Y, Moriya H, Tokunaga M, Tsuji Y. Chem Commun. 2003:2820–2821. doi: 10.1039/b310565b. [DOI] [PubMed] [Google Scholar]; (b) Rassadin VA, Six Y. Tetrahedron. 2014;70:787–794. [Google Scholar]; For the use of n-BuLi en route to Ti–imine complexes, see:; (c) Tarselli MA, Micalizio GC. Org Lett. 2009;11:4596–4599. doi: 10.1021/ol901870n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.