

Abstract
Objective
Enzyme-modified LDL (ELDL) is present in human atherosclerotic lesions. Our objective is to understand the mechanisms of ELDL uptake and its effects on vascular smooth muscle cells.
Approach and Results
Transformation of murine aortic SMC into foam cells in response to ELDL was analyzed. ELDL, but not acetylated or oxidized LDL, was very potent in inducing SMC foam cell formation. Inhibitors of macropinocytosis (LY294002, wortmannin, amiloride) attenuated ELDL uptake. In contrast, inhibitors of receptor mediated endocytosis (dynasore, sucrose) and inhibitor of caveolae/lipid raft mediated endocytosis (filipin) had no effect on ELDL uptake in SMC, suggesting that macropinocytosis is the main mechanism of ELDL uptake by SMC. Receptor for advanced glycation end products (RAGE) is not obligatory for ELDL induced SMC foam cell formation, but primes SMC for the uptake of oxidized LDL in a RAGE-dependent manner. ELDL increased intracellular reactive oxygen species (ROS), cytosolic calcium, and expression of lectin like oxidized LDL receptor (LOX-1) in wild type SMC but not in RAGE−/− SMC. The macropinocytotic uptake of ELDL is regulated predominantly by intracellular calcium since ELDL uptake was completely inhibited by pretreatment with the calcium channel inhibitor lacidipine in wild type and RAGE−/− SMC. This is in contrast to pretreatment with PI3K inhibitors which completely prevented ELDL uptake in RAGE−/− SMC, but only partially in wild type SMC.
Conclusions
ELDL is highly potent in inducing foam cells in murine SMC. ELDL endocytosis is mediated by calcium dependent macropinocytosis. Priming SMC with ELDL enhances the uptake of oxidized LDL.
Keywords: Enzyme-modified LDL, foam cells, smooth muscle cells, macropinocytosis, atherosclerosis
Graphical abstract
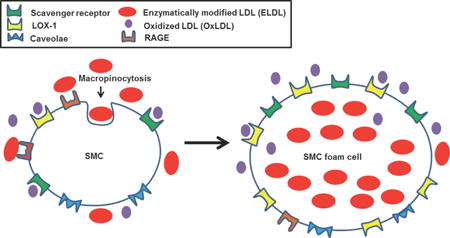
Introduction
Foam cell formation is the central feature in the initiation and development of atherosclerotic lesions. Since foam cells express several macrophage markers, it is thought that foam cells arise primarily from monocyte-derived macrophages. Despite the understandable emphasis on macrophage foam cells, there is evidence for the participation also of smooth muscle cells (SMCs) in forming foam cells. This is particularly relevant in human arteries which are enriched in SMC 1–3. Scavenger receptors account for lipid accumulation in macrophages primarily through uptake of modified LDLs such as oxidized LDL (OxLDL) 4, 5. Smooth muscle cells also express scavenger receptors 6, 7 and acquire macrophage-like phenotypes upon lipid loading, expressing macrophage markers and suppression of SMC markers8. SMC-derived foam cells in human atherosclerotic lesions are well documented 9–12, with recent studies showing as many as 50% of foam cells in human and murine lesions as may be SMC derived 10, 13.
The characteristics of SMC-derived foam cells are less well established. Recent work has suggested that there are significant distinctions in gene expression and modes of lipid loading between macrophages and SMC. For example, lipid loaded macrophages express high levels of cholesterol transporter ABCA114, scavenger receptors CD36 and SRA115, 16 and undergo apoptosis upon excess free cholesterol loading17. In contrast, SMC-derived foam cells express significantly less ABCA19, 10 and are apparently more resistant to free cholesterol toxicity18. Studies in vitro have shown lipid loading of SMC by cholesterol8, OxLDL19, and enzyme modified non oxidized LDL (ELDL)20, but mechanisms by which SMC take up lipids in vivo are mostly unknown.
ELDL differs from OxLDL in that it lacks oxidized lipids21. ELDL has been detected in human atherosclerotic lesions and has been generated in vitro by the joint action of trypsin and cholesteryl ester hydrolase 21–24. While cholesteryl ester hydrolase is present in the human atherosclerotic vessels at concentrations high enough for direct detection by immunostaining 25, 26, there are a number of protease candidates that could act on vessel wall LDL in vivo. Chymase, plasmin, matrix metalloproteinases (MMPs), and cathepsins are highly expressed in atherosclerotic plaques 24, 26–32, and potentially could act on LDL as modifying proteases. Plasmin and MMPs were reported to co-localize with ELDL in human atherosclerotic lesions29.
Here we report that ELDL uptake by SMC occurs via macropinocytosis rather than by scavenger receptor-dependent mechanisms. Interestingly, we found that lectin like oxidized LDL receptor (LOX-1), a major scavenger receptor for OxLDL 33, 34, is significantly upregulated by ELDL in SMC and this upregulation is dependent on the receptor for advanced glycation end products (RAGE).
Materials and Methods
Materials and Methods are available in the online-only Data Supplement. Briefly, ELDL was prepared from native LDL upon digestion with trypsin and cholesterol esterase followed by dialysis and sterile filtration which resulted in ELDL particle size between 100 KD and 2500 KD.
Results
1. ELDL, but not AcLDL or OxLDL, induces foam cells in mouse aortic smooth muscle cells in vitro
ELDL has been shown to be a more efficient inducer of macrophage foam cells than are other modified LDLs such as AcLDL or OxLDL 21. Nevertheless, the comparative ability of these modified LDLs to induce foam cell formation in SMC has not been studied. Therefore, SMC and macrophages were cultured in the presence of 10 μg/ml of a modified LDL (AcLDL, OxLDL or ELDL) and lipid content was visualized by staining with Oil Red O. The accumulation of lipid was evident as early as 4h after incubation with modified LDLs. As expected, ELDL, OxLDL and AcLDL at 10 μg/ml, induced significant foam cell formation in macrophages over 24h in contrast to SMC, where only ELDL induced foam cells (Fig. 1A). This was confirmed by quantitative assessment of cellular cholesterol accumulation (Fig. 1B). ELDL at 10, 25 and 50 μg/ml, induced dose dependent accumulation of lipid in SMC (Fig. 1C and D). Although at 100 μg/ml, significant foam cell formation was observed, cell viability was less than 50%, probably due to cholesterol toxicity (data not shown). Substantially higher concentrations were required for SMC foam cell formation by AcLDL (500 μg/ml) and OxLDL (200 μg/ml) as shown in figure 1C and 1D. The small amount of intracellular lipid accumulation induced by AcLDL (500 μg/ml) was associated with significant amount of cell death as evidenced by trypan blue staining (approx.75%, data not shown). Increased cell death was also observed in SMC treated with OxLDL at concentrations above 200 μg/ml perhaps due to the presence of modified sterols in these LDLs (data not shown). Previously it has been shown that the concentration of LDL in the intimal fluid of normal and atherosclerotic lesions is 2 to 4 times higher than that in circulation35.
Figure 1. ELDL, but not acetylated or oxidized LDL, induces foam cells in cultured mouse aortic SMC.

A. Peritoneal macrophages (top row) or aortic SMC (bottom row) were incubated with 10 μg/ml (based on lipoprotein protein) of native LDL, acetylated LDL (AcLDL), oxidized LDL (OxLDL) or Enzyme-modified LDL (trypsin and cholesterol esterase modified LDL, ELDL) for 24h. The cells were stained for lipids with Oil Red O. B. Total cellular cholesterol/protein from experiments shown in A. Data are represented as mean ± SD. (** p<0.01, AcLDL, OxLDL & ELDL vs. native LDL) C. Aortic SMC were incubated with 10, 25 and 50 μg/ml (based on protein) of ELDL, 2000 μg/ml native LDL, 500 μg/ml AcLDL, and 200 μg/ml OxLDL and stained for lipids with Oil Red O. D. Total cellular cholesterol/protein from experiments shown in C. Data are represented as mean ± SD. p<0.01 ELDL (all tested concentrations of 10, 25 and 50 μg/ml) vs. native LDL (2000 μg/ml) or OxLDL (200 μg/ml). p< 0.01 ELDL (50 μg/ml) vs AcLDL(500 μg/ml), p<0.05 AcLDL (500 μg/ml) vs LDL (2000 μg/ml) or OxLDL (200 μg/ml). P<0.05 for native LDL 2000 μg/ml vs 0 or 10 μg/ml native LDL.
Since the normal concentration of LDL in circulation is about 1 mg/ml, the concentration of LDL in the intimal fluid may be at least 2 mg/ml. We found that native LDL when used at 2 mg/ml induced foam cells in SMC even though it was significantly less than that by ELDL at 10 μg/ml (Fig. 1C and D). Since we aimed to quantify the uptake of modified LDLs by SMC as whole particles, we normalized loading of cells with LDL, ELDL, AcLDL and OxLDL based on lipoprotein protein rather than cholesterol, since modified LDL-uptake by cells is often a protein (receptor) based phenomenon. As shown in supplementary Fig. IA, the cholesterol to protein ratio differs among these modified lipoproteins. Nevertheless, the experiments shown in Figure 1C and D, even when recalculated on the basis of lipoprotein cholesterol exposure, showed that ELDL was much more effective in SMC lipid loading than any of the other three tested lipoprotein preparations (supplementary Fig. IB). LDL-receptor (LDLR) mediates the uptake of modest amounts of native LDL with the consequent modification of the regulatory pool of sterol which tightly regulates LDLR mRNA expression36. We measured LDLR mRNA expression in SMC incubated with native or modified LDLs at 10 μg protein/ml. LDLR mRNA was down regulated in LDL and ELDL loaded SMCs (~ 25 and 75% decrease respectively) compared to BSA-treated SMC, while AcLDL or OxLDL showed no such effect (supplementary Fig. II). By this measure too, ELDL delivered much more cholesterol to the sterol regulatory pool of SMC than did AcLDL or OxLDL at an experimental concentration of 10 μg/ml.
2. Aortic smooth muscle cells endocytose the larger LDL particles in ELDL preparations
Particle size of native LDL is approximately 25 nm, while ELDL preparations were shown to contain a heterogeneous mixture of particles with sizes ranging from 10 to 200 nm 21. Enzyme action results in the fusion of many LDL particles forming larger particles in ELDL preparations 21. This was confirmed by us in agarose gel electrophoresis of native LDL versus modified LDLs. ELDL had a lower electrophoretic mobility than native LDL indicating a higher mass or less negative charge for ELDL (Fig. 2A). Additionally, we found that tryptic proteolysis without cholesterol esterase induced a similar pattern of lower electrophoretic mobility. In contrast, exposure to cholesterol esterase alone did not result in slower moving particles. As expected, OxLDL and AcLDL showed higher electrophoretic mobility than native LDL37. Taken together, tryptic proteolysis of LDL alters electrophoretic mobility of LDL particels, likely reflecting changes in mass and/or charge. In accordance with earlier reports21, we found SMC foam cell formation by ELDL modified by trypsin or cholesterol esterase alone was much less efficient than SMC foam cell formation with ELDL modified by both trypsin and cholesterol esterase (data not shown). Aiming to identify the components in ELDL preparations that were responsible for foam cell formation in SMC, we employed fast protein liquid chromatography (FPLC) to separate ELDL particles according to particle sizes. ELDL showed two protein peaks; one peak corresponding to the FPLC elution profile of human plasma VLDL, and another peak corresponding to that of human plasma LDL (Fig. 2B, upper panel). Even though both peaks contained proteins, only the fractions in the first peak (VLDL-sized peak) contained cholesterol and phospholipid. The second peak representing smaller proteins did not have any measurable amount of cholesterol and contained a small amount of phospholipid (Fig. 2B, lower panels). ELDL preparations also appear to contain phospholipid containing vesicles that are apparently devoid of protein as noted in fractions 14 and 16 in Fig. 2B. Importantly, when FPLC fractions from both peaks were incubated with wild type aortic SMC, only the larger size fractions (fraction 8–10) induced foam cell formation (Fig. 2C, and quantified in Fig. 2D). Taken together, this indicates that the larger particles within ELDL preparations were responsible for foam cell formation in mouse aortic SMC. However, it is not clear whether the activity of the large particles is attributable to its much higher cholesterol content or whether particle size per se also played a role.
Figure 2. Aggregated/fused ELDL is preferentially taken up by aortic SMC.

A. Agarose gel electrophoresis of native LDL, ELDL (trypsin and cholesterol esterase modified LDL), CLDL (cholesterol esterase modified LDL), TLDL (trypsin modified LDL), AcLDL and OxLDL. Arrows indicate a LDL with reduced mobility that is indicative of fused LDLs or lower negative charge. B. Protein, cholesterol and phospholipid content in ELDL fractions after separation by FPLC. C. Foam cell formation in aortic SMC incubated with 5 μg/ml of the indicated FPLC fractions; fraction numbers are shown in the panels. Cells were stained for lipids with Oil Red O. D. Total cellular cholesterol/mg protein from experiments shown in C. Data are represented as mean ± SD (** p<0.01, Fractions 8 &10 vs 16, 18, 22 and 24).
3. RAGE dependent up regulation of OxLDL receptor-1 (LOX1) expression by ELDL in aortic SMC
Scavenger receptor dependent endocytosis is the principle route for modified LDL uptake by cells, with CD36, SRA1, and LOX-1 as the major scavenger receptors involved in this process38. ELDL uptake in macrophages has been suggested to be partly through CD36 21, 23, while the mechanisms of ELDL uptake in vascular SMC is currently unknown. We measured mRNA expression of CD36, SRA1 and LOX-1 in mouse aortic SMC in response to incubation with ELDL, AcLDL and OxLDL using real-time RT PCR. In contrast to the known up regulation of CD36 and SRA1 expression in macrophages upon lipid loading 38, we found no change in CD36 and SRA1 mRNA in SMC upon incubation with the modified LDLs (Fig. 3A). Interestingly, mRNA for the receptor for oxidized LDL (LOX-1), a major scavenger receptor in arterial cells39, was increased ~ 4-fold in SMC upon incubation with ELDL (p<0.001), and to a lesser extent (~20%, p<0.05) upon incubation with OxLDL (Fig. 3A). Protein levels of LOX-1 were also increased significantly in SMC upon incubation with ELDL, but did not change in response to native LDL, AcLDL or OxLDL (Fig. 3B). Native LDL even at 2 mg/ml did not show any changes in LOX-1 mRNA or protein in the SMC (data not shown).
Figure 3. ELDL up regulates expression of oxidized LDL receptor (LOX-1) in aortic SMC and promotes uptake of OxLDL.

A. qRT PCR analysis of scavenger receptor gene expression in mouse aortic SMCs; wild type SMC were incubated with 10 μg/ml of native LDL, ELDL, OxLDL, or AcLDL for 24h (expression is relative to levels in cells incubated with native LDL). Data are represented as mean ± SD, for LOX-1 * p<0.05 for OxLDL vs native LDL, ** p<0.01 for ELDL vs native LDL. B. Semi-quantitative expression of LOX-1 protein expression in SMC treated as indicated in Panel A, ** p<0.01 for ELDL vs native LDL. C. LOX-1 protein expression in WT and RAGE−/− SMC upon pretreatment with ELDL as indicated and densitometric analysis. ** p<0.01 either 50, 25 or 10 μg/ml vs 0 μg/ml. D. qRT PCR analysis of mRNA in WT and RAGE−/− SMC incubated with 10 μg/ml ELDL versus control (10 μg/ml native LDL) ** p<0.01 for ELDL vs native LDL. E. Fluorescent images of WT or RAGE−/− SMC pretreated with bovine serum albumin (BSA) or ELDL for 24h, followed by incubation for 24h with DiI-labeled LDL (red color) or DiI-labeled OxLDL (red color). ELDL was removed prior to adding DiI-labelled LDL or OxLDL. Nuclei were stained in blue with Hoechst. F. Fluorescence intensity from experiments in E was quantified using Image J as a function of total pixel density per total area of cells in frame (refer methods for details). G. Foam cell formation assessed by Oil red O staining in RAGE−/− SMC incubated for 24h with 10 μg/ml ELDL or native LDL (negative control). H. Total cellular cholesterol/protein from experiments shown in G.
Oligomerization of ligands is known to enhance ligand binding and activation of RAGE40. Since our data suggests that ELDL consists of aggregated or fused LDL molecules as shown in Fig. 2A, we queried whether RAGE might play a role in the up regulation of LOX-1 by ELDL. We employed aortic SMC isolated from RAGE deficient mice (RAGE−/− SMC) and from mice with intact RAGE signaling (WT SMC). LOX-1 protein was dose dependently up regulated in wild type SMC incubated for 24h with up to 25 μg/ml ELDL, while higher concentrations of ELDL (50 μg/ml) showed a decrease in LOX-1 protein expression (Fig. 3C). On the other hand, surprisingly, we found ELDL did not up regulate LOX-1 protein or mRNA expression in RAGE−/− SMC (Fig. 3C and D). OxLDL is known to bind RAGE on account of its AGE epitopes41. Although ELDL so far has not been described as a RAGE-binding molecule, it is possible that ELDL too has AGE-like epitopes that could mediate binding to RAGE. However, binding of ELDL to RAGE is indirectly suggested by our experiments as shown in supplementary Fig. III. When SMC were incubated with recombinant human RAGE or with recombinant human RAGE pre-complexed with ELDL (hRAGE/ELDL), hRAGE was immunodetected only in the lysates of cells incubated with hRAGE (lane 2), and not in the lysates of cells incubated with the hRAGE/ELDL complex (lane 4). This suggests that hRAGE may be binding to integrins, collagens and other cell membrane structures as previously described42. The inability to detect hRAGE when cells were incubated with hRAGE/ELDL complexes is possibly related to a rapid uptake of the complexes into the cytosol and subsequent lysosomal/proteolytic degradation of hRAGE. If this were the case it implies that hRAGE is carried into the cells by ELDL i.e. that the two molecules do form a bimolecular complex. Alternatively, the preformed complexes may obstruct the association of hRAGE with the cells and be washed away prior to preparation of the SMC lysate. At present, we cannot distinguish between these possibilities, nevertheless, these observations indirectly suggest that ELDL binds RAGE. Taken together, ELDL up regulates LOX-1 in aortic SMC in a RAGE dependent manner; however, the details of this interaction and whether ELDL binds directly to RAGE requires future investigation. It is also unknown at this point whether LOX-1 upregulation is dependent upon any cytoplasmic component in ELDL- RAGE signaling.
OxLDL uptake by SMC is low as shown in Figure 1. However, given the ability of ELDL to induce LOX-1 mRNA and protein in SMC, we next examined whether preincubation with ELDL would increase uptake of OxLDL by the SMC. We preincubated WT and RAGE−/− SMC with ELDL (10 μg/ml) for 24h followed by incubation with 10 μg/ml DiI-labeled OxLDL or DiI-labeled native LDL (as control) for 24h. ELDL was removed prior to adding DiI labelled lipoproteins. We found that in WT SMC, ELDL significantly increased uptake of DiI-labeled OxLDL but not the uptake of DiI-LDL (Fig. 3E and quantified in Fig. 3F). However, in RAGE−/− SMC, pre-treatment with ELDL did not promote the uptake of DiI-OxLDL. This suggests, at least in culture, that ELDL in a RAGE-dependent manner primes SMC for enhanced uptake of OxLDL possibly via up regulation of LOX-1. However, despite these findings suggesting a role for RAGE in facilitating uptake of OxLDL, RAGE is not required for ELDL induced SMC foam cell formation since robust lipid accumulation was seen in RAGE−/− SMC upon incubation with ELDL (Fig. 3G and H). In other words, the loading of SMC with cholesterol derived from ELDL can occur independent of RAGE, but up-regulation of LOX-1 receptor and its promotion of OxLDL uptake are regulated via RAGE.
4. ELDL endocytosis by aortic smooth muscle cells is not receptor/clathrin mediated or lipid raft/caveolae dependent, but mediated by macropinocytosis
Aiming to identify the route of ELDL endocytosis in murine aortic SMC, we focused on the three major endocytic pathways that have been studied extensively43, namely the receptor mediated (clathrin dependent) pathway, lipid raft/caveolin dependent pathway, and macropinocytosis. In receptor mediated endocytosis, the separation of the budding clathrin-coated vesicle from plasma membrane relies on the GTPase, dynamin44. Dynasore, a pharmacological inhibitor of dynamin, is known to completely block receptor mediated endocytosis44. However, as shown in Figure 4A, dynasore (100 μM) did not inhibit ELDL induced foam cell formation in aortic SMC as shown by Oil Red O staining (Fig. 4A and quantified in 4D). Similarly, SMC cultured in hypertonic media containing 0.1M sucrose, a known inhibitor of receptor mediated endocytosis43, showed intact foam cell formation in response to treatment with ELDL (Fig. 4A and quantified in 4D). As expected, treatment of SMC with dynasore (100 μM) or sucrose (0.1 M), attenuated the uptake of transferrin, a classical ligand for receptor mediated endocytosis43 (supplementary Fig. IV). Two potential candidate receptors for uptake of ELDL by SMC could be LOX-1 and RAGE. However neither appears to be directly involved in the uptake of ELDL, since antibody specific to LOX-1 did not attenuate ELDL uptake and foam cell formation in SMC (supplementary Fig. V). Moreover, SMC lacking the receptor RAGE exhibited normal uptake of ELDL and foam cell formation as shown in Fig. 3G and H. These results collectively suggest that receptor-mediated endocytosis is not critically involved in the uptake of ELDL in SMC.
Figure 4. ELDL endocytosis by aortic SMC is mediated by macropinocytosis and is not receptor/clathrin mediated or lipid raft/caveolae dependent.

A. Foam cell formation assessed by Oil red O staining in aortic SMC incubated for 24h with 10 μg/ml ELDL (positive control) or native LDL as negative control. Prior to incubation with lipoproteins, cells were pretreated for 1h with inhibitors of receptor mediated endocytosis (100 μM dynasore or 0.1M sucrose). Pharmacologic inhibitors were not removed from media until end of experiment. B. Aortic SMC pre-incubated for 1h in serum free medium with 5 μM filipin to inhibit lipid raft/caveolae mediated endocytosis followed by incubation with 10 μg/ml ELDL or native LDL for 24h. C. Aortic SMC were pre-incubated for 1h with 3 mM amiloride to inhibit macropinocytosis, followed by incubation with 10 μg/ml ELDL or native LDL for 24h (amiloride was not removed from the media prior to adding ELDL). D. Total cellular cholesterol/protein from experiments shown in A, B and C. Data are represented as mean ± SD (** p<0.01 ELDL untreated vs. ELDL amiloride treated)
Next, we examined whether ELDL uptake by SMC depends on lipid raft or caveolins. Caveolae are specialized membrane lipid rafts enriched with the protein, caveolin-1, and caveolae contributes to a type of clathrin-independent endocytosis43. We used filipin, a polyene antibiotic and a selective inhibitor of lipid raft/caveolae-mediated endocytosis43. Aortic SMC were incubated in medium containing 5 μM filipin for 1h followed by incubation with ELDL for 24h. Filipin treatment did not attenuate ELDL-induced SMC foam cell formation (Fig. 4B and quantified in 4D). Control experiments in WT SMC using cholera-toxin B sub unit, a selective caveolae dependent endocytic marker43, showed filipin inhibiting the uptake of Alexa Fluor- labelled cholera-toxin B sub unit (supplementary Fig. VI). In summary, our results suggest that ELDL endocytosis in aortic SMC is not lipid raft or caveolae dependent.
We then focused on macropinocytosis, a process by which bulk cargo such as dissolved molecules and large volumes of extracellular fluid find their way into the cell in discrete vacuoles called macropinosomes45. The Na+/H+ exchange inhibitors amiloride and its derivative 5-(N-ethyl-Nisopropyl) amiloride (EIPA) are classical inhibitors of macropinocytosis43. Importanly, amiloride at 3 mM in the media for 1h prior to incubating with ELDL abolished lipid uptake and foam cell formation in WT SMC (Fig. 4C and quantified in 4D). For control experiments, we used high molecular weight dextrans which were previously shown to be taken up by macropinocytosis45. WT SMCs preincubated for 1h with amiloride at 3 mM inhibited the uptake of FITC-labelled dextran as shown in supplementary Fig. VII. Additionally, fluorescently labeled latex beads (30 nm) were employed as cargo for macropinocytic uptake, since latex beads sized less than 100 nm have been previously identified as specific macropinocytic markers45. WT SMCs preincubated for 1h with amiloride at 3 mM also inhibited the uptake of fluorescently-labelled latex beads (supplementary Fig. VII). Taken together, this indicates that ELDL uptake by SMCs is through a mechanism involving macropinocytosis.
5. PIP-independent macropinocytotic uptake of ELDL in WT SMC
Macropinosomes are derived from the F-actin driven ruffles on the cell surface that forms into a cup fusing to engulf a volume of surrounding medium and this process is regulated by the phosphoinositide PIP3 (phosphatidylinositol 3,4,5-trisphosphate). The role of PIP3 in macropinocytosis has been increasingly elaborated in recent times46, 47. PIP3 is produced when PIP2 (phosphatidylinositol 4,5-bisphosphate) is phosphorylated at the 3rd carbon of inositol group by PI3 Kinase (PI3K). The conversion of PIP2 to PIP3 by PI3K promotes proper closure of nascent macropinosomes and their subsequent trafficking. Macropinocytic membrane is highly enriched in PIP3; however, exact mechanism by which PIP3 participates in macropinocytosis is still unknown46. Pharmacological inhibitors of PI3K (wortmannin and LY294002) are known inhibitors of macropinocytosis, and LY294002 has been previously shown to inhibit macropinocytosis in cells including macrophages at concentrations less than 50 μM43. To further explore the role of macropinocytosis in the uptake of ELDL by SMC, we incubated WT aortic SMC with wortmannin or LY294002 at different concentrations for 1h followed by ELDL for 24h. When treated with 50 μM-200 μM LY294002, we found an approximately 50% reduction in foam cell formation in WT SMC, while at 300 μM LY294002 ELDL uptake and foam cell formation was completely inhibited (Fig. 5A and quantified in 5C). Cell viability was not affected by treatment with LY294002 at any of the tested doses. Furthermore, wortmannin, a potent inhibitor of PI3K and previously shown to inhibit macropinocytosis in vitro at concentrations less than 0.2 μM43, reduced ELDL-mediated foam cell formation of SMC at concentrations of 2 μM (Fig. 5A and quantified in Fig. 5C). Concentrations of wortmannin higher than 2 μM caused significant cell death in aortic SMC (data not shown). Taken together, complete inhibition of ELDL uptake and foam cell formation in SMC requires high concentrations of the PI3K inhibitors while LY294002 (50μM) and wortmannin (1μM) readily inhibited the uptake of dextran (500KD) and latex beads (30 nm) in wildtype SMC (data not shown). This suggests that PIP3 only partically regulates ELDL uptake in SMC’s, however, true involvement of PI3K in macropinocytosis of ELDL or the possibility of off target effects of the inhibitors in SMC can not be excluded.
Figure 5. PIP-independent macropinocytotic uptake of ELDL in WT SMC.

Foam cell formation assessed by Oil red O staining in A. WT SMC and B. RAGE−/− SMC incubated for 24h with 10 μg/ml ELDL or native LDL (negative control). The cells were pretreated for 1h with PI3K inhibitors (20 to 300 μM LY294002 or 1 to 2 μM wortmannin) prior to adding ELDL; inhibitors were not removed from media until end of experiment. C. Total cellular cholesterol/protein in cells from panels A and B. Data are represented as mean ± SD.
As shown in Fig. 3G and H, foam cell formation in response to ELDL is preserved in RAGE deficient SMC suggesting that RAGE is not required for uptake of ELDL. We next examined the effects of PI3K inhibitors on foam cell formation in RAGE−/− SMC and found that RAGE−/− SMC are more sensitive to LY294002 and wortmannin than wild type cells. Both inhibitors, at a concentration that were only partially effective in wild type cells (50%), completely suppressed ELDL uptake and foam cell formation in RAGE−/− SMC (Fig. 5B and quantified in Fig. 5C). This suggests that RAGE modulates PI3K activity. It was previously reported that RAGE signaling down regulates Akt, an upstream determinant of membrane PIP3 levels48, and therefore we queried whether ELDL-RAGE interaction down regulates PIP3 levels. We found reduced pAkt (pAkt Ser 473) formation, suggesting reduced PIP3 levels in WT SMC, but not in RAGE−/− SMC upon stimulation with ELDL. Baseline pAkt Ser 473 did not differ between SMC with intact or absent RAGE (supplementary Fig. VIII). Taken together, our results suggest that ELDL is taken up by macropinocytosis in WT SMC through non-PIP3 dependent mechanisms. Apparently, ELDL-RAGE axis suppresses PIP3 activity in WT SMC and also activates the non-PIP3 dependent macropinocytosis of ELDL. In the absence of RAGE, PIP3-mediated macropinocytosis appears to be a major determinant of ELDL uptake in RAGE−/− SMC.
6. Macropinocytotic uptake of ELDL by SMC: a role for ROS and intracellular calcium
In view of the differential sensitivity of WT and RAGE−/− SMCs to PI3K inhibitors we examined other features of macropinocytosis that could be regulated by RAGE. RAGE activation has been shown to upregulate cellular levels of both calcium and reactive oxygen species (ROS)49, 50. ROS and calcium positively regulate the intracellular levels of each other as there is considerable cellular cross talk between them 51. ROS has not been directly implicated in macropinocytosis unlike calcium which is known to regulate macropinocytosis 45, 52, therefore we analyzed both intracellular calcium and ROS in ELDL treated SMC. We incubated ELDL (10 μg/ml) with WT and RAGE−/− SMC and analyzed intracellular ROS by live cell imaging using H2DCFDA at various time points over 24h. We found that ROS in WT SMC treated with ELDL steadily increased until 4h and then declined. Conversely, RAGE−/− SMC showed no increase in ROS production throughout the experimental period (Fig. 6A and quantified in Fig. 6C). These results indicate that ELDL-RAGE interaction is involved in the upregulation of ROS in WT cells. We next analyzed intracellular calcium upon incubation with ELDL in WT and RAGE−/− SMC by live cell imaging using Fluo-8AM at various time points over 24h. In response to ELDL, WT SMC calcium peaked at 6h and then declined, while in RAGE−/− SMC, the intracellular calcium remained at baseline (Fig. 6B and quantified in Fig. 6D). The increase in calcium levels in WT SMC upon stimulation with ELDL was associated positively with increased cytosolic peroxide production as shown in Fig. 6A and quantified in Fig. 6C. In contrast, RAGE−/−SMC did not show increased calcium or H2O2 production upon stimulation with ELDL, suggesting that ELDL-RAGE signaling regulates ROS and calcium levels in a coordinated manner. Additionally, we found that amiloride, a known inhibitor of macropinocytosis 43 which completely inhibited uptake of ELDL in WT SMC (Fig. 4C), prevented calcium upregulation in WT SMC after 6h of incubation with ELDL (Fig. 6B/D). This strongly suggests that calcium upregulation in cells is associated with macropinocytosis-mediated uptake of ELDL in SMC. Lastly, we queried whether reducing of cytosolic calcium could block ELDL-macropinocytosis in SMC. We incubated ELDL with WT and RAGE−/− SMC pretreated with the calcium channel blocker lacidipine (30 μM for 1h) and analyzed foam cell formation after 4h and 24h. ELDL mediated foam cell formation was significantly blocked at 4h and 24h in WT, and in RAGE−/− SMC at 4h, upon pre-treatment with lacidipine (Fig. 6E and quantified in Fig. 6F). Taken together, our data shows that ELDL macropinocytosis in SMC is mostly a function of increased cytosolic calcium that is regulated, at least in part, by interaction of ELDL with RAGE as schematically summarized in Fig. 6G. However, since calcium is known to regulate cytosolic ROS levels and vice versa51, it is uncertain whether ELDL-macropinocytosis in SMC is primarily regulated by cytosolic calcium or ROS dependent mechanisms.
Figure 6. Macropinocytotic uptake of ELDL by SMC: role for ROS and intracellular calcium.

A. WT and RAGE−/− SMC were incubated with 10 μg/ml ELDL, and ROS expression was analyzed in live cells by staining with H2DCFDA at time points as indicated. B. WT and RAGE−/− SMC were incubated with 10 μg/ml ELDL and cytosolic calcium was analyzed in live cells by staining with Fluo-8AM at time points as indicated. WT SMC were also pretreated with amiloride for 1h prior to adding ELDL (amiloride was not removed prior to the addition of ELDL). C/D. Fluorescence intensity from A and B quantified using Image J. Data are represented as mean ± SD. E. WT and RAGE−/− SMC were incubated with 10 μg/ml ELDL after pretreated for 1h with the calcium channel inhibitor lacidipine (30 μM). Foam cell formation was analyzed by Oil Red O staining at 4h and 24h after ELDL loading. F. Total cellular cholesterol/protein from experiments shown in E. Data are represented as mean ± SD. (** p<0.01,* p<0.05). G. Schematic model of the role of RAGE in ELDL endocytosis in mouse aortic smooth muscle cells. PIP2 and PIP3 are plasma membrane inositides. PIP2 is phosphorylated to PIP3 by PI3K, and blocking PIP3 formation with PI3K inhibitors (LY294002, wortmannin) are known strategies to impair macropinocytosis. PI3K inhibitors partially prevented macropinocytosis of ELDL in SMC in contrast to the complete inhibition of macropinocytosis of ELDL in SMC deficient for RAGE treated with the PI3K inhibitors. ELDL-RAGE signaling down regulates PIP3; nevertheless, ROS and cellular Ca2+ levels are upregulated by ELDL in the SMC. Ca2+ is also an important determinant of macropinocytosis whose mechanism of action is unknown. Despite PIP3 inhibition by ELDL, we see unhindered macropinocytosis of ELDL, which suggests elevated cellular Ca2+ is potent enough to override the inhibition of PIP3 by ELDL via some unknown mechanisms.
Discussion
Early atherogenesis is characterized by the accumulation of intimal foam cells, which are often thought to be derived from the inflowing blood monocytes that differentiate into macrophages. But intimal foam cells may also be derived from SMC. The studies reported in this manuscript have focused exclusively on cultured primary murine SMC which become foam cells in response to enzyme-modified lipoproteins such as ELDL. We suspect that these mechanisms may also be operative in vivo.
The contribution of SMC to foam cells of the atheromatous plaque has been subject of recent studies. Identification of lesional SMC derived foam cell is problematic because lipid loaded SMC assume aspects of macrophage like cells. Even an excellent recent review did not appear to resolve this issue because of the difficulty of positively identifying the cell of origin of foam cells53. However, with the markers, miRNA 143/145 and myocardin, Vengrenyuk and colleagues were able to indicate that SMCs probably account for about 40% of the foam cells in mature plaques 13. This is consistent with relative contribution suggested by Allahverdian and colleagues10.
Given the likely importance of SMC foam cell formation in vivo, we have here employed culture experiments to explore how such foam cell formation may occur. We show that at low concentration, LDL modified by proteolytic and esterolytic enzymes (ELDL) is able to induce SMC foam cells, a response not seen in SMC with LDL, AcLDL or OxLDL, the latter two being modified LDLs readily taken up by macrophages via scavenger receptors. The enzymes used for the production of ELDL are trypsin and cholesterol esterase. Trypsin here is a model protease. If this process occurs in vivo, plasmin and metalloprotease, which have been detected in the vessel wall, are much more likely to be the operative enzymes. Cathepsins and chymase are also possible candidates. There are several candidates for the esterase in vivo such as the secretory pancreatic carboxyl ester lipase54 which has cholesterol esterolytic activity, and cholesterol ester lipase secreted by endothelial cells stimulated by OxLDL 55. Additionally, Maxfield and colleagues have demonstrated that an extracellular acid compartment allows for the hydrolysis of aggregated LDL by macrophages employing enzymes derived from lysosomes 56. These compartments are apparently dynamic allowing the release of some or all of their components, including esterase, protease and partially hydrolyzed LDL, that could provide the modified LDL similar to that studied here. That ELDL may be demonstrated in lesions was suggested by Torzewski and colleagues who raised monoclonal antibodies that react only with proteolysed apo B 22. This antibody recognized products in the vicinity of SMC in human coronary arteries.
When comparing aortic SMC and peritoneal macrophages, we found that ELDL uptake by SMC was not through mechanisms involving scavenger receptors in contrast to the typical scavenger receptor-mediated uptake of modified LDL by macrophages. Although, LOX-1, a major macrophage scavenger receptor for OxLDL was significantly up regulated in SMC treated with ELDL, it was not obligatorily required for ELDL uptake by the SMC. Normal SMC has low abundance of receptors for OxLDL57, 58. Interestingly, it was reported that only intimal SMC isolated from lesions but not medial SMC showed scavenger receptor activity58, 59 suggesting that scavenger receptor activity of intimal SMC might stem from local factors within the lesions. The causative factors are currently unknown, and our data suggest that ELDL could potentially mediate the increased expression of at least one scavenger receptor.
A primary conclusion of our work is that ELDL uptake by SMC occurs via macropinocytosis. Specific inhibitors have been employed to show that receptor mediated endocytosis, and caveolar function are not required for ELDL uptake. Also the concentration dependence of ELDL uptake is consistent with a fluid phase mechanism such as macropinocytosis. Macropinocytosis has been well established to account for the uptake of normal LDL by macrophages60 which we have shown in this study to also occur in aortic SMC. But higher concentrations (>2mg/ml) of normal LDL are required for these pathways than is required for the uptake of ELDL by SMC, for which concentrations as low as 10 μg/ml are sufficient. However, limitations of our study is the use of pharmacological inhibitors to various forms of ELDL uptake, and as with all experiments using pharmacological compounds, off target effects, known or unknown, cannot be excluded. Moreover, other potential mechanisms operating in SMC for the uptake of ELDL such as patocytosis, which was suggested as a mechanism for ELDL uptake in macrophages by Kruth et al61, can not be exluded since specific inhibitors of patocytosis are not available.
Our knowledge of macropinocytosis mechanisms is evolving. Calcium is suggested to be an inducer of macropinocytosis45, 52. In our studies, calcium channel blocker lacidipine abolished ELDL uptake in the WT SMC. Lacidipine (compared to atenolol) was previously shown to decrease an carotid artery atherosclerosis in the European Lacidipine Study on Atherosclerosis with a greater efficiency on carotid intima–media thickness and number of plaques per patient, despite a smaller reduction in ambulatory blood pressure. This suggests an anti-atherogenic action of lacidipine independent of its anti-hypertensive action 62. Inhibition of ELDL-induced SMC foam cells could be one reason for the reduced IMT and carotid atherosclerosis observed in this clinical trial with lacidipine.
RAGE is not required for the uptake of ELDL. However, RAGE does seem to influence some of the signals that participate in the regulation of factors thought to be involved in the macropinocytosis pathway. This is schematized in Fig. 6G. PIP3 is known to be involved macropinocytosis 46, 47, 63, but like RAGE, it is apparently not obligatory for ELDL-macropinocytosis. We demonstrate that pharmacological inhibition of PI3K, the enzyme responsible for PIP3 formation, was partially effective in preventing the uptake of ELDL in wild type SMC. On the other hand, ELDL uptake was abolished by PI3K inhibitors in RAGE deficient SMC, suggesting that RAGE regulates PI3K and PIP3 levels. PIP3 has pleiotropic functions beyond regulating macropinocytosis. For example, PIP3 is an important plasma membrane phospholipid that is involved in the phosphorylation and activation of Akt64. RAGE signaling has been shown to activate PI3K/PIP3/Akt pathway in some studies 65, 66, while other studies have shown RAGE inactivating Akt 48, 67. Apparently, different ligands activate RAGE differently with regard to its downstream Akt activation. In our studies, ELDL uptake by wild type SMC down regulates phosphorylated Akt (pAkt-Ser473) similar to that by LY294002 (PI3K inhibitor), but does not down regulate Akt in RAGE−/− SMC. Experiments with SMC pre-treated with 2 μM triciribine, a global inhibitor of Akt, indicated that Akt activity was not required for the normal uptake of ELDL and foam cell formation (supplementary Fig. IX). It is clear that the relationship between macropinocytosis of ELDL by SMC and RAGE influence on the PI3K/PIP3/Akt axis is a complex one that will depend on further exploration for clarification.
Although RAGE is not required for the macropinocytosis of ELDL, RAGE modifies the cellular responses to ELDL. We showed that upregulation of LOX-1 and uptake of oxLDL is primed by ELDL in a RAGE-dependent manner in SMC. RAGE is a multi-ligand receptor with a propensity for increased binding affinity for several ligands that oligomerize40. ELDL particles consist of fused and larger LDLs and may therefore bind and activate RAGE similar to what was previously shown for OxLDL68. While OxLDL binds RAGE on account of its AGE epitopes, ELDL differs significantly from OxLDL21 and it is not known whether ELDL has AGE epitopes. Given the mechanisms here explored, we can conclude that RAGE is not functioning as an endocytic receptor for ELDL uptake, but rather influences uptake as a cell surface signaling molecule. RAGE is strongly implicated in the pathology of atherosclerosis. Studies with Apoe, Rage- double knockout mice had shown significant protection from high fat diet induced atherosclerosis 41, 69 and diabetes induced atherosclerosis 70. Our data suggest that impaired intimal SMC foam cell formation could be one underlying mechanism for the reduced atherosclerosis seen in Rage null/Apoe null mice. In view of the complex interaction between macropinocytosis and RAGE participation, one possibility that should be entertained is that there are two pathways for the macropinocytosis of ELDL, one of which is influenced by RAGE. For example in the macropinocytosis of normal LDL by macrophages, Kruth and colleagues have suggested that this may be by either a microtubule dependent or independent pathway60.
In summary, there is compelling in vitro evidence that ELDL, of the modified LDLs studied, is the preferred cholesterol-containing particle taken up by SMC causing formation of foam cells. Whether ELDL induces the same changes in intimal SMC in vivo, and thereby possibly accelerates atherosclerosis, is not known. Conditional knockouts of gene participants in the pathways here described could provide further insight on the contribution of each of these cells. A better understanding of these processes may lead to potential therapeutic options to inhibit ELDL uptake and smooth muscle foam cell formation.
Supplementary Material
Highlights.
Cultured SMC are readily converted into foam cells by incubation with LDL that has been modified by proteolysis and cholesterol esterolysis (enzymatically modified LDL, ELDL).
ELDL uptake in SMC is dependent on macropinocytosis rather than receptors.
Exposure of SMC to ELDL enhances their uptake of oxidized LDL, which is not readily taken up on its own. Thus, these various modifications of LDL in the vessel wall may synergize to load SMC with lipid.
Although there is a good deal of evidence that the vessel wall contains enzymatically modified LDL (ELDL), future studies are needed to investigate the quantitatively contribution of SMC to the complement of foam cells in atherosclerotic lesons.
Acknowledgments
The RAGE null mice were a generous gift from Dr. Ann Marie Schmidt, NYU, New York, NY.
Source of Funding: This work was supported by the National Health Lung and Blood Institute (1R01HL4821 to MAHB).
Non-standard Abbreviations and Acronyms
- ELDL
Enzyme-Modified Low Density Lipoprotein
- SMC
Smooth Muscle Cells
- RAGE
Receptor for Advanced Glycosylation End Products
- AcLDL
Acetylated Low Density Lipoprotein
- OxLDL
Oxidized Low Density Lipoprotein
- MMP
Matrix Metalloproteinases
- LOX1
Lectin Like Oxidized Low Density Lipoprotein Receptor1
- H2DCFDA
2′,7′-dichlorodihydrofluorescein diacetate
- DiI-LDL
DiI Labeled Low Density Lipoprotein
Footnotes
Disclosures: None, and no conflicts of interest.
Contributor Information
Bijoy Chellan, Department of Medicine, University of Chicago, IL 60637.
Catherine A. Reardon, Department of Pathology, University of Chicago, IL 60637
Godfrey S. Getz, Department of Pathology, University of Chicago, IL 60637
Marion A. Hofmann Bowman, Department of Medicine, University of Chicago, IL 60637
References
- 1.Nakashima Y, Wight TN, Sueishi K. Early atherosclerosis in humans: Role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res. 2008;79:14–23. doi: 10.1093/cvr/cvn099. [DOI] [PubMed] [Google Scholar]
- 2.Orekhov AN, Andreeva ER, Krushinsky AV, Novikov ID, Tertov VV, Nestaiko GV, Khashimov KA, Repin VS, Smirnov VN. Intimal cells and atherosclerosis. Relationship between the number of intimal cells and major manifestations of atherosclerosis in the human aorta. Am J Pathol. 1986;125:402–415. [PMC free article] [PubMed] [Google Scholar]
- 3.Bauriedel G, Hutter R, Welsch U, Bach R, Sievert H, Lüderitz B. Role of smooth muscle cell death in advanced coronary primary lesions: Implications for plaque instability. Cardiovasc Res. 1999;41:480–488. doi: 10.1016/s0008-6363(98)00318-6. [DOI] [PubMed] [Google Scholar]
- 4.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linton MF, Fazio S. Macrophages, inflammation, and atherosclerosis. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S35–40. doi: 10.1038/sj.ijo.0802498. [DOI] [PubMed] [Google Scholar]
- 6.Mietus-Snyder M, Gowri MS, Pitas RE. Class a scavenger receptor up-regulation in smooth muscle cells by oxidized low density lipoprotein. Enhancement by calcium flux and concurrent cyclooxygenase-2 up-regulation. J Biol Chem. 2000;275:17661–17670. doi: 10.1074/jbc.275.23.17661. [DOI] [PubMed] [Google Scholar]
- 7.Li W, Febbraio M, Reddy SP, Yu DY, Yamamoto M, Silverstein RL. Cd36 participates in a signaling pathway that regulates ros formation in murine vsmcs. J Clin Invest. 2010;120:3996–4006. doi: 10.1172/JCI42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi HY, Rahmani M, Wong BW, Allahverdian S, McManus BM, Pickering JG, Chan T, Francis GA. Atp-binding cassette transporter a1 expression and apolipoprotein a-i binding are impaired in intima-type arterial smooth muscle cells. Circulation. 2009;119:3223–3231. doi: 10.1161/CIRCULATIONAHA.108.841130. [DOI] [PubMed] [Google Scholar]
- 10.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 11.Katsuda S, Boyd HC, Fligner C, Ross R, Gown AM. Human atherosclerosis. Iii. Immunocytochemical analysis of the cell composition of lesions of young adults. Am J Pathol. 1992;140:907–914. [PMC free article] [PubMed] [Google Scholar]
- 12.Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998;97:2307–2315. doi: 10.1161/01.cir.97.23.2307. [DOI] [PubMed] [Google Scholar]
- 13.Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, Fisher EA. Cholesterol loading reprograms the microrna-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546. doi: 10.1161/ATVBAHA.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the abc1 promoter by the liver x receptor/retinoid x receptor. J Biol Chem. 2000;275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 15.Feng J, Han J, Pearce SF, Silverstein RL, Gotto AM, Hajjar DP, Nicholson AC. Induction of cd36 expression by oxidized ldl and il-4 by a common signaling pathway dependent on protein kinase c and ppar-gamma. J Lipid Res. 2000;41:688–696. [PubMed] [Google Scholar]
- 16.Hayden JM, Brachova L, Higgins K, Obermiller L, Sevanian A, Khandrika S, Reaven PD. Induction of monocyte differentiation and foam cell formation in vitro by 7-ketocholesterol. J Lipid Res. 2002;43:26–35. [PubMed] [Google Scholar]
- 17.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 18.Rong JX, Kusunoki J, Oelkers P, Sturley SL, Fisher EA. Acyl-coenzymea (coa):Cholesterol acyltransferase inhibition in rat and human aortic smooth muscle cells is nontoxic and retards foam cell formation. Arterioscler Thromb Vasc Biol. 2005;25:122–127. doi: 10.1161/01.ATV.0000148202.49842.3b. [DOI] [PubMed] [Google Scholar]
- 19.Ricciarelli R, Zingg JM, Azzi A. Vitamin e reduces the uptake of oxidized ldl by inhibiting cd36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation. 2000;102:82–87. doi: 10.1161/01.cir.102.1.82. [DOI] [PubMed] [Google Scholar]
- 20.Klouche M, Rose-John S, Schmiedt W, Bhakdi S. Enzymatically degraded, nonoxidized ldl induces human vascular smooth muscle cell activation, foam cell transformation, and proliferation. Circulation. 2000;101:1799–1805. doi: 10.1161/01.cir.101.15.1799. [DOI] [PubMed] [Google Scholar]
- 21.Bhakdi S, Dorweiler B, Kirchmann R, Torzewski J, Weise E, Tranum-Jensen J, Walev I, Wieland E. On the pathogenesis of atherosclerosis: Enzymatic transformation of human low density lipoprotein to an atherogenic moiety. J Exp Med. 1995;182:1959–1971. doi: 10.1084/jem.182.6.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torzewski M, Klouche M, Hock J, Messner M, Dorweiler B, Torzewski J, Gabbert HE, Bhakdi S. Immunohistochemical demonstration of enzymatically modified human ldl and its colocalization with the terminal complement complex in the early atherosclerotic lesion. Arterioscler Thromb Vasc Biol. 1998;18:369–378. doi: 10.1161/01.atv.18.3.369. [DOI] [PubMed] [Google Scholar]
- 23.Kapinsky M, Torzewski M, Büchler C, Duong CQ, Rothe G, Schmitz G. Enzymatically degraded ldl preferentially binds to cd14(high) cd16(+) monocytes and induces foam cell formation mediated only in part by the class b scavenger-receptor cd36. Arterioscler Thromb Vasc Biol. 2001;21:1004–1010. doi: 10.1161/01.atv.21.6.1004. [DOI] [PubMed] [Google Scholar]
- 24.Han SR, Momeni A, Strach K, Suriyaphol P, Fenske D, Paprotka K, Hashimoto SI, Torzewski M, Bhakdi S, Husmann M. Enzymatically modified ldl induces cathepsin h in human monocytes: Potential relevance in early atherogenesis. Arterioscler Thromb Vasc Biol. 2003;23:661–667. doi: 10.1161/01.ATV.0000063614.21233.BF. [DOI] [PubMed] [Google Scholar]
- 25.Sakurada T, Orimo H, Okabe H, Noma A, Murakami M. Purification and properties of cholesterol ester hydrolase from human aortic intima and media. Biochim Biophys Acta. 1976;424:204–212. [PubMed] [Google Scholar]
- 26.Hakala JK, Oksjoki R, Laine P, Du H, Grabowski GA, Kovanen PT, Pentikäinen MO. Lysosomal enzymes are released from cultured human macrophages, hydrolyze ldl in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2003;23:1430–1436. doi: 10.1161/01.ATV.0000077207.49221.06. [DOI] [PubMed] [Google Scholar]
- 27.Kremen M, Krishnan R, Emery I, Hu JH, Slezicki KI, Wu A, Qian K, Du L, Plawman A, Stempien-Otero A, Dichek DA. Plasminogen mediates the atherogenic effects of macrophage-expressed urokinase and accelerates atherosclerosis in apoe-knockout mice. Proc Natl Acad Sci U S A. 2008;105:17109–17114. doi: 10.1073/pnas.0808650105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossignol P, Luttun A, Martin-Ventura JL, Lupu F, Carmeliet P, Collen D, Anglès-Cano E, Lijnen HR. Plasminogen activation: A mediator of vascular smooth muscle cell apoptosis in atherosclerotic plaques. J Thromb Haemost. 2006;4:664–670. doi: 10.1111/j.1538-7836.2005.01765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torzewski M, Suriyaphol P, Paprotka K, Spath L, Ochsenhirt V, Schmitt A, Han SR, Husmann M, Gerl VB, Bhakdi S, Lackner KJ. Enzymatic modification of low-density lipoprotein in the arterial wall: A new role for plasmin and matrix metalloproteinases in atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:2130–2136. doi: 10.1161/01.ATV.0000144016.85221.66. [DOI] [PubMed] [Google Scholar]
- 30.Sukhova GK, Zhang Y, Pan JH, Wada Y, Yamamoto T, Naito M, Kodama T, Tsimikas S, Witztum JL, Lu ML, Sakara Y, Chin MT, Libby P, Shi GP. Deficiency of cathepsin s reduces atherosclerosis in ldl receptor-deficient mice. J Clin Invest. 2003;111:897–906. doi: 10.1172/JCI14915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oörni K, Sneck M, Brömme D, Pentikäinen MO, Lindstedt KA, Mäyränpää M, Aitio H, Kovanen PT. Cysteine protease cathepsin f is expressed in human atherosclerotic lesions, is secreted by cultured macrophages, and modifies low density lipoprotein particles in vitro. J Biol Chem. 2004;279:34776–34784. doi: 10.1074/jbc.M310814200. [DOI] [PubMed] [Google Scholar]
- 32.Kovanen PT. Chymase-containing mast cells in human arterial intima: Implications for atherosclerotic disease. Heart Vessels. 1997;(Suppl 12):125–127. [PubMed] [Google Scholar]
- 33.Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, Masaki T. An endothelial receptor for oxidized low-density lipoprotein. Nature. 1997;386:73–77. doi: 10.1038/386073a0. [DOI] [PubMed] [Google Scholar]
- 34.Morawietz H. Lox-1 and atherosclerosis: Proof of concept in lox-1-knockout mice. Circ Res. 2007;100:1534–1536. doi: 10.1161/CIRCRESAHA.107.101105. [DOI] [PubMed] [Google Scholar]
- 35.Smith EB. Transport, interactions and retention of plasma proteins in the intima: The barrier function of the internal elastic lamina. Eur Heart J. 1990;11(Suppl E):72–81. doi: 10.1093/eurheartj/11.suppl_e.72. [DOI] [PubMed] [Google Scholar]
- 36.Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X. Srebp-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci U S A. 1993;90:11603–11607. doi: 10.1073/pnas.90.24.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhaliwal BS, Steinbrecher UP. Cholesterol delivered to macrophages by oxidized low density lipoprotein is sequestered in lysosomes and fails to efflux normally. J Lipid Res. 2000;41:1658–1665. [PubMed] [Google Scholar]
- 38.Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11:3061–3072. doi: 10.2174/1381612054865064. [DOI] [PubMed] [Google Scholar]
- 39.Murphy JE, Vohra RS, Dunn S, Holloway ZG, Monaco AP, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Oxidised ldl internalisation by the lox-1 scavenger receptor is dependent on a novel cytoplasmic motif and is regulated by dynamin-2. J Cell Sci. 2008;121:2136–2147. doi: 10.1242/jcs.020917. [DOI] [PubMed] [Google Scholar]
- 40.Herold K, Moser B, Chen Y, Zeng S, Yan SF, Ramasamy R, Emond J, Clynes R, Schmidt AM. Receptor for advanced glycation end products (rage) in a dash to the rescue: Inflammatory signals gone awry in the primal response to stress. J Leukoc Biol. 2007;82:204–212. doi: 10.1189/jlb.1206751. [DOI] [PubMed] [Google Scholar]
- 41.Yan SF, Ramasamy R, Schmidt AM. The rage axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–853. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, Rutledge R, Lin B, Amoscato AA, Zeh HJ, Lotze MT. Rage (receptor for advanced glycation endproducts), rage ligands, and their role in cancer and inflammation. J Transl Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivanov AI. Pharmacological inhibition of endocytic pathways: Is it specific enough to be useful? Methods Mol Biol. 2008;440:15–33. doi: 10.1007/978-1-59745-178-9_2. [DOI] [PubMed] [Google Scholar]
- 44.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Falcone S, Cocucci E, Podini P, Kirchhausen T, Clementi E, Meldolesi J. Macropinocytosis: Regulated coordination of endocytic and exocytic membrane traffic events. J Cell Sci. 2006;119:4758–4769. doi: 10.1242/jcs.03238. [DOI] [PubMed] [Google Scholar]
- 46.Hoeller O, Bolourani P, Clark J, Stephens LR, Hawkins PT, Weiner OD, Weeks G, Kay RR. Two distinct functions for pi3-kinases in macropinocytosis. J Cell Sci. 2013;126:4296–4307. doi: 10.1242/jcs.134015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veltman DM, Lemieux MG, Knecht DA, Insall RH. Pip3-dependent macropinocytosis is incompatible with chemotaxis. J Cell Biol. 2014;204:497–505. doi: 10.1083/jcb.201309081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu P, Lai D, Lu P, Gao J, He H. Erk and akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. 2012;29:613–618. doi: 10.3892/ijmm.2012.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basta G, Lazzerini G, Del Turco S, Ratto GM, Schmidt AM, De Caterina R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler Thromb Vasc Biol. 2005;25:1401–1407. doi: 10.1161/01.ATV.0000167522.48370.5e. [DOI] [PubMed] [Google Scholar]
- 50.Kook SY, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I. Aβ1-42-rage interaction disrupts tight junctions of the blood-brain barrier via ca2+-calcineurin signaling. J Neurosci. 2012;32:8845–8854. doi: 10.1523/JNEUROSCI.6102-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gordeeva AV, Zvyagilskaya RA, Labas YA. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry (Mosc) 2003;68:1077–1080. doi: 10.1023/a:1026398310003. [DOI] [PubMed] [Google Scholar]
- 52.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. Amiloride inhibits macropinocytosis by lowering submembranous ph and preventing rac1 and cdc42 signaling. J Cell Biol. 2010;188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shamir R, Johnson WJ, Morlock-Fitzpatrick K, Zolfaghari R, Li L, Mas E, Lombardo D, Morel DW, Fisher EA. Pancreatic carboxyl ester lipase: A circulating enzyme that modifies normal and oxidized lipoproteins in vitro. J Clin Invest. 1996;97:1696–1704. doi: 10.1172/JCI118596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li F, Hui DY. Synthesis and secretion of the pancreatic-type carboxyl ester lipase by human endothelial cells. Biochem J. 1998;329( Pt 3):675–679. doi: 10.1042/bj3290675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haka AS, Grosheva I, Chiang E, Buxbaum AR, Baird BA, Pierini LM, Maxfield FR. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol Biol Cell. 2009;20:4932–4940. doi: 10.1091/mbc.E09-07-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mietus-Snyder M, Friera A, Glass CK, Pitas RE. Regulation of scavenger receptor expression in smooth muscle cells by protein kinase c: A role for oxidative stress. Arterioscler Thromb Vasc Biol. 1997;17:969–978. doi: 10.1161/01.atv.17.5.969. [DOI] [PubMed] [Google Scholar]
- 58.Gong Q, Pitas RE. Synergistic effects of growth factors on the regulation of smooth muscle cell scavenger receptor activity. J Biol Chem. 1995;270:21672–21678. doi: 10.1074/jbc.270.37.21672. [DOI] [PubMed] [Google Scholar]
- 59.Li H, Freeman MW, Libby P. Regulation of smooth muscle cell scavenger receptor expression in vivo by atherogenic diets and in vitro by cytokines. J Clin Invest. 1995;95:122–133. doi: 10.1172/JCI117628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, Combs CA, Malide D, Zhang WY. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2005;280:2352–2360. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- 61.Kruth HS, Zhang WY, Skarlatos SI, Chao FF. Apolipoprotein b stimulates formation of monocyte-macrophage surface-connected compartments and mediates uptake of low density lipoprotein-derived liposomes into these compartments. J Biol Chem. 1999;274:7495–7500. doi: 10.1074/jbc.274.11.7495. [DOI] [PubMed] [Google Scholar]
- 62.Zanchetti A, Bond MG, Hennig M, Neiss A, Mancia G, Dal Palù C, Hansson L, Magnani B, Rahn KH, Reid JL, Rodicio J, Safar M, Eckes L, Rizzini P investigators ELSoA. Calcium antagonist lacidipine slows down progression of asymptomatic carotid atherosclerosis: Principal results of the european lacidipine study on atherosclerosis (elsa), a randomized, double-blind, long-term trial. Circulation. 2002;106:2422–2427. doi: 10.1161/01.cir.0000039288.86470.dd. [DOI] [PubMed] [Google Scholar]
- 63.Posor Y, Eichhorn-Gruenig M, Puchkov D, Schöneberg J, Ullrich A, Lampe A, Müller R, Zarbakhsh S, Gulluni F, Hirsch E, Krauss M, Schultz C, Schmoranzer J, Noé F, Haucke V. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature. 2013;499:233–237. doi: 10.1038/nature12360. [DOI] [PubMed] [Google Scholar]
- 64.Song G, Ouyang G, Bao S. The activation of akt/pkb signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rai V, Touré F, Chitayat S, Pei R, Song F, Li Q, Zhang J, Rosario R, Ramasamy R, Chazin WJ, Schmidt AM. Lysophosphatidic acid targets vascular and oncogenic pathways via rage signaling. J Exp Med. 2012;209:2339–2350. doi: 10.1084/jem.20120873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng CL, Tang Y, Zheng Z, Liu X, Ye ZC, Wang C, Lou TQ. Advanced glycation end-products activate the renin-angiotensin system through the rage/pi3-k signaling pathway in podocytes. Clin Invest Med. 2012;35:E282. doi: 10.25011/cim.v35i5.18701. [DOI] [PubMed] [Google Scholar]
- 67.Hou X, Hu Z, Xu H, Xu J, Zhang S, Zhong Y, He X, Wang N. Advanced glycation endproducts trigger autophagy in cadiomyocyte via rage/pi3k/akt/mtor pathway. Cardiovasc Diabetol. 2014;13:78. doi: 10.1186/1475-2840-13-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun L, Ishida T, Yasuda T, Kojima Y, Honjo T, Yamamoto Y, Yamamoto H, Ishibashi S, Hirata K, Hayashi Y. Rage mediates oxidized ldl-induced pro-inflammatory effects and atherosclerosis in non-diabetic ldl receptor-deficient mice. Cardiovasc Res. 2009;82:371–381. doi: 10.1093/cvr/cvp036. [DOI] [PubMed] [Google Scholar]
- 69.Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM. Vascular and inflammatory stresses mediate atherosclerosis via rage and its ligands in apoe−/− mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soro-Paavonen A, Watson AM, Li J, Paavonen K, Koitka A, Calkin AC, Barit D, Coughlan MT, Drew BG, Lancaster GI, Thomas M, Forbes JM, Nawroth PP, Bierhaus A, Cooper ME, Jandeleit-Dahm KA. Receptor for advanced glycation end products (rage) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.