

Abstract
Objective
CD36 is a scavenger and antiangiogenic receptor that is important in atherothrombotic diseases, diabetes, cancer and obesity. Lysophosphatidic acid (LPA), a phospholipid signaling mediator, abolishes endothelial cell responses to antiangiogenic proteins containing thrombospondin type 1 homology domains by down-regulating endothelial CD36 transcription via protein kinase D1 (PKD-1) signaling. We aimed to understand mechanisms by which LPA-mediated angiogenic signaling is integrated to regulate CD36 transcription and endothelial cell function via a nuclear transcriptional complex.
Approach and Results
Microvascular endothelial cells (MVECs) expressing CD36 were used for studying angiogenic signaling and CD36 transcription. Gene transfection and transduction, RT-qPCR, avidin–biotin-conjugated DNA-binding assay, chromatin immunoprecipitation assay, co-immunoprecipitation, proximal ligation assay and immunofluorescence microscopy showed that LPA-mediated CD36 transcriptional repression involved PKD-1 signaling mediated formation of FoxO1-HDAC7 complex in the nucleus. Unexpectedly, turning off CD36 transcription initiated reprogramming MVECs to express ephrin B2 (EFNB2), a critical “molecular signature” involved in angiogenesis and arteriogenesis. Spheroid-based angiogenesis and in vivo Matrigel angiogenesis assays indicated that angiogenic branching morphogenesis and in vivo angiogenesis were dependent on PKD-1 signaling. A mouse tumor angiogenesis model revealed enhanced PKD-1 signaling and expression of EFNB2 and smooth muscle actin (SMA) in neovessels of Lewis Lung Carcinomas, along with low CD36 expression or CD36 deficiency.
Conclusions
LPA/PKD-1 signaling leads to nuclear accumulation of HDAC7, where it interacts with FoxO1 to suppress endothelial CD36 transcription and mediates silencing of antiangiogenic switch, resulting in proangiogenic and proarteriogenic reprogramming. Targeting this signaling cascade could be a novel approach for ischemic cardiovascular disease and cancer.
Introduction
A dynamic balance between pro- and anti-angiogenic signals determines neo-vessel formation during physiological and pathological conditions. Broadly and constitutively expressed in microvascular beds, CD36 functions as a receptor that can initiate an antiangiogenic response by interaction with thrombospondin-1 and related proteins containing thrombospondin type 1 repeat (TSR) domains1–4. TSR binding to CD36 provides a “switch” that blunts vascular endothelial growth factor receptor signals5, 6 and converts growth factor–mediated proangiogenic responses into antiangiogenic, proapoptotic responses1–4, 6–9. Diminishing CD36 expression, for example in tumor vasculature, may tip the balance in favor of proangiogenic responses under certain pathological circumstances.
CD36 transcription in monocyte/macrophages, adipocytes and myocytes is tightly regulated by various cellular stimuli including cytokines, lipids and lipoproteins, reactive oxygen species, and differentiation and adhesion events10. The expression is also regulated post-transcriptionally. Accelerated atherosclerosis in diabetics has been linked to increased translation of macrophage CD36 transcripts under high glucose conditions11. Despite the biological importance of CD36 in angiogenesis, little is known regarding how endothelial cell-specific CD36 transcription is regulated or how that might relate to endothelial cell heterogeneity and function. In retinal MVECs, CD36 transcription is up-regulated in response to hypoxia via HIF-1 transcription factor12. The antiangiogenic pathway triggered by HIF-1 signaling counteracts hypoxia-driven proangiogenic pathways, providing a “homeostatic brake” to prevent pathological neovascularization. Recently we showed that LPA, a phospholipid signaling mediator and angiogenic factor13–19, led to long-term CD36 transcriptional repression via PKD-1 signaling in MVECs to promote angiogenesis7. The precise mechanisms, however, by which LPA mediated PKD-1 signaling regulates CD36 transcription and functions remain largely unknown.
PKD-1 is a serine/threonine protein kinase that is classified as a member of the calcium/calmodulin-dependent protein kinase family. PKD-1-initiated signaling is essential for angiogenesis, inflammatory responses, cardiac hypertrophy and tumor progression, and related to its functions in regulating cell proliferation and death, migration, and differentiation7, 20–27. We have hypothesized that PKD-1 signaling triggered by LPA receptors initiates assembly of a nuclear transcriptional complex, which suppresses CD36 transcription and thereby turns off the anti-angiogenic switch. We found that Forkhead box protein O1(FoxO1) controls basal MVEC CD36 transcription and that PKD-1 signaling induces recruitment of a co-repressor, nuclear receptor co-repressor 1 (NCoR1), and a specific histone deacetylase (HDAC), HDAC7, to the FoxO1 nuclear complex, resulting in CD36 transcriptional repression. Intriguingly and unexpectedly, down-regulation of CD36 transcription was associated with reprogramming of MVEC transcription towards pro-angiogenic and arteriogenic gene expression, which is partially dependent on CD36 expression. Furthermore, the activation of this signaling pathway in the tumor vasculature of mice could be associated with CD36 deficiency and pro-angiogenic and pro-arteriogenic responses in the tumor microenvironment. Our study suggests that there is a plasticity in EC differentiation and postnatal angiogenesis, which is regulated by the LPA/PKD-1-FoxO1-CD36 signaling pathway. Targeting this pathway could provide a novel therapeutic strategy in ischemic heart and vascular diseases, cancer, and other disorders such as diabetes and obesity.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
CD36 transcription in endothelial cells is regulated by histone deacetylase 7
HMVECs transfected with a luciferase reporter plasmid (pGL3-CD36-luc) encoding a 3519 bp sequence of the CD36 gene immediately proximal to the transcriptional start site12 showed significant basal luciferase activity (Figure 1A) compared to cells transfected with the control pGL3 plasmid. Activity was decreased by over 80% after LPA exposure for 24 hrs (p < 0.01). Moreover, CD36 transcriptional repression was long lasting; removal of LPA from MVEC cultures after 24 hrs followed by reconstitution in complete media for 48 hrs did not restore the CD36 transcription (Figure 1B). There was also a significant down-regulation of CD36 in primary human cardiac MVEC (HMVEC-C), which was not seen in cells treated with a pharmacologic inhibitor of the G protein coupled LPA receptors LPA1 and LPA3 (Figure 1C). To determine whether this long lasting effect was associated with chromatin remodeling, we assessed CD36 promoter specific histone acetylation status by chromatin immunoprecipitation analyses and found that nucleosome histone H3K9 enrichment in the CD36 promoter was reduced in response to LPA for 24 hrs (Figure 1D), suggesting that CD36 transcriptional repression by LPA may be regulated by histone deacetylases.
Figure 1.
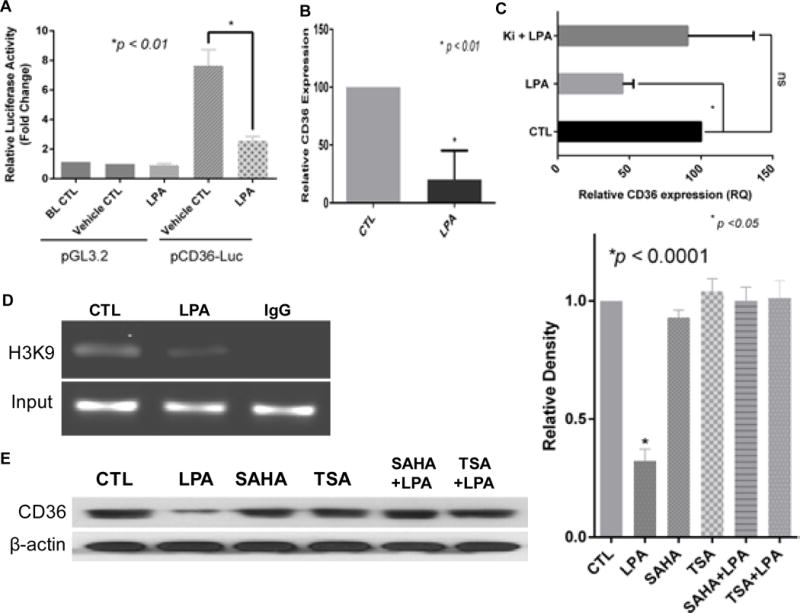
LPA-mediated sustained CD36 transcriptional repression may be associated with histone deacetylase activity in endothelial cells. A, LPA exposure suppresses CD36 promoter activity. Primary HMVECs were transfected with reporter constructs containing the CD36 proximal promoter fused to the pGL3-basic (pCD36-luc) for 24 hrs with co-transfection of pRL Renilla luciferase for normalization. The cells were then exposed to LPA for 24 hrs before measurement of luciferase activity with Dual-Luciferase® Reporter Assay System. Promoter activity was expressed as the fold-change in relative luciferase activity compared with pGL3.2 basic control (mean ± S.E. of three independent experiments). B, LPA mediates sustained CD36 mRNA downregulation. Primary HMVECs were exposed to LPA (10 μM) for 24 hrs, and then LPA was removed and replaced with complete media for 48 hrs. The mRNA level was assayed with quantitative real time RT-PCR. Triplicate experiments were performed (p <0.01). C, LPA signaling significantly reduces CD36 mRNA expression in cardiac microvascular endothelial cells (HMVEC-C). Primary HMVEC-Cs (passage 3) were exposed to LPA (10 μM) for 24 hrs or pretreated with LPA receptor1,3 antagonist Ki16425 (2 μM) for 30 min followed by LPA (10 μM) for 24 hrs. Total RNA was obtained for quantitative real time RT-PCR. D, ChIP analyses targeting the CD36 proximal promoter in LPA-treated (10 μM, 24 hrs) HMVECs using antibodies against AcH3 (K9). Input shows DNA isolated from the lysate before immunoprecipitation (10% of the total chromatin was used for the PCR reactions). Lane labeled “IgG” is ChIP performed with a non-immune IgG. E, CD36 expression in primary HMVECs treated with LPA and histone deacetylase inhibitors suberanilohydroxamic acid (SAHA) or trichostatin (TSA). The HMVECs were pre-exposed to 500 nM of SAHA or 500 ng/ml of TSA for 30 min followed with 10 μM of LPA for 24 hrs. Cell lysates were subjected to Western blots for CD36 protein expression (right panel). Densitometry was performed and relative expression levels calculated for triplicate experiments (left panel).
Class IIa HDACs including HDAC5 and HDAC7 are signal-dependent modulators of transcription28 and previous studies indicated that HDAC529 and HDAC730 play roles in PKD signaling and gene transcription that regulates EC functions30, 31. We hypothesized that LPA suppression of CD36 transcription was via either one or both of these HDACs. To test this hypothesis we pretreated HMVECs with class IIa HDAC inhibitors, suberoylanilide hydroxamic acid or trichostatin A, and then exposed them to LPA. Both inhibitors abolished LPA-mediated down-regulation of CD36 protein expression (Figure 1E). By using immunofluorescence microscopy and biochemical cell fractionation we found that LPA exposure increased the nuclear accumulation of HDAC7 but not HDAC5 (Figure 2A).
Figure 2.
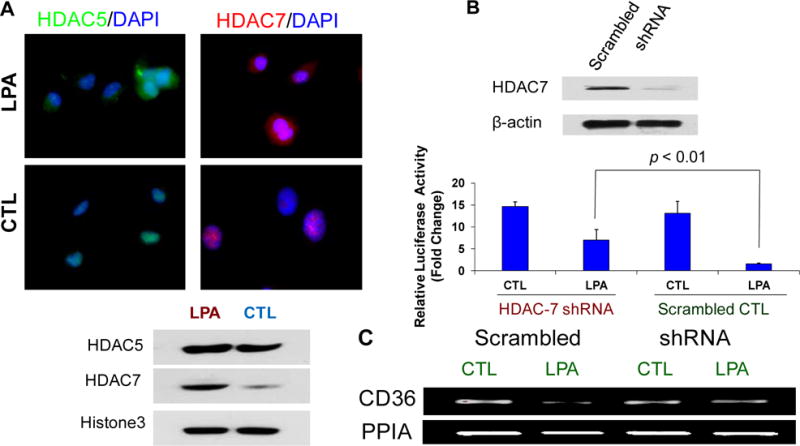
HDAC7 regulates CD36 transcription in endothelial cells. A, Nuclear HDAC7 translocation is increased in response to LPA exposure. MVECs were exposed to LPA (10 μM) for 24 hrs and then incubated with antibodies to HDAC5 or HADC7 followed by Alexa fluor® conjugated secondary antibodies. Cells were co-stained with DAPI to mark nuclei. In some studies cells were fractionated and the nuclear fractions isolated and subjected to Western blots for HDAC5 or HDAC7 expression. Representative results are shown from triplicate experiments. B., Knocking down endogenous HDAC7 attenuates LPA-mediated CD36 transcriptional repression in HMVECs. Western blots showed that HDAC 7 shRNA plasmids transfection effectively reduced the endogenous HDAC7 expression (upper panel). Cells transfected with HDAC7 shRNA plasmids or scrambled control were co-transfected with pGL3-CD36-luc and Renilla plasmids for 48 hrs. The cells were exposed to LPA (10 μM) or vehicle control for 24 hrs and luciferase activity measured as in Figure 1A (p < 0.01). C, HMVECs were transfected with a pool of HDAC7 shRNA plasmids for 28 hrs, and the cells were exposed to LPA (10 μM) for 18 hrs. The cells were harvested and total RNA was isolated for RT-PCR for CD36 mRNA expression. The image shows a representative gel from triplicate experiments.
To define the role of specific HDACs in LPA-mediated CD36 transcriptional repression, we knocked down HDAC7 expression in human MVECs transfected with pGL3-CD36-luc by using a specific shRNA (Figure 2B upper panel) and showed that LPA-inhibited CD36 promoter activity was partially attenuated (Figure 2B lower panel; p < 0.01). Moreover, LPA-mediated CD36 mRNA down-regulation was blocked by transfection with shRNA plasmids targeting HDAC7 (Figure 2C). Knockdown of HDAC5 had no effect in these assays (data not shown).
Transcription factor FoxO1 is required for constitutive CD36 transcription in MVECs
We next sought to identify transcription factors responsible for endothelial cell (EC) CD36 transcription. The analysis of the CD36 gene promoter region revealed three putative FoxO-responsive elements (FHRE) located between −3500 to −250 bp relative to transcription start site (Figure 3A upper panel). FoxO1, a member of Forkhead transcription factor family, is known to be important in vascular development and postnatal angiogenesis32, 33. We deleted proximal promoter sequences containing FHRE at 258 bp relative to transcriptional start site in the pGL3-CD36-luc plasmid. The deletion of this regulatory element significantly reduced basal promoter activity in HMVECs (Figure 3A lower panel). Similar results were seen when this FHRE was subjected to site-directed mutagenesis (Figure 3A lower panel). Chromatin immunoprecipitation analyses revealed that endogenous FoxO1 in HMVECs bound specifically to a sequence within regulatory regions of the CD36 gene (Figure 3B upper panel). Whereas antibody to the transcription factor the nuclear receptor subfamily 4 group A member 1 used as a control did not precipitate CD36 promoter sequences in these experiments, demonstrating specificity.
Figure 3.
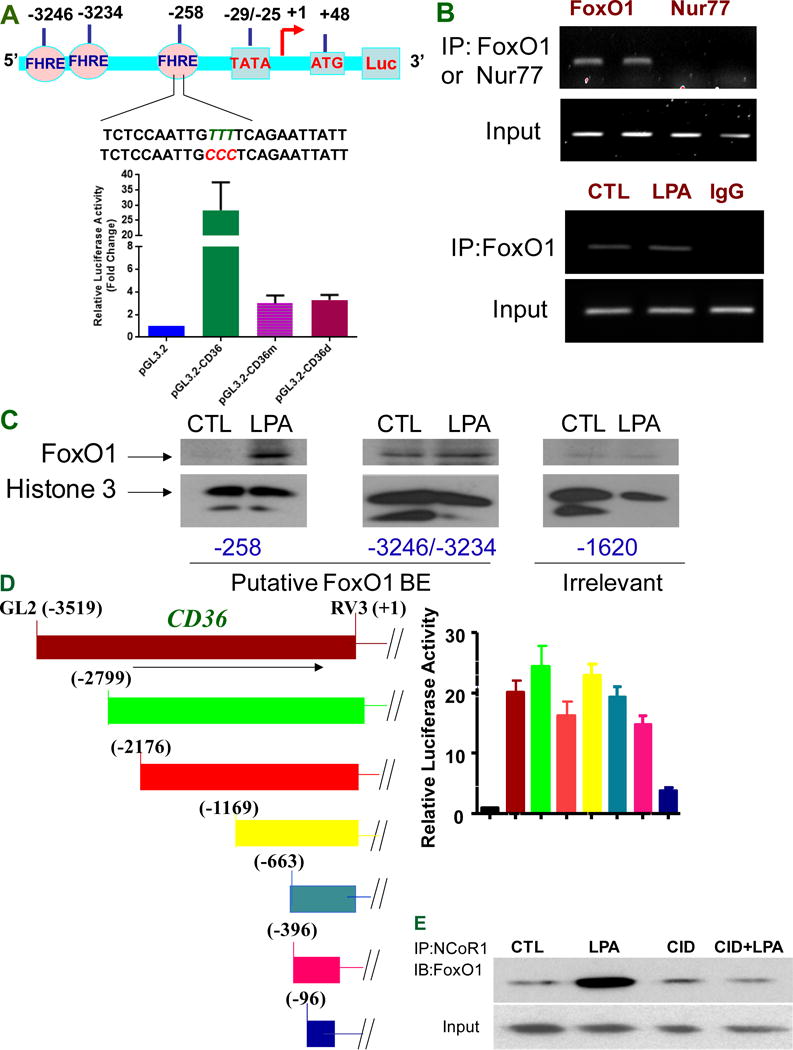
FoxO1 transcriptional activities are associated with CD36 transcription. A, FoxO1 regulates activity of the CD36 promoter through recognition of FoxO1 responsive element (FHRE) at the proximal promoter. The promoter analysis shows three putative forkhead response elements (FHREs), respectively located at −258, −3234, and −3246 relative to the transcription start site. The TTT were mutated into CCC in FHRE sequence contained at proximal promoter (upper panel). Proximal promoter mutagenesis at −258 (pGL.3.2-CD36m) or deletion of proximal promoter (pGL3.2-CD36d) attenuated CD36 promoter activity (lower panel). Data represent mean ± SD of three independent experiments. B, FoxO1 binds CD36 proximal promoter (upper panel) but LPA does not enhance this binding in vivo (lower panel). HMVECs were exposed to LPA (10 μM) for 24 hrs and subjected to Chromatin immunoprecipitation analyses. Nur77 (nuclear receptor subfamily 4 group A member 1) is used as a control. Representative results are shown in triplicate experiments. C, LPA increases the interactions of FoxO1 FHRE with the CD36 proximal promoter. Based on putative binding sites for FoxO1 at CD36 promoter, biotin-labeled probes targeting FHRE respectively at proximal, distal promoter or irrelevant site were designed for detecting FoxO1 interaction with FHREs. HMVECs were exposed to LPA (5 μM) for 24 hrs, and the nuclear components were isolated and subjected to an avidin-biotin conjugated DNA binding assay. D, Distal promoter deletion does not alter CD36 promoter activity. The CD36 promoter variants in the luciferase reporter system were prepared as shown in left panel and the basal CD36 promoter activity with various deletion mutations were assayed in the HMVECs as in Figure 1A (right panel). E, PKD-1 signaling regulates recruitment of co-repressor NCoR1 to FoxO1 complex in response to LPA. HMVECs were exposed to LPA (10 μM) for 24 hrs or pre-exposed to a selective PKD inhibitor CID 755673 (25 μM) for 30 min followed with LPA (10 μM) for 24 hrs before the cell lysates were collected and subjected to co-immunoprecipitation (IP). Representative result is shown.
To investigate mechanisms by which FoxO1 might play a role in LPA-mediated CD36 repression, ChIP analyses were performed to determine whether the interaction between FoxO1 and the CD36 gene was altered in response to LPA. Unexpectedly, no significant change was seen (Figure 3B lower panel). However, using an alternative avidin-biotin conjugated DNA and protein binding assay we found that LPA induced increased FoxO1 interaction with the CD36 proximal promoter but not distal promoters in vitro (Figure 3C). This is consistent with the data (Figure 3A) showing that deletion or mutation in the proximal FHRE abrogated basal promoter activity. Further analyses revealed that deletions in the distal promoter removing sequences from −2799 to −396 did not have a significant impact on promoter activity in MVECs (Figure 3D). These results suggest other mechanisms are involved in the regulation of FoxO1 transcriptional activity by LPA.
Having shown that the proximal FHRE is essential for basal CD36 promoter activity in HMVECs and that LPA actually increased FoxO1 interaction with the promoter we reasoned that the inhibitory effect of LPA was due to recruitment of a transcriptional repressor. Indeed LPA treatment of HMVECs induced interaction of the co-repressor NCoR1 with FoxO1. Whereas, this interaction was attenuated by pharmacologic inhibition of PKD-1 (Figure 3E), the kinase that we showed was downstream of LPA signaling in the CD36 transcriptional repression pathway7.
LPA/PKD-1 signaling mediates FoxO1-HDAC7 interaction
To explore potential mechanisms by which LPA/PKD-1 signaling contributes to endothelial CD36 transcription, we generated lentiviral vectors encoding green fluorescence protein (GFP), wild type PKD-1 (PKD-WT) or constitutively active PKD (PKD-CA) fused to GFP (Figure 4A)34 using both tetracycline inducible and constitutive systems. Tumor associated endothelial cells (TAECs) that express abundant CD36 were transduced by the inducible PKD-CA lentivirus and then exposed to doxycycline. The doxycycline treated cells showed down-regulated CD36 protein expression (Figure 4A) to levels similar to that seen in HMVECs exposed to LPA.
Figure 4.
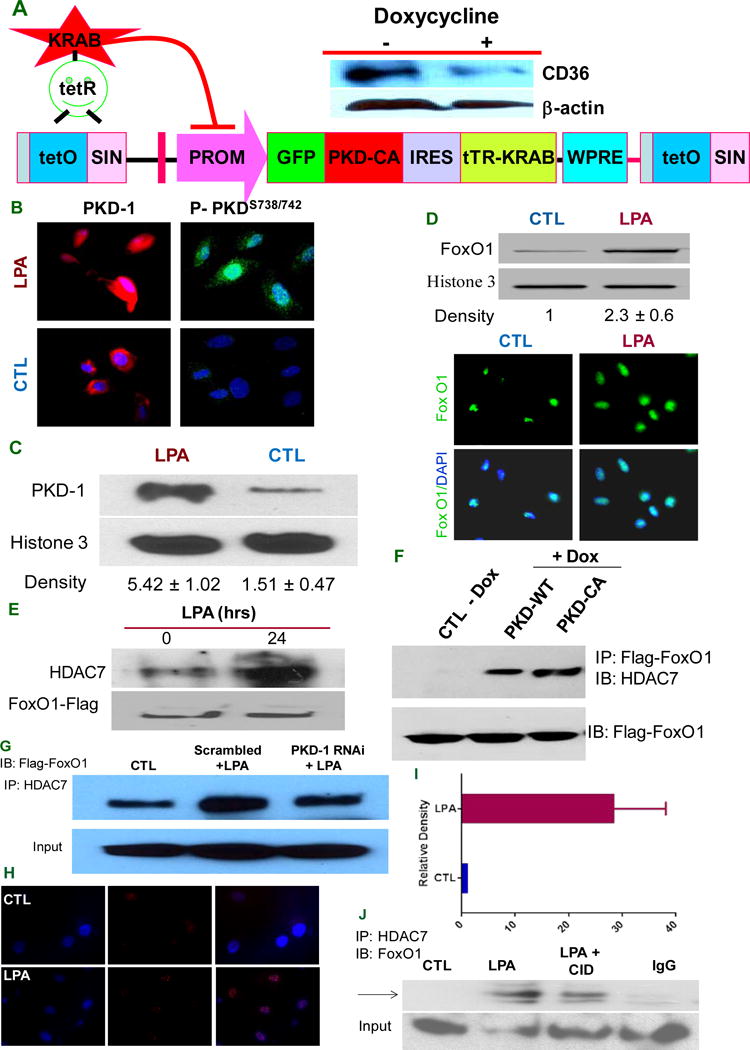
PKD-1 signals are required for HDAC7/FoxO1/interactions to regulate CD36 expression. A, PKD-CA expression inhibits CD36 expression in TAECs. A doxycycline-controllable single Lentiviral vector system containing pLVET-GFP: PKD-CA was established. TAECs were transduced with the vector and GFP positive clones were selected. Clone 3 cells were grown in the presence of 1 μg/ml doxycycline for 36 hrs and cell lysates were processed and subjected to Western blots. B, PKD-1 is translocated to the nucleus in response to LPA. HMVECs were exposed to LPA (5 μM) or vehicle for 24 hrs (left panels) or for 30 min (right panels) and incubated with anti-PKD-1 or anti-phospho-PKD-1 antibodies followed by Alexa Fluor® 2nd antibody. Images were obtained with a immunofluorescence microscopy or confocal microscopy. C, HMVECs were exposed to LPA (5 μM) for 24 hrs, the nuclear components were isolated and subjected to Western blots for total PKD-1 and histone 3 antibodies were used to determine nuclear protein loading (left panel). The relative PKD-1 level was determined by densitometry for triplicate experiments (right panel). D, FoxO1 level increases in the nucleus with LPA exposure. After LPA (10 μM) treatment for 24 hrs, ECs were subjected to immunofluorescence microscopy using anti-FoxO1 antibody (lower panel). In some studies nuclear components were isolated from the cells and subjected to Western blots for FoxO1 protein expression and relative density was calculated from triplicate experiments (upper panel). E, LPA promotes HDAC7-FoxO1 interaction. HMVECs were transfected with FoxO1-flag for 36 hrs and then exposed to LPA (5 μM) for 18 hrs. Lysates were immunoprecipitated with HDAC7 antibody and re-probed with anti-Flag antibody. Total cell lysate was subjected to Western blots for Flag-tag. F, Overexpressing PKD-1 increases HDAC7 interaction with FoxO1. TAECs transduced with pLVET-GFP: PKD1-WT or pLVET-GFP: PKD1-CA plasmids were co-transfected with FoxO1-Flag in the absence or presence of doxycycline (1 μg/ml) for 36 hrs. Cell lysates were collected and immunoprecipitated with anti-Flag antibody, followed by Western blot analyses using anti-HDAC7 or anti-Flag antibody. G, PKD-1 is required for HDAC7-FoxO1 interactions. HMVECs transfected with scrambled RNAi plasmids (Scramble), or pSUPER PKD-1 RNAi and co-transfected with FoxO1 for 36 hrs were treated with LPA (5 μM) for 18 hrs and analyzed by Co-IP for FoxO1-HDAC7 interactions as in panel E. Representative results are shown in the figure. H, LPA exposure promotes interaction of endogenous HDAC7 and FoxO1 in the nucleus of HMVEC-C. In response to LPA (10 μM), there was a significant increase in the HDAC7-FoxO1 interaction in early passages of HMVEC-C (P3) detected as bright red dots in the cell nuclei by fluorescence microscopy using the Proximity Ligation Assay (PLA). I, Increased relative density of red fluorescent dots in the PLA indicating HDAC7-FoxO1 interactions in HMVEC-C exposed to LPA (10 μM). J, LPA-induced HDAC7-FoxO1 interaction was partially attenuated by PKD inhibitor. HMVEC-C (P3) wasexposed to LPA (10 μM), LPA (10 μM) plus a selective PKD inhibitor CID755673 (25 μM) or buffer control and the nuclei were isolated and subjected to immunoprecipitation with anti-HDAC7 followed by immunoblot with anti-FoxO1 to detect endogenous interactions.
Since specialized roles of PKD-1 may depend on both its localization and substrate proximity27 we examined PKD-1 intracellular localization by fluorescence microscopy (Figure 4B left panels). LPA exposure increased nuclear PKD-1 expression and the phosphorylated PKD-1 was also increased in the nucleus in response to LPA treatment by fluorescence confocal laser scanning microscopy (Figure 4B right panels). To confirm the microscopy data we performed cell fractionation to isolate nuclear components. Western blots of cell fractions showed that LPA increased nuclear accumulation of PKD-1 (Figure 4C). Similarly, LPA exposure increased nuclear FoxO1 levels in HMVECs as determined by Western blots and immunofluorescence microscopy (Figure 4D).
Having shown that PKD-1 and its substrate HDAC720, 35 as well as FoxO1 accumulate in the MVEC nuclei in response to LPA, we hypothesized that PKD-1 mediates formation of a nuclear signaling complex to regulate CD36 transcription. HMVECs were thus transfected with Flag-tagged FoxO1 to assess FoxO1-HDAC7 interactions in response to LPA. Co-immunoprecipitation experiments showed that FoxO1 interacted with endogenous HDAC7 at a basal level and that LPA exposure enhanced the interaction (Figure 4E). Furthermore, inducing expression of either PKD-WT or PKD-CA by doxycycline promoted FoxO1-HDAC7 interactions, but PKD-CA promoted interaction to a greater extent (Figure 4F). To determine whether PKD-1 is essential for this interaction, we knocked down PKD-1 expression in the cells with RNAi plasmids. FoxO1-HDAC7 interactions in response to LPA were significantly attenuated with PKD-1 knockdown (Figure 4G). To assess interactions of endogenous FoxO1 and HDAC7 in HMVEC-C we utilized a fluorescence-based immunologic cross linking assay (Proximity Ligation Assay/PLA) that uses antibodies to the 2 proteins derived from different animal species and detects proteins in close proximity (<40nm) microscopically as bright red fluorescent dots. As seen in Figure 4H, multiple bright dots were seen in cell nuclei after LPA treatment, but not in the control untreated cells (Figure 4I, p < 0.01). As an additional control, no signal was detected using antibodies to FoxO1 and HDAC5 (Supplemental Figure I). Co-immunoprecipitation confirmed the increased interaction between endogenous HDAC7 and FoxO1 (Figure J) and this interaction was attenuated partially by treating the cells with a pharmacological PKD inhibitor (Figure 4J).
Regulation of pro-arteriogenic and angiogenic responses by LPA/PKD-1 signaling
To determine whether the repression of CD36 transcription induced by LPA/PKD-1 signaling is part of a broader program of angiogenic gene regulation, we performed a focused angiogenic gene expression array using real-time qPCR. In response to LPA exposure the expression levels of 20 angiogenic genes were up-regulated over 3–fold, including EFNB2, neuropilin 1 and flt1 (Supplemental Figure II, A and B). These data suggest that down-regulation of the anti-angiogenic receptor CD36 was linked to the up-regulation of specific pro-angiogenic genes. EFNB2 is an arterial endothelial cell marker that is important in angiogenesis and arteriogenesis36–38. To validate the qPCR profiling data we showed significant increased expression of EFNB2 mRNA by qPCR and of protein by western blots in the PKD-CA transduced HMVEC-C and mouse MVEC lines (Figure 5A & 5B). Moreover, over-expressing PKD-CA in HMVEC-C significantly increased expression of additional pro-arteriogenic genes including delta-like ligand 4 and HEY2, but not neuropilin1 (Figure 5C). Over-expressing CD36 down-regulated EFNB2 expression in HMVEC-C and LPA treatment significantly reversed CD36-mediated down-regulation (Figure 5D). Intriguingly, CD36 overexpression did not significantly change delta-like ligand 4, HEY2 or neuropilin1 expression (Figure 5E).
Figure 5. LPA/PKD-1-CD36 signaling axis regulates the expression of angiogenic and arteriogenic genes.
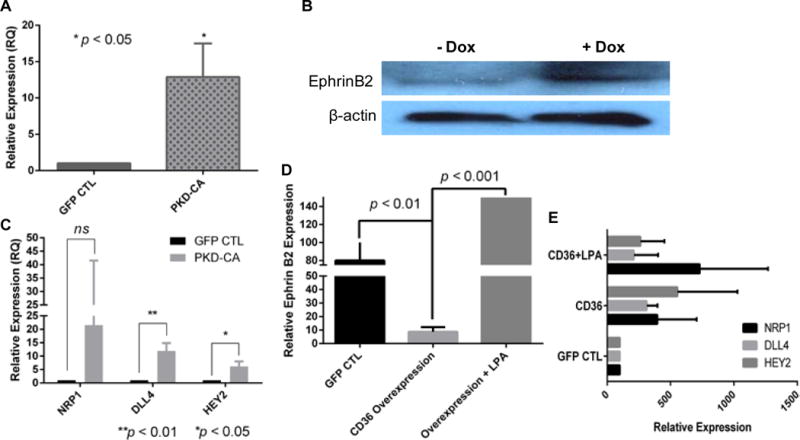
A, Overexpression of PKD-CA increases the expression of EFNB2 mRNA level. HMVEC-C (passage 2) transduced with GFP or PKD-1:GFP were processed for total RNA and mRNA expression by quantitative real time RT-PCR. B, Clone 3 TAEC cells were processed as in Figure 4A and cell lysates collected and subjected to Western blots with anti-ephrinB2 antibody. Shown is a representative result from duplicate experiments. C, The impact of PKD-CA overexpression on angiogenic gene expression. HMVEC-C transduced with GFP or PKD-1:GFP were processed for total RNA and mRNA expression of NRP1, DLL4 and HEY2 by quantitative real time RT-PCR. D, Overexpression of human CD36 downregulates EFNB2 expression in HMVEC-C. HMVEC-C (passage 2) transduced with GFP or hCD36:GFP were processed for total RNA and mRNA expression by quantitative real time RT-PCR. E, Overexpression of human CD36 does not impact the expression of NRP1 (neuropilin1), DLL4 (delta like ligand 4) and HEY2 in HMVEC-C. HMVEC-C (passage 2) was processed as described in D for quantitative real time RT-PCR.
To show functional significance of this transcriptional program switch we studied the impact of LPA on angiogenic differentiation in mouse heart MVECs expressing CD36. Exposure to LPA promoted endothelial sprout formation in vitro as measured in a three-dimensional spheroid assay; the spheroids showed robust branching morphogenesis compared to the control (p<0.01, Figure 6A) and this was partially attenuated by addition of a selective PKD inhibitor (p>0.05, Figure 6A). Furthermore, an in vivo Matrigel assay showed that pharmacologic inhibition of PKD signaling reduced the inhibitory effect of LPA on TSR/CD36-mediated anti-angiogenic effects in response to VEGF in vivo (Figure 6B left panel). LPA, when incorporated into VEGF containing Matrigel plugs induced formation of functional vascular networks in vivo as demonstrated by the presence of hematopoietic cells within the neo-vessels (Figure 6B upper right panel). Pharmacologic inhibition of LPA signaling did not significantly change microvascular density (Figure 6B lower right panel). However, LPA significantly promoted microvascular remodeling as shown by increased number of cells expressing alpha SMA (Figure 6C, upper and lower left panel, p < 0.05). Furthermore, conditioned media from LPA-treated HMVECs significantly increased proliferation of VSMCs in culture compared to cells grown in standard media (Figure 6C, lower right panel, p < 0.05). Vascular smooth muscle cell (VSMC) proliferation in the presence of conditioned media from HMVECs treated with PKD inhibitor was not different from cells treated with standard media alone (Figure 6C, lower right panel, p>0.05). These data suggest the LPA signaling through PKD pathway in MVECs can impact VSMC function via secreted factors. We also examined tumor angiogenesis and relevant signaling pathways in a syngeneic tumor transplant model using Lewis Lung Carcinoma cells in wild type and cd36 null mice. Immunofluorescence microscopy revealed extensive angiogenesis within the tumors detected with anti-VEGFR2 antibody (Figure 6D and 6E lower panels). The angiogenic response was associated with lower levels of expression of CD36 in the neo-vessels compared to nearby cutaneous vessels (Figure 6D and 6E) and with expression of phospho-PKD-1, EFNB2 and alpha SMA in the tumor vasculature (Figure 6E, 6F and 6G). There was no significant difference in the number of alpha SMA expressing cells in tumor vessels in wild type and cd36 null mice (Figure 6G right panel, p > 0.05).
Figure 6. PKD-1 signaling promotes proangiogenic and proarteriogenic responses.
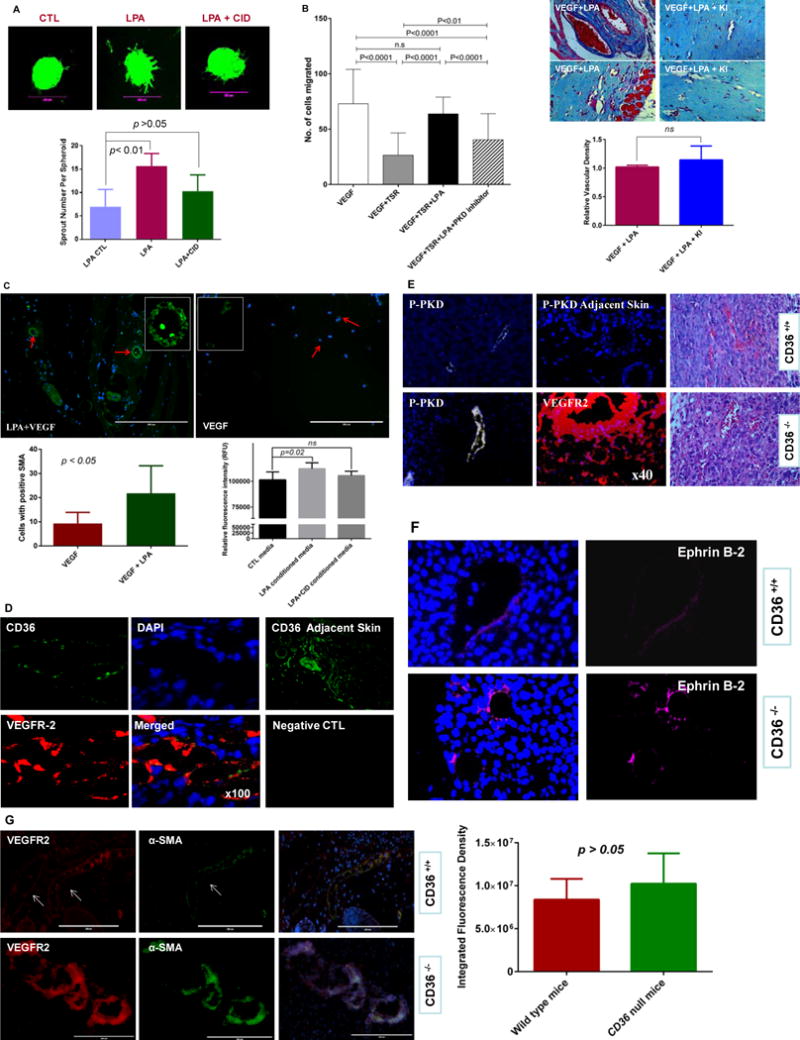
A, PKD-1 signaling is associated with LPA-stimulated sprouting morphogenesis in primary MHECs. MHECs were exposed to LPA and used in a three-dimensional spheroid assay. LPA (10 μM) was added to the media for 48 hrs to induce branching morphogenesis, and a selective PKD inhibitor CID755673 (25 μmol/L) was added in a group with LPA treatment with vehicle as control. Branching morphogenesis was observed under fluorescence microscope and pictures were taken with a digital camera linked to MetaMorph® 7.0 software and representative pictures are shown, and scale bar is 200 μm (upper panel). Sprouting described in B was quantified microscopically by counting sprouts per spheroid at 48 hrs in response to LPA (lower panel). B, LPA inhibition of TSR-induced inhibition of vascular invasion is partially dependent on PKD-1 signaling. Matrigel plugs mixed with a combination of VEGF (50 ng/ml), recombinant TSR (10 nM), LPA (10 μM) and CID 755673 (25 μmol/L) were injected into mice and analyzed histologically after 10d. Bar graph shows the relative angiogenic responses and vascular invasion in the Matrigel plugs (left panel). Additionally, LPA signaling promotes vascular remodeling. Matrigel plugs mixed with a combination of VEGF (50 ng/ml) and LPA (10 μM) or VEGF and LPA plus LPA receptor 1,3 antagonist Ki16426 (2 μM) were respectively injected into mice and analyzed histologically after 10d. Representative Masson’s Trichrome stained images are shown and bar graph shows vascular invasion relative to VEGF stimulation (right panel; p >0.05; bar 50 μm). Images were acquired with a Nikon Eclipse E600 microscope and NIH Image J was used for the analysis of vascular invasion. ns: no significant statistical difference. C, LPA stimulates the formation of SMA positive vessels. Matrigel plugs were sectioned and examined with an EVOS®FL imaging system using antibodies to alpha SMA (green) and nuclei was stained with DAPI (blue), and representative images are shown, bar = 200 μM. Images with positive SMA staining were randomly collected and the positive cells counted with NIH image J. The average cell number was calculated in each visual field and compared between VEGF and LPA plus VEGF groups (p <0.05, upper and lower left panel). Additionally, VSMCs were exposed to the conditioned media from HMVECs treated with LPA or LPA and PKD inhibitor, and cell proliferation was assessed by an alamarBlue® Cell viability assay (lower right panel). Two independent experiments were performed, and the cell proliferation is represented as relative fluorescence intensity. LPA conditioned media significantly increases VSMC proliferation (p<0.05; ns: no significant statistical difference). D, Tumors harvested from C57Bl and cd36+/+ mice after transplantation of Lewis Lung carcinoma cells were sectioned and examined by immunofluorescence microscopy using antibodies to VEGFR2 (red) and CD36 (green). Nuclei were stained with DAPI (blue) (100×). CD36 expression in skin adjacent to the tumors and sections incubated with CD36 antibodies or non-immune IgG are shown as positive and negative controls. Representative results are shown. E, Tumor vessels as in Panel D were examined by immunofluorescence using antibodies to phospho-PKD-1 (white) in wild type or the cd36 null mice, and no PKD-1 phosphorylation was shown in the skin adjacent to the tumor tissues. VEGFR-2 (red) staining was shown in the tumor tissues to visualize the angiogenic response in the cd36 null mice. H&E staining is used to show tumor tissues in both wild type and cd36 deficiency mice (40×). Representative images are shown. F, Tumor vessels were examined by immunofluorescence using antibodies to EFNB2 (magenta) in wild type or the cd36 null mice. Representative images are shown. G, Tumor vessels were examined by immunofluorescence using antibodies to alpha SMA and VEGFR2 and DAPI was used for staining the nuclei (VEGFR2 red, SMA green, DAPI blue) in wild type or the cd36 null mice. Images were randomly acquired with a Zeiss Axioskop Microscope with Photometrics Cool SNAP ES Camara system using Metamorph v7 software or EVOS®FL cell imaging system, and representative images are shown (left panel). NIH Image J was used to analyze the integrated fluorescence density (IFD) of SMA positive cells (green) in the hypervascular areas of the tumor microenvironment. The average IFD was used to assess the relative number of SMA positive cells in the neovessels and compared between wild type and cd36 null mice. No significant statistical difference was found between these two groups (p > 0.05, right panel).
Discussion
CD36 is an important angiostatic receptor on MVECs that interacts with TSP-1 and other anti-angiogenic proteins containing TSR domains1–4 to modulate VEGF signaling and promote endothelial cell apoptosis. Our previous work demonstrated that LPA promotes angiogenesis in the presence of antiangiogenic TSR-containing proteins by down-regulating CD36 gene transcription via a protein kinase PKD-1 pathway7. In the current study we made the novel observations that the transcription factor FoxO1 is required for constitutive CD36 expression in MVECs and, as shown in the model in Figure 7, that LPA-mediated PKD-1 activation and nuclear translocation results in recruitment of the class II histone deacetylase HDAC7 to the CD36 promoter driving formation of an HDAC7/NCoR1/FoxO1 nuclear complex and subsequently triggering CD36 transcriptional repression. Furthermore, turning off CD36 transcription was associated with transcriptional reprogramming that up-regulated expression of arterial genes, including EPHB2. On a functional level, our data suggest that activating this pathway in vivo may promote microvascular remodeling and functional stability (Figure 6), and in murine tumor model systems absence of CD36 was associated with increased expression of the arterial marker in the tumor microvasculature.
Figure 7.
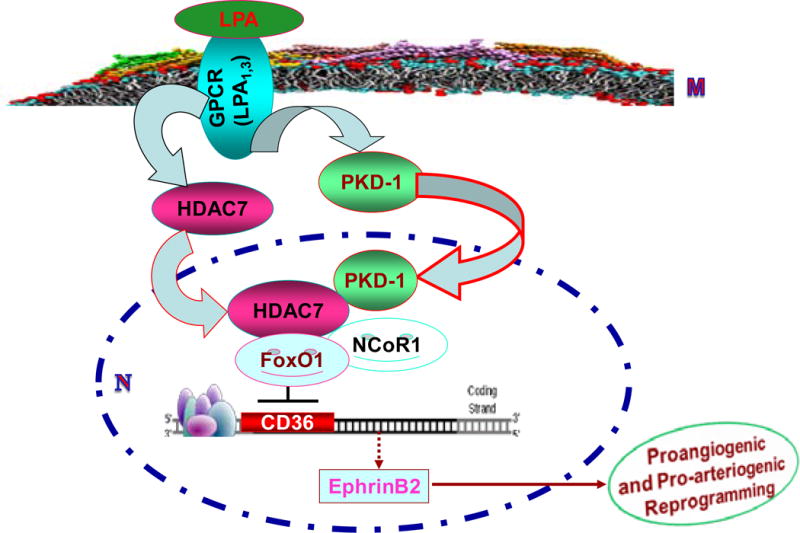
Model showing potential mechanism by which an LPA/PKD-1-HDAC7-FoxO1 signaling axis regulates EC CD36 transcription and angiogenesis. LPA signaling in MVEC is mediated by G-protein coupled LPA receptors 1 and 3 at the EC membrane (M) which activate protein kinase PKD-1, leading to PKD-1 and HDAC7 translocation into the nucleus (N) where they interact with the transcription factor FoxO1 and the co-repressor NCoR1. This results in CD36 transcriptional repression and reprogramming EC to express ephrinB2 which promotes angiogenesis, arteriogenic differentiation and microvascular remodeling.
These results also support the concept that terminally differentiated MVECs are developmentally plastic, and in the setting of specific environmental cues can be re-programmed39–42 towards an arteriogenic phenotype that includes change in expression of surface proteins and secretion of factor(s) that support VSMC proliferation and migration. LPA is likely to be an important such cue as it is often generated in angiogenic microenvironments associated with inflammation, ischemia, platelet activation, cancer and obesity. Consistent with previous reports43, 44, our results show unsaturated LPA promoted SMC entry into Matrigel plugs in vivo. While our data suggest that LPA in the microenvironment of lung cancers may reduce MVEC CD36 expression and contribute to microvascular remodeling comparable to that seen in the cd36 null mice at the late stages of tumor development examined in these studies, we did not detect differences in VSMC investment in neovessels, as detected by anti-alpha SMA staining. Our data also suggest that CD36 transcriptional status might be more than just a marker for the loss of capillary endothelial phenotype. Forced expression of CD36 in MVECs abrogated the arteriogenic “switch” that may be turned on by LPA, perhaps implicating CD36 in the regulatory pathway.
PKD-1-mediated signaling has been shown to regulate cardiovascular functions including myocardial responses to ischemia, angiogenesis, and arterial remodeling,7, 20–24, 42, 45 and the transcription factor FoxO1 is well known to be involved in vascular development, postnatal angiogenesis,32, 33 and regulation of EPHB2 expression46. Our study now links the PKD-1 signaling pathway initiated by specific LPA receptors to FoxO1 in MVECs and demonstrates a critical mechanistic role for nuclear accumulation of activated PKD-1 in recruiting a specific class II histone deacetylase, HDAC7, to the FoxO1 transcriptional regulatory complex. HDAC7, which is known to be highly expressed in ECs45, 47, was previously shown by Chang and colleagues31 to be important in maintenance of vascular integrity during embryonic development via a mechanism that involves inhibition of a transcriptional activator, myocyte enhancer factor-2 (MEF-2). This interaction results in transcriptional repression of the matrix metalloproteinase-10 gene and promotes stability of the extracellular matrix. Thus these studies and our new data show that HDAC7 interacts with specific EC transcription factors (FoxO1 and MEF-2) that function in at least two pathways related to vascular development and stability. Our studies using pharmacologic HDAC inhibitors or specific shRNA (Figures 1E, 2B and 2C) also suggest that HDAC7 could be a therapeutic target to de-stabilize angiogenesis and modulate EC differentiation48.
CD36 is a multifunctional transmembrane protein that on phagocytic cells serves as a scavenger receptor for endogenous danger-associated molecular patterns (e.g. oxidized low density lipoproteins) and exogenous pathogen-associated molecular patterns (e.g. staphylococcus lipoteichoic acid)3. On fat, muscle, and gut epithelial cells CD36 functions as a transporter of free fatty acids. Interestingly, CD36 transcriptional regulation in MVECs is very different than in these other cell types, in which expression is regulated both positively and negatively by metabolic factors, cytokines, bacterial components, reactive oxygen species, and adhesion3, 10. Our studies have not revealed any response of CD36 expression in MVECs to those factors that up-regulated CD36 in phagocytic cells, fat and muscle (e.g. PPARγ agonists and cytokines) or that down-regulated expression in these cells (e.g. bacterial endotoxin), suggesting that the expression of CD36 in MVECs may have evolved for a different function; i.e. as a receptor for TSR proteins and a regulator of both angiogenesis and arteriogenesis38, rather than as a scavenger receptor or fatty acid transporter. Further study of the precise mechanisms linking CD36 expression to EC differentiation and of the in vivo relevance of this regulatory pathway may aid in discovering novel therapeutic targets against ischemic diseases, cancer, obesity and diabetes.
Supplementary Material
Significance.
FoxO1, a transcription factor targeting genes in angiogenesis, vascular remodeling and homeostasis, may be required for endothelial cell CD36 transcription whereas a novel LPA/PKD-1-HDAC7-FoxO1 signaling axis is involved in CD36 transcriptional repression in microvascular endothelial cells. Intriguingly, the endothelial cells are reprogrammed to express arteriogenic genes once CD36 transcription is turned off. Our study defines a novel mechanistic pathway of CD36 transcriptional repression and implicates the potential importance of this repression in proarteriogenic responses and microvascular remodeling under ischemic conditions. Our study also suggests the plasticity of angiogenic gene transcription during postnatal angiogenesis, and this plasticity could be controlled via cellular context-dependent transcriptional switch of FoxO1 regulated by LPA/PKD1-HDAC7 signaling. PKD-1-FoxO1 signaling could serve as a reprogramming factor for modulating arterial development and arteriogenesis.
Acknowledgments
We thank Dr. Randall Peterson at the Cardiovascular Research Center of Massachusetts General Hospital and Harvard Medical School for critical reading of this manuscript. We also thank the help from Mrs. Rong Yuan at Blood Research Institute, Blood Center of Wisconsin.
Sources of Funding. This work was supported by a National Scientific Development Grant from the American Heart Association (BR), an Institutional Research Grant (# 86-004-26) from American Cancer Society (BR) and a Career Development Award from The Central Society of Clinical and Translational Research (BR), and the National Institutes of Health (HL085718 to RLS and CA140182 to PS).
Abbreviations
- FHRE
FoxO-responsive elements
- PKD-CA
constitutively active protein kinase D1
- TAEC
tumor associated endothelial cell
- HMVEC-C
cardiac human microvascular endothelial cells
- PKD-WT
wild type PKD-1
- TSR
thrombospondin type 1 repeat
Footnotes
Disclosures. None.
References
- 1.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–8. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 2.Ren B, Yee KO, Lawler J, Khosravi-Far R. Regulation of tumor angiogenesis by thrombospondin-1. Biochimica et biophysica acta. 2006;1765:178–88. doi: 10.1016/j.bbcan.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ren B, Song K, Parangi S, Jin T, Ye M, Humphreys R, Duquette M, Zhang X, Benhaga N, Lawler J, Khosravi-Far R. A double hit to kill tumor and endothelial cells by TRAIL and antiangiogenic 3TSR. Cancer Res. 2009;69:3856–65. doi: 10.1158/0008-5472.CAN-08-2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu LY, Ramakrishnan DP, Silverstein RL. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood. 2013;122:1822–32. doi: 10.1182/blood-2013-01-482315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kazerounian S, Duquette M, Reyes MA, Lawler JT, Song K, Perruzzi C, Primo L, Khosravi-Far R, Bussolino F, Rabinovitz I, Lawler J. Priming of the vascular endothelial growth factor signaling pathway by thrombospondin-1, CD36, and spleen tyrosine kinase. Blood. 2011;117:4658–66. doi: 10.1182/blood-2010-09-305284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren B, Hale J, Srikanthan S, Silverstein RL. Lysophosphatidic acid suppresses endothelial cell CD36 expression and promotes angiogenesis via a PKD-1-dependent signaling pathway. Blood. 2011;117:6036–45. doi: 10.1182/blood-2010-12-326017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie L, Duncan MB, Pahler J, Sugimoto H, Martino M, Lively J, Mundel T, Soubasakos M, Rubin K, Takeda T, Inoue M, Lawler J, Hynes RO, Hanahan D, Kalluri R. Counterbalancing angiogenic regulatory factors control the rate of cancer progression and survival in a stage-specific manner. Proc Natl Acad Sci U S A. 2011;108:9939–44. doi: 10.1073/pnas.1105041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren B. Endothelial cells: a key player in angiogenesis and lymphangiogenesis. MOJ Cell Sci Report. 2015;2:00015. [Google Scholar]
- 10.Olagnier D, Lavergne RA, Meunier E, Lefevre L, Dardenne C, Aubouy A, Benoit-Vical F, Ryffel B, Coste A, Berry A, Pipy B. Nrf2, a PPARgamma alternative pathway to promote CD36 expression on inflammatory macrophages: implication for malaria. PLoS Pathog. 2011;7:e1002254. doi: 10.1371/journal.ppat.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffin E, Re A, Hamel N, Fu C, Bush H, McCaffrey T, Asch AS. A link between diabetes and atherosclerosis: Glucose regulates expression of CD36 at the level of translation. Nat Med. 2001;7:840–6. doi: 10.1038/89969. [DOI] [PubMed] [Google Scholar]
- 12.Mwaikambo BR, Yang C, Chemtob S, Hardy P. Hypoxia up-regulates CD36 expression and function via hypoxia-inducible factor-1- and phosphatidylinositol 3-kinase-dependent mechanisms. J Biol Chem. 2009;284:26695–707. doi: 10.1074/jbc.M109.033480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature reviews Cancer. 2003;3:401–10. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 14.Rivera-Lopez CM, Tucker AL, Lynch KR. Lysophosphatidic acid (LPA) and angiogenesis. Angiogenesis. 2008;11:301–10. doi: 10.1007/s10456-008-9113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Ramakrishnan DP, Ren B. Regulation of angiogenesis by phospholipid lysophosphatidic Acid. Front Biosci. 2013;18:852–61. doi: 10.2741/4148. [DOI] [PubMed] [Google Scholar]
- 16.Smyth SS, Cheng HY, Miriyala S, Panchatcharam M, Morris AJ. Roles of lysophosphatidic acid in cardiovascular physiology and disease. Biochim Biophys Acta. 2008;1781:563–70. doi: 10.1016/j.bbalip.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris AJ, Panchatcharam M, Cheng HY, Federico L, Fulkerson Z, Selim S, Miriyala S, Escalante-Alcalde D, Smyth SS. Regulation of blood and vascular cell function by bioactive lysophospholipids. J Thromb Haemost. 2009;7(Suppl 1):38–43. doi: 10.1111/j.1538-7836.2009.03405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeon ES, Lee IH, Heo SC, Shin SH, Choi YJ, Park JH, Park do Y, Kim JH. Mesenchymal stem cells stimulate angiogenesis in a murine xenograft model of A549 human adenocarcinoma through an LPA1 receptor-dependent mechanism. Biochim Biophys Acta. 2010;1801:1205–13. doi: 10.1016/j.bbalip.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Chuang YW, Chang WM, Chen KH, Hong CZ, Chang PJ, Hsu HC. Lysophosphatidic acid enhanced the angiogenic capability of human chondrocytes by regulating Gi/NF-kB-dependent angiogenic factor expression. PloS one. 2014;9:e95180. doi: 10.1371/journal.pone.0095180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avkiran M, Rowland AJ, Cuello F, Haworth RS. Protein kinase d in the cardiovascular system: emerging roles in health and disease. Circulation research. 2008;102:157–63. doi: 10.1161/CIRCRESAHA.107.168211. [DOI] [PubMed] [Google Scholar]
- 21.Ha CH, Jin ZG. Protein kinase D1, a new molecular player in VEGF signaling and angiogenesis. Mol Cells. 2009;28:1–5. doi: 10.1007/s10059-009-0109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rozengurt E. Protein kinase D signaling: multiple biological functions in health and disease. Physiology. 2011;26:23–33. doi: 10.1152/physiol.00037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong C, Jin ZG. Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. The Journal of biological chemistry. 2005;280:33262–9. doi: 10.1074/jbc.M503198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin L, Zeng H, Zhao D. Requirement of protein kinase D tyrosine phosphorylation for VEGF-A165-induced angiogenesis through its interaction and regulation of phospholipase Cgamma phosphorylation. The Journal of biological chemistry. 2006;281:32550–8. doi: 10.1074/jbc.M604853200. [DOI] [PubMed] [Google Scholar]
- 25.Fu Y, Rubin CS. Protein kinase D: coupling extracellular stimuli to the regulation of cell physiology. Embo Reports. 2011;12:785–796. doi: 10.1038/embor.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eiseler T, Doppler H, Yan IK, Goodison S, Storz P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009;11:R13. doi: 10.1186/bcr2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. The Journal of biological chemistry. 2005;280:13205–8. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 28.Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, Shaw RJ. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–21. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ha CH, Wang W, Jhun BS, Wong C, Hausser A, Pfizenmaier K, McKinsey TA, Olson EN, Jin ZG. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. The Journal of biological chemistry. 2008;283:14590–9. doi: 10.1074/jbc.M800264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ha CH, Jhun BS, Kao HY, Jin ZG. VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arteriosclerosis Thrombosis and Vascular Biology. 2008;28:1782–1788. doi: 10.1161/ATVBAHA.108.172528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126:321–34. doi: 10.1016/j.cell.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 32.Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Ikeda K, Motoyama N, Mori N. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279:34741–9. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- 33.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115:2382–92. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szulc J, Wiznerowicz M, Sauvain MO, Trono D, Aebischer P. A versatile tool for conditional gene expression and knockdown. Nat Methods. 2006;3:109–16. doi: 10.1038/nmeth846. [DOI] [PubMed] [Google Scholar]
- 35.Dequiedt F, Van Lint J, Lecomte E, Van Duppen V, Seufferlein T, Vandenheede JR, Wattiez R, Kettmann R. Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J Exp Med. 2005;201:793–804. doi: 10.1084/jem.20042034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simons M, Eichmann A. Molecular controls of arterial morphogenesis. Circulation research. 2015;116:1712–24. doi: 10.1161/CIRCRESAHA.116.302953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–6. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 38.Ren B, Deng Y, Mukhopadhyay A, Lanahan AA, Zhuang ZW, Moodie KL, Mulligan-Kehoe MJ, Byzova TV, Peterson RT, Simons M. ERK1/2-Akt1 crosstalk regulates arteriogenesis in mice and zebrafish. The Journal of clinical investigation. 2010;120:1217–28. doi: 10.1172/JCI39837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliver G, Srinivasan RS. Endothelial cell plasticity: how to become and remain a lymphatic endothelial cell. Development. 2010;137:363–72. doi: 10.1242/dev.035360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464:549–53. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miquerol L, Thireau J, Bideaux P, Sturny R, Richard S, Kelly RG. Endothelial plasticity drives arterial remodeling within the endocardium after myocardial infarction. Circ Res. 2015;116:1765–71. doi: 10.1161/CIRCRESAHA.116.306476. [DOI] [PubMed] [Google Scholar]
- 42.Evans IM, Zachary IC. Protein kinase D in vascular biology and angiogenesis. IUBMB Life. 2011;63:258–63. doi: 10.1002/iub.456. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida K, Nishida W, Hayashi K, Ohkawa Y, Ogawa A, Aoki J, Arai H, Sobue K. Vascular remodeling induced by naturally occurring unsaturated lysophosphatidic acid in vivo. Circulation. 2003;108:1746–52. doi: 10.1161/01.CIR.0000089374.35455.F3. [DOI] [PubMed] [Google Scholar]
- 44.Cheng Y, Makarova N, Tsukahara R, Guo H, Shuyu E, Farrar P, Balazs L, Zhang C, Tigyi G. Lysophosphatidic acid-induced arterial wall remodeling: requirement of PPARgamma but not LPA1 or LPA2 GPCR. Cellular signalling. 2009;21:1874–84. doi: 10.1016/j.cellsig.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altschmied J, Haendeler J. A new kid on the block: PKD1: a promising target for antiangiogenic therapy? Arterioscler Thromb Vasc Biol. 2008;28:1689–90. doi: 10.1161/ATVBAHA.108.174250. [DOI] [PubMed] [Google Scholar]
- 46.Papanicolaou KN, Izumiya Y, Walsh K. Forkhead transcription factors and cardiovascular biology. Circ Res. 2008;102:16–31. doi: 10.1161/CIRCRESAHA.107.164186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mottet D, Bellahcene A, Pirotte S, Waltregny D, Deroanne C, Lamour V, Lidereau R, Castronovo V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circulation Research. 2007;101:1237–1246. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 48.Yan MS, Marsden PA. Epigenetics in the Vascular Endothelium: Looking From a Different Perspective in the Epigenomics Era. Arterioscler Thromb Vasc Biol. 2015;35:2297–306. doi: 10.1161/ATVBAHA.115.305043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.