

Abstract
Exemestane is an aromatase inhibitor drug used for the treatment of hormone-dependent breast cancer. 17-hydroexemestane, the major and biologically active metabolite of exemestane in humans is eliminated via glucuronidation by the polymorphic UGT2B17 phase II drug metabolizing enzyme. Previous microsomal studies have shown that UGT2B17 gene deletion affects the intrinsic hepatic clearances of 17-hydroexemestane in vitro. In this open-label study, we set out to assess the effect of UGT2B17 gene deletion on the pharmacokinetics of 17-hydroexemestane in healthy female volunteers with and without UGT2B17. To achieve this goal, 14 healthy postmenopausal women (eight carriers of the homozygous UGT2B17 wild-type allele and six carriers of the homozygous UGT2B17 gene deletion allele) were enrolled and invited to receive a single 25 mg oral dose of exemestane. Pharmacokinetics was assessed over 72 hours post-dosing. Our results showed that there were statistically significant differences in plasma 17-hydroexemestane AUC0-∞ (p = 0.0007) and urine 17-hydroexemestane C24hr (p = 0.001) between UGT2B17 genotype groups. Our data suggest that UGT2B17 gene deletion influences 17-hydroexemestane pharmacokinetics in humans.
Keywords: 17-hydroexemestane, exemestane, UGT2B17 gene deletion, pharmacokinetics and pharmacogenetics, breast cancer
INTRODUCTION
Exemestane is a drug used for the treatment of hormone-receptor positive breast cancers. It works by covalently and irreversibly inhibiting aromatase, the rate limiting enzyme responsible for the peripheral conversion of androgens (testosterone and androstenedione) into estrogens (estradiol and estrone) in postmenopausal women1-3. The major metabolite of exemestane in humans is 17-hydroexemestane. Preclinical studies have shown that 17-hydroexemestane is biologically active4-8. It has bone-protective activities in rats and is a potent aromatase inhibitor in vitro 4-8.
17-hydroexemestane is a substrate of UGT2B17 and is cleared via glucuronidation by this polymorphic phase II drug metabolizing enzyme 9,10. A recent microsomal study demonstrated that UGT2B17 gene deletion affects the intrinsic hepatic clearance of 17-hydroexemestane in vitro 9,10. In their study, Sun et al. found that 1) the rate of 17hydroexemestane-glucuronide formation was shown to be significantly (P<0.001) decreased (14-fold) in human liver microsomes (HLMs) exhibiting the UGT2B17(*2/*2) deletion genotype versus wild-type UGT2B17(*1/*1) HLMs; 2) a 36-fold lower intrinsic clearance (Vmax/Km) (P=0.023) was observed in UGT2B17(*2/*2) versus UGT2B17(*1/*1) HLMs; and 3) a significant (P<0.0001, r2=0.72) correlation was observed between HLM-mediated 17-hydroexemestane glucuronide formation and liver UGT2B17 mRNA expression 9,10.
Given the high frequency of the UGT2B17 gene deletion (~30% prevalence in Caucasians and African-Americans) 11-13 and its potential significant importance in the metabolism of 17-hydroexemestane, this study was designed to assess the impact of UGT2B17 gene deletion on 17-hydroexemestane pharmacokinetics in healthy postmenopausal female volunteers.
METHODS
Study subjects
Study subjects were enrolled with written informed consent under a protocol that was reviewed and approved by the Institutional Review Board at Harding University. All participants were healthy, postmenopausal female volunteers judged to be in good medical condition based on history, physical exam, and blood and urine chemistry results. Postmenopausal status was confirmed by an FSH level between 23-116 mU. Inclusion criteria included: 1) age greater than 45 years at the time of enrollment, 2) postmenopausal as defined by absence of menstruation for 12 months or 6 months if subject had bilateral oophorectomy, 3) healthy with no medical conditions requiring the use of prescription medications, and 4) normal liver function tests (LFTs) (AST, ALT ≤ 75 U/L) as well as normal values for hemoglobin (12-15 gm/dL), platelet count (150-400 × 103/μL), and thyroid-stimulating hormone (0.4 – 4.2 μU/mL). Participants were ineligible for participation with any of the following: 1) hypersensitivity to exemestane or related compounds, 2) use of prescription medications, 3) history of alcohol or drug abuse, 4) blood donation within the last 2 months, 5) BMI of less than 18.5 Kg/m2 or more than 35 Kg/m2, 6) inability to give informed consent 7) enrollment in a study within the past 2 months, and 8) smoking.
Study design
This was a nonrandomized, open-label study with subjects selected based on their UGT2B17 genotype. This study was designed and powered on the basis of a previous preclinical study10. To detect a statistically significant 2-fold change between UGT2B17*1/*1 and UGT2B17*2/*2 genotyping groups in the pharmacokinetic parameters of 17-hydroexemestane with a power of 85% (α-level 5%), 14 participants (eight with the UGT2B17*1/*1 genotype and six with the UGT2B17*2/*2 genotype) were found to be sufficient and were enrolled in this investigation. In preparation for the study, subjects were asked to abstain from the use of alcoholic beverages, garlic, citrus products (especially grapefruit and grapefruit juice) as well as apple and grape foodstuffs for at least one week prior to the study. Volunteers were required to stop all over-the-counter medications, caffeinated beverages, herbal or dietary supplements 2 days prior to drug administration. All subjects were pre-screened no more than 6 weeks before drug administration. Pre-screening was comprised of a complete history and physical examination, and laboratory studies including a complete blood count, comprehensive metabolic panel, urinalysis, and UGT2B17 genotyping. On study day 1, baseline blood was collected via upper extremity peripheral venipuncture and a urine sample was collected prior to dosing. After a breakfast consisting of approximately 30% fat, 45% carbohydrates, 25% protein, and 300 ml orange juice, subjects were then dosed with exemestane 25 mg within 15-30 min. Blood samples were taken via intravenous catheterization at 0.5, 1, 2, 4, 6, and 8 hr after drug administration on study day 1. Catheters were removed and subjects were allowed to go home. Subjects returned to provide urine and blood samples for pharmacokinetic and pharmacodynamic analyses on study days 2 and 4. Subjects were required to return within 24 hours (+/− 2 hours) of the initial dosing time of the study drug on study day 1. Ten milliliters of blood were collected at each visit and used for quantitation of exemestane and 17-hydroexemestane concentrations. Urine samples were also used to assess exemestane and 17-hydroexemestane concentrations. All blood samples were collected in green top (sodium heparin) vacutainer blood collection tubes. Collected samples in tubes were placed on ice immediately after collection and centrifuged at high speed for 10 min within 15 min of collection. Plasma samples were removed and stored at −80°C until analysis. For each study participant, the total amount of blood drawn during the entire study was less than 70 ml. An exit examination, performed at the end of the study was comprised of a complete history and physical examination as well as safety laboratory studies including full blood count, urea, electrolytes, liver function tests (bilirubin, alkaline phosphatase, transaminases, albumin, and total protein), serum lipids (cholesterol and triglycerides), and serum glucose.
UGT2B17 genotyping
Genomic DNA was extracted from whole blood using a Promega's Maxwell biorobot (Madison, WI, USA). DNA quantity and purity was determined photometrically at 260 nm and 280 nm using the Thermo Scientific Nanodrop 2000 C spectrophotometer (Wilmington, DE, USA) according to the manufacturer's instructions. UGT2B17 deletion genotyping was carried out as described previously by Gallagher et al. using real-time polymerase chain reaction (RT-PCR) with allelic discrimination14. Each reaction well included two primers and one FAM-labeled probe to amplify exon 1 of UGT2B17, and two primers and one 6-JOE-labeled probe (spanning the deletion cut site) that amplify only in the presence of UGT2B17 deletion. The primer and probe sequences for the UGT2B17 genotyping assay were as follows: exon 1 (forward) 5’-TGAAAATGTTCGATAGATGGACATATAAGTA-3’, exon 1 (reverse) 5’-GACATCAAATTTTGACTCTTGTAGTTTTC-3’, exon 1 (probe) 5’FAMTACATTTTGGTCATATTTTTCACAACTACAAGAATTGT-3’BHQ1, deletion (forward) 5’-TTTAATGTTTTCTGCCTTATGCCAC-3’, deletion (reverse) 5’-AGCCTATGCAATTTTCATTCAACATAG-3’, and deletion (probe) 5’-6-JOEACTACACTGAGATTTACAAAAGAATTCTGTCAGGATATAG-3’BHQ1. All primers and probes were obtained from GenScript (Piscataway, NJ, USA). Positive control samples for UGT2B17*1/*1 (NA11993), UGT2B17*1/*2 (NA10861), and UGT2B17*2/*2 (NA12057) genotypes were provided by Coriell. Allelic discrimination was accomplished by RT-PCR amplification performed with the Roche LightCycler 480 detection system (Indianapolis, IN, USA) with the following conditions: 50°C for 2 min, then 95°C for 15 min, followed by 50 cycles of amplification at 95°C for 1 min and 60°C for 90 sec. Data were analyzed with the LightCycler 480 software version 1.5 (Indianapolis, IN, USA).
Determination of exemestane and 17-hydroexemestane concentrations
Previously described LC-MS/MS methodologies were used to quantify exemestane and 17-hydroexemestane in plasma and urine samples of our healthy postmenopausal female volunteers9,15. In order to assess the potential role of glucuronidation in the metabolism of exemestane and 17-hydroexemestane, plasma and urine samples were assayed with and without beta-glucuronidase treatment to reflect total (unconjugated + conjugated) and free (unconjugated) concentrations, respectively. To a 100 μl (200 μl) of subject plasma (urine), 100 μl (200 μl) of 200 units/ml beta-glucuronidase in 75 mM phosphate buffer (pH 7.4) was added. In the absence of beta-glucuronidase, 75 mM phosphate buffer alone was added to plasma or urine samples. The mixture was then allowed to incubate at 37°C for 24 hr. Thereafter, 100 μl (200 μl) of 300 ng/ml internal standard (norgestrel) and 100 μl (200 μl) of acetonitrile were added, vortex-mixed and centrifuged at 6000 g for 10 min in a Sorvall ST 16R centrifuge (Osterode, Germany). The supernatant was then removed and injected onto an LC-MS/MS instrument consisting of an Agilent 1100 HPLC pump (Tokyo, Japan), a CTC Analytics HTCPAC autosampler (Zwingen, Switzerland), and an API 4000 QTRAP MS/MS system (Toronto, Canada). The separation system included a Kinetex 2.6 μm 150 × 4.6 mm column (Phenomenex, Torrance, CA, USA), a security 4 × 3 mm guard column, and a mobile phase consisting of 50% acetonitrile and 50% water at a flow rate of 500 μl/min. The mass spectrometer was operated using electrospray ionization with an ion spray voltage of 5000 V and the temperature was set at 700°C. The positive ion multiple reaction monitoring (MRM) mode was used for analysis and was performed using nitrogen as the collision gas. The nebulizer gas (GS1) and the turbo heater gas (GS2) were set at 20V and 50V, respectively. Declustering potential was 50V for both exemestane and 17-hydroexemestane. A dwell time of 200 ms and a pause time of 5 ms between scans were used to monitor precursor/product ion pairs. Two positive transitions (a quantifier MRM and a qualifier MRM) were used for each analyte. Plasma and urine concentrations of exemestane and 17-hdyroexemestane were quantified using the ratio of peak area of exemestane and 17-hydroexemestane to that of the internal standard and calibration curves that were constructed by spiking blank plasma and urine with known amounts of exemestane and 17-hydroexemestane. Data collection and processing was performed using the Analyst software package (version 1.6, AB SCIEX, Ontario, Canada).
Plasma and urine concentrations of exemestane and its major active metabolite 17-hydroexemestane were quantitated using a sensitive and validated LC-MS/MS method9,15. An API 4000 QTRAP instrument was used for quantitation using the positive electrospray ionization technique and multiple reaction monitoring mode. The monitored precursor to product-ion transitions for exemestane, 17-hydroexemestane, and Norgestrel (the internal standard) were 297->121, 299->135, and 313->109, respectively. Peak areas for exemestane and 17-hydroexemestane were linear from 0 to 200 ng/ml (r2 = 0.998). The lower limit of quantitation for exemestane and 17-hydroexemestane were 0.2 and 0.1 ng/ml, respectively. The between-run imprecision (n = 10) was 6.45%. The total chromatographic run time was 13 min. The retention times of 17-hydroexemestane, exemestane, and internal standard were 7.5, 9.5, and 10.7 min, respectively. A representative chromatogram of exemestane, 17-hydroexemestane, and internal standard is shown in Fig. 1
Fig. 1.
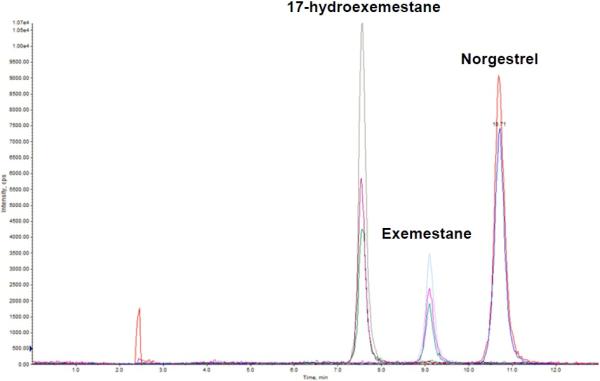
Representative Multiple Reaction Monitoring (MRM) chromatogram of exemestane, 17-hydroexemestane, and norgestrel (internal standard) of a spiked plasma sample. The retention times of 17-hydroexemestane, exemestane, and internal standard were 7.5, 9.5, and 10.7 min, respectively.
Chemicals
17-hydroexemestane was obtained from Toronto Research Chemicals Inc. (Toronto, Canada). Exemestane, norgestrel (internal standard), beta-glucuronidase, and phosphate buffer were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents were of HPLC grade or of the highest grade commercially available.
Data analysis
Pharmacokinetic and statistical analyses were performed using Microsoft Excel (version 2013) and Prism (version 6, Graph Pad software, San Diego, CA), respectively. Cmax and Tmax were obtained by inspection. AUC0-8 was calculated by the linear trapezoidal method for the observed values. AUC0-∞ was determined by extrapolation to infinity. The demographics (BMI, age, and weight) and the pharmacokinetic (Cmax, Tmax, AUC0-8, and AUC0-∞) were compared with the subjects’ genotype using a two-sided unpaired nonparametric Mann-Whitney U Test. A value of p ≤0.05 was considered statistically significant.
RESULTS
Subject characteristics and UGT2B17 genotyping data
14 healthy, postmenopausal female volunteers were selected to participate in an open-label nonrandomized exemestane pharmacokinetic clinical study. The subject characteristics of these healthy female volunteers are described in Table 1. Eight women carried the UGT2B17*1/*1 genotype and the remaining six were homozygous for the UGT2B17*2 allele. There were no statistically significant differences in body mass index, age, and weight between genotype groups (Table 1). Thirteen participants completed the study without adverse events. One subject complained of severe headaches prior to the start of the study due to caffeine withdrawal. The subject's symptoms were resolved within the next 24 hours. All participants completed the study.
Table 1.
Descriptive data of study participants.
|
UGT2B17 Genotype |
|||
|---|---|---|---|
| Demographics | *1/*1 (n = 8) | *2/*2 (n =6) | P values |
| BMI Kg/m2 (SD) | 26.2 (4.9) | 27.5 (5.8) | 0.9 |
| Age, years (SD) | 57.5 (6.2) | 54.0 (4.0) | 0.4 |
| Weight, Kg (SD) | 70.3 (13.6) | 74.2 (15.5) | 0.9 |
SD, standard deviation.
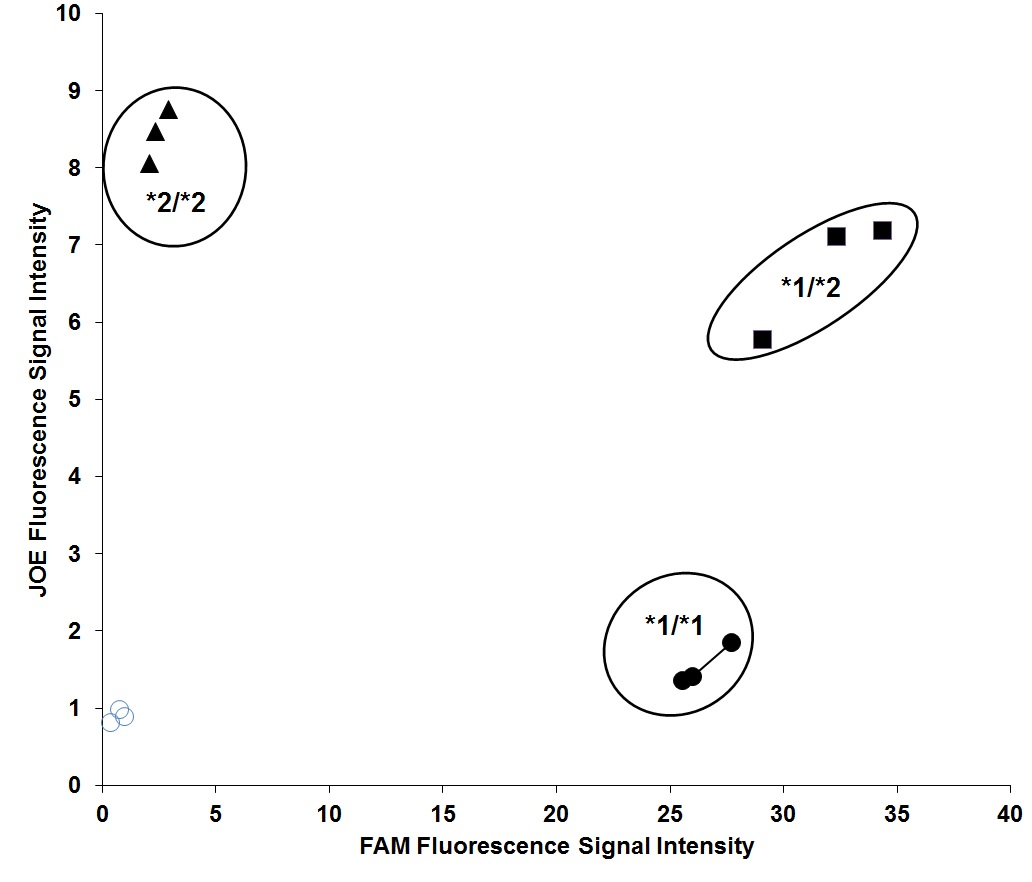
UGT2B17 genotyping was carried out using a previously described fluorescent-based assay14,16. A representative plot of UGT2B17 genotyping is shown in Supplemental Figure F1.
Impact of UGT2B17 Gene Deletion on Exemestane pharmacokinetics
The area under the plasma concentration-time curve of exemestane (EXE AUC0-8) and 17-hydroexemestane (17-HEXE AUC0-8) were calculated based on plasma concentrations at predose and at 0.5, 1, 2, 4, 6, and 8 hours after exemestane administration in the presence and absence of beta-glucuronidase using the linear trapezoidal rule. Plasma concentrations at 24 and 72 hours after exemestane administration were not calculated because they were below the lowest limit of quantification. Fig. 2 shows the plasma concentration time profiles of exemestane and 17-hydroexemestane of 14 healthy postmenopausal female volunteers who received a single oral 25 mg dose of exemestane. 24 hr urine concentrations of exemestane (EXE C24hr) and 17-hydroexemestane (17-HEXE C24hr) were determined using LC-MS/MS as described in the METHODS section. The impact of UGT2B17 gene deletion on exemestane pharmacokinetics was assessed using a two-sided unpaired nonparametric Mann-Whitney U Test.
Fig. 2.

Pharmacokinetic of exemestane and 17-hydroexemestane stratified by UGT2B17 genotype. Concentration vs. time plots of exemestane and 17-hydroexemestane was measured with a validated LC-MS/MS assay in subject plasma samples up to 72 hr after administration of a single oral 25 mg exemestane dose. Plasma samples were analyzed with beta-glucuronidase (+BG) and without incubation with beta-glucuronidase (−BG). A (Exemestane +BG), B (17-hydroexemestane +BG), C (exemestane –BG), and D (17-hydroexemestane –BG). Circles and squares represent UGT2B17*1/*1 and UGT2B17*2/*2 genotypes, respectively. The bars depict standard deviations of the mean.
In the absence of beta-glucuronidase (−BG), there were no significant differences in the pharmacokinetics (AUC and Cmax) of exemestane and 17-hydroexemestane between genotype groups (Tables 2A &3A and Fig. 2C &2D).
Table 2.
Pharmacokinetic (PK) of exemestane and 17-hydroexemestane of serum samples processed in the absence (A) and presence (B) of beta-glucuronidase. The data are stratified by UGT2B17 genotype.
| A. Without beta-glucuronidase | ||||||
|---|---|---|---|---|---|---|
| Exemestane | 17-hydroexemestane | |||||
| PK Parameter, unit | UGT2B17 *1/*1 (n = 8) | UGT2B17 *2/*2 (n = 6) | P values | UGT2B17 *1/*1 (n = 8) | UGT2B17 *2/*2 (n = 6) | P values |
| AUC0-∞, ng × hr/ml (SD) | 22.7 (10.5) | 19.7 (3.8) | 0.7 | 1.2 (0.8) | 1.3 (0.3) | 0.4 |
| Cmax, ng/ml (SD) | 5.6 (10.5) | 5.3 (1.4) | 0.7 | 0.2 (0.1) | 0.2 (0.1) | 0.9 |
| Tmax, hr (SD) | 3.1 (1.2) | 1.7 (1.2) | 0.1 | 3.6 (1.5) | 2 (1.1) | 0.06 |
| B. With beta-glucuronidase | ||||||
|---|---|---|---|---|---|---|
| Exemestane | 17-hydroexemestane | |||||
| PK Parameter, unit | UGT2B17 *1/*1 (n = 8) | UGT2B17 *2/*2 (n = 6) | P values | UGT2B17 *1/*1 (n = 8) | UGT2B17 *2/*2 (n = 6) | P values |
| AUC0-∞, ng × hr/ml (SD) | 19.8 (5.5) | 19.0 (3.3) | 0.5 | 11.7 (7.1) | 1.6 (0.7) | 0.0007 |
| Cmax, ng/ml (SD) | 4.4 (1.6) | 5.2 (1.5) | 0.3 | 1.7 (1.5) | 0.2 (0.1) | 0.0007 |
| Tmax, hr (SD) | 3.1 (1.2) | 1.9 (1.2) | 0.1 | 3.5 (0.9) | 2 (1.1) | 0.02 |
AUC, area under the plasma concentration-time curve; SD, standard deviation;
Table 3.
24 hr concentrations of exemestane and 17-hydroexemestane of urine samples processed in the presence and absence of beta-glucuronidase. The results are stratified by UGT2B17 genotype.
| A. Without beta-glucuronidase | ||||||
|---|---|---|---|---|---|---|
| Exemestane | 17-hydroexemestane | |||||
| PK Parameter, unit | UGT2B17 *1/*1 (n = 8) | UGT2B17 *2/*2 (n = 6) | P values | UGT2B17 *1/*1 (n = 8) | UGT2B17 *2/*2 (n = 6) | P values |
| C24 hr, ng/ml (SD) | 1.9 (0.9) | 2.5 (2.1) | 0.7 | 0.4 (0.2) | 0.4 (0.3) | 0.9 |
| B. With beta-glucuronidase | ||||||
|---|---|---|---|---|---|---|
| Exemestane | 17-hydroexemestane | |||||
| PK Parameter, unit | UGT2B17 *1/*1 (n = 8) | UGT2B17 *2/*2 (n = 6) | P values | UGT2B17 *1/*1 (n = 8) | UGT2B17 *2/*2 (n = 6) | P values |
| C24 hr, ng/ml (SD) | 1.7 (1.0) | 2.1 (1.6) | 0.8 | 2.3 (1.2) | 0.5 (0.3) | 0.001 |
SD, standard deviation; C24 hr, 24-hr urine concentration.
In the presence of beta-glucuronidase (+BG), EXE AUC0-8, EXE AUC0-∞, and urine EXE C24hr did not differ significantly between UGT2B17*1/*1 and UGT2B17*2/*2 carriers (EXE AUC0-8 19.8 ± 5.5 vs 19.0 ± 0.5 ng × hr/ml, p = 0.5; EXE AUC0-8 16.2 ± 3.5 vs 15.5 ± 4.2 ng × hr/ml, p = 0.7; EXE C24hr 1.7 ± 1.0 vs 2.1 ± 1.6 ng/ml, p = 0.8) (Tables 2B & 3B and Fig. 2A). However there were statistically significant differences in 17-HEXE AUC0-∞, 17-HEXE AUC0-8 and 17-HEXE C24hr between genotype groups (17-HEXE AUC0-∞, 11.7 ± 7.1 vs 1.6 ± 0.7 ng × hr/ml, p = 0.0007; 17-HEXE AUC0-8 7.3 ± 2.6 vs 0.9 ± 0.2 ng × hr/ml, p = 0.0007; 17-HEXE C24hr 2.3 ± 1.2 vs 0.5 ± 0.3 ng/ml, p = 0.001) (Tables 2B & 3B and Fig. 2B).
The distribution of pharmacokinetic parameters of exemestane and 17-hydroexemestane based on UGT2B17 genotype in the presence and absence of beta-glucuronidase is shown in Fig. 3A-D and Fig. 4A-D for Cmax and AUC0-8, respectively. We also assessed the influence of UGT2B17 gene deletion on parent drug to metabolite ratio. Our analysis showed that exemestane to 17-hydroexemestane AUC0-∞ ratio differed significantly and statistically between UGT2B17*1/*1 and *2/*2 carriers in the presence of beta-glucuronidase (Fig. 5B, p = 0.0007).
Fig. 3.

Cmax of exemestane and 17-hydroexemestane stratified by UGT2B17 genotype. Plasma samples were analyzed with beta-glucuronidase (+BG) and without incubation with beta-glucuronidase (−BG). A (Exemestane −BG), B (exemestane +BG), C (17-hydroexemestane −BG), and D (17-hydroexemestane +BG). Circles and squares represent UGT2B17*1/*1 and UGT2B17*2/*2 genotypes, respectively.
Fig. 4.

AUC0-∞ of exemestane and 17-hydroexemestane stratified by UGT2B17 genotype. Plasma samples were analyzed with beta-glucuronidase (+BG) and without incubation with beta-glucuronidase (−BG). A (Exemestane −BG), B (exemestane +BG), C (17-hydroexemestane −BG), D (17-hydroexemestane +BG). Circles and squares represent UGT2B17*1/*1 and UGT2B17*2/*2 genotypes, respectively.
Fig. 5.

Ratio of AUC0-∞ of exemestane over AUC0-∞ of 17-hydroexemestane stratified by UGT2B17 genotype. Plasma samples were analyzed with beta-glucuronidase (+BG) and without incubation with beta-glucuronidase (−BG). A (Exemestane AUC0-∞ / 17-hydroexemestane AUC0-∞ −BG) and B (Exemestane AUC0-∞ / 17-hydroexemestane AUC0-∞ +BG). Circles and squares represent UGT2B17*1/*1 and UGT2B17*2/*2 genotypes, respectively.
DISCUSSION
This pilot, clinical pharmacogenetic study reports for the first time an association between UGT2B17 gene deletion and exemestane metabolism in humans in vivo. In our study, we enrolled healthy volunteers that either expressed (n = 8 *1/*1) or did not express (n = 6 *2/*2) UGT2B17. Plasma and urine samples were assayed with and without beta-glucuronidase treatment to reflect total (unconjugated + conjugated) and free (unconjugated) concentrations, respectively. Our results showed that there were statistically significant differences in the total plasma 17-hydroexemestane AUC0-∞ (p = 0.0007), AUC0-8 (p = 0.0007) and urine 17-hydroexemestane C24hr (p = 0.001) between UGT2B17 genotype groups. These findings suggest that UGT2B17 gene deletion influences 17-hydroexemestane pharmacokinetics in humans.
Several phase I exemestane single dose pharmacokinetic clinical studies in postmenopausal volunteers have been conducted in the past. Most of these studies were designed to assess the efficacy and safety of exemestane following a single oral 25 mg dose. Others were carried out to study the effects of hepatic and renal impairment, or of concomitant raloxifene administration on the pharmacokinetics of exemestane 17-19. In these studies, the pharmacokinetic parameters of exemestane and its major metabolite 17-hydroexemestane were calculated. They found that plasma concentrations over time (AUC, ng × hr/ml) and maximal plasma concentrations (Cmax, ng/mL) of exemestane and 17-hydroexemestane ranged from 25.6 to 98.1 ng × hr/ml and 2.46 to 20.89 ng × hr/ml, and from 3.0 to 16.1 ng/ml and 0.22 to 2.44 ng/ml, respectively. In our pharmacokinetic study, AUC and Cmax of exemestane and 17-hydroexemestane ranged from 9.2 to 40.6 ng × hr/ml and 0.7 to 26.9 ng × hr/ml, and from 2.2 to 13.3 ng/ml and 0.1 to 2.7 ng/ml, respectively. This indicates that our pharmacokinetic data concur with previously reported studies 17-19.
In 2010, Sun et al. observed a significant correlation between 17-hydroexemestane glucuronide formation and UGT2B17 expression in vitro 10. Using a panel of 110 human liver microsomes with different levels of UGT2B17 mRNA (64 high expressers or *1/*1; 34 intermediate expressers or *1/*2; and 12 poor expressers or *2/*2), Sun et al. showed that there was a 36-fold difference in the intrinsic clearance of 17-hydroexemestane via UGT-mediated deactivation between high and poor expressers10. We validated this in vitro finding in our human pharmacogenetic study in vivo, as we observed an almost 8-fold difference in the AUC and Cmax of conjugated 17-hydroexemestane between high and poor expressers.
Previous research studies have demonstrated a relationship between UGT2B17 gene deletion and human physiology and pathophysiology. UGT2B17 is known to be involved in the metabolism of endogenous estrogens and androgens, such as estradiol and testosterone 20,21. Juul et al. showed that UGT2B17 strongly affects urinary excretion patterns of androgen metabolites in pubertal boys. They observed that subjects with a homozygous deletion of UGT2B17 had significantly lower urinary levels of testosterone and 5alpha- and 5beta-androstanediol. A similar trend was seen in a larger case-control genome-wide copy number variation study, which aimed at identifying the genetic markers associated with the pathogenesis of osteoporosis 21. In their study involving subjects consisting of 689 Chinese and 1000 Caucasians, Yang et al. demonstrated that individuals with zero copies of UGT2B17 (poor expressers) had significantly higher serum testosterone and estradiol concentrations. They also showed that Caucasian and Chinese individuals, with either one or two copies of UGT2B17, had lower bone mineral density and increased risk of osteoporosis. These studies suggest that UGT2B17 may play a significant role in human growth and disease. In our current study, we report for the first time a link between UGT2B17 gene deletion and exemestane disposition in vivo.
What are the clinical implications of our findings? It is generally accepted that aromatase inhibitor drugs exemestane, anastrozole, and letrozole are clinically equivalent. Consequently, oncologists have used these drugs interchangeably with the assumption that they exhibit similar risk-to-benefit ratios 3,22-24. This may not be appropriate because it has been observed in clinical practice that patients who complain of serious adverse events, such as arthralgia, with one aromatase inhibitor can tolerate another. This suggests that there are potentially significant differences between aromatase inhibitors and that there is significant interindividual variability in response to this pharmacological class of agents 25-28. Some studies have attempted to address this by demonstrating that distinct pharmacological differences exist between steroidal (exemestane) and nonsteroidal aromatase inhibitors (letrozole and anastrozole) 4-6,29-32. Preclinical studies in ovariectomized rats have shown that exemestane is less toxic to bone than nonsteroidal aromatase inhibitors 4-6. In these preclinical studies, 17-hydroexemestane was identified as the primary molecule responsible for exemestane-induced bone protection. Some clinical studies have validated these animal findings by demonstrating that women receiving non-steroidal aromatase inhibitors have significantly lower bone mineral densities than those treated with exemestane29-32. However, several other clinical studies have not replicated these observations. We hypothesize that the failure to demonstrate superiority or inferiority of exemestane over nonsteroidal aromatase inhibitors in larger clinical trials may be due to significant inter-individual variability in the formation of 17-hydroexemestane. In this pilot study, we showed that the interindividual variability in 17-hydroexemestane metabolism and disposition is dependent on UGT2B17 genotype status. Given the high frequency of the UGT2B17 gene deletion (UGT2B17*1/*1: 48-53% versus UGT2B17*1/*2: 39-40% and UGT2B17*2/*2: 8-12%) 11-13, future pharmacogenetic studies aimed at assessing the differences in exemestane pharmacokinetics and pharmacodynamics as well as breast cancer outcomes among these three UGT2B17 genotyping groups are now warranted.
Supplementary Material
{kind=link}
Acknowledgments
This publication was made possible by the 2011 Young Investigator Award supported by grant funding from the American Society for Clinical Pharmacology and Therapeutics (ASCPT) and by the Arkansas INBRE program, supported by grant funding from the National Institute of Health (NIH) National Institute of General Medical Sciences (NIGMS) (P20 GM103429). We thank Mr. David Crouch for helping with subject recruitment. We are very thankful to study volunteers, White County Medical Center, students, faculty and staff of the Harding community for their participation and contribution. We are especially grateful to Drs. Kathryn Momary, Susan Rahman, and Reggie Ewesuedo for the mentorship they exhibited throughout the conduct of this clinical study.
Footnotes
Disclosure: The authors have no conflict of interest to disclose.
REFERENCES
- 1.Buzdar AU, Coombes RC, Goss PE, Winer EP. Summary of aromatase inhibitor clinical trials in postmenopausal women with early breast cancer. Cancer. 2008 Feb 1;112(3 Suppl):700–709. doi: 10.1002/cncr.23193. [DOI] [PubMed] [Google Scholar]
- 2.Coombes RC, Kilburn LS, Snowdon CF, et al. Survival and safety of exemestane versus tamoxifen after 2-3 years' tamoxifen treatment (Intergroup Exemestane Study): a randomised controlled trial. Lancet. 2007 Feb 17;369(9561):559–570. doi: 10.1016/S0140-6736(07)60200-1. [DOI] [PubMed] [Google Scholar]
- 3.Robinson A. A review of the use of exemestane in early breast cancer. Therapeutics and clinical risk management. 2009 Feb;5(1):91–98. [PMC free article] [PubMed] [Google Scholar]
- 4.Ariazi EA, Leitao A, Oprea TI, et al. Exemestane's 17-hydroxylated metabolite exerts biological effects as an androgen. Molecular cancer therapeutics. 2007 Nov;6(11):2817–2827. doi: 10.1158/1535-7163.MCT-07-0312. [DOI] [PubMed] [Google Scholar]
- 5.Goss PE, Qi S, Cheung AM, Hu H, Mendes M, Pritzker KP. Effects of the steroidal aromatase inhibitor exemestane and the nonsteroidal aromatase inhibitor letrozole on bone and lipid metabolism in ovariectomized rats. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004 Sep 1;10(17):5717–5723. doi: 10.1158/1078-0432.CCR-04-0438. [DOI] [PubMed] [Google Scholar]
- 6.Goss PE, Qi S, Josse RG, et al. The steroidal aromatase inhibitor exemestane prevents bone loss in ovariectomized rats. Bone. 2004 Mar;34(3):384–392. doi: 10.1016/j.bone.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Kamdem L, Flockhart D, Desta Z. 17-HYDROEXEMESTANE: A POTENT INHIBITOR OF CYP19 AND SUBSTRATE OF CYP3A. Paper presented at: CLINICAL PHARMACOLOGY & THERAPEUTICS. 2011 [Google Scholar]
- 8.Landry K, David F, Zeruesenay D. 17-Hydroexemestane: A Potent Inhibitor of CYP19 (Aromatase) and Substrate of CYP3A. J Drug Metab Toxicol. 2014;5(171):2. [Google Scholar]
- 9.Kamdem LK, Flockhart DA, Desta Z. In vitro cytochrome P450-mediated metabolism of exemestane. Drug metabolism and disposition: the biological fate of chemicals. 2011 Jan;39(1):98–105. doi: 10.1124/dmd.110.032276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun D, Chen G, Dellinger RW, Sharma AK, Lazarus P. Characterization of 17-dihydroexemestane glucuronidation: potential role of the UGT2B17 deletion in exemestane pharmacogenetics. Pharmacogenetics and genomics. 2010 Oct;20(10):575–585. doi: 10.1097/FPC.0b013e32833b04af. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen G, Giambrone NE, Jr., Dluzen DF, et al. Glucuronidation genotypes and nicotine metabolic phenotypes: importance of functional UGT2B10 and UGT2B17 polymorphisms. Cancer research. 2010 Oct 1;70(19):7543–7552. doi: 10.1158/0008-5472.CAN-09-4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallagher CJ, Kadlubar FF, Muscat JE, Ambrosone CB, Lang NP, Lazarus P. The UGT2B17 gene deletion polymorphism and risk of prostate cancer. A case-control study in Caucasians. Cancer detection and prevention. 2007;31(4):310–315. doi: 10.1016/j.cdp.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson W, 3rd, Pardo-Manuel de Villena F, Lyn-Cook BD, et al. Characterization of a common deletion polymorphism of the UGT2B17 gene linked to UGT2B15. Genomics. 2004 Oct;84(4):707–714. doi: 10.1016/j.ygeno.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Gallagher CJ, Muscat JE, Hicks AN, et al. The UDP-glucuronosyltransferase 2B17 gene deletion polymorphism: sex-specific association with urinary 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol glucuronidation phenotype and risk for lung cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2007 Apr;16(4):823–828. doi: 10.1158/1055-9965.EPI-06-0823. [DOI] [PubMed] [Google Scholar]
- 15.Corona G, Elia C, Casetta B, et al. A liquid chromatography-tandem mass spectrometry method for the simultaneous determination of exemestane and its metabolite 17-dihydroexemestane in human plasma. Journal of mass spectrometry : JMS. 2009 Jun;44(6):920–928. doi: 10.1002/jms.1566. [DOI] [PubMed] [Google Scholar]
- 16.Balliet RM, Chen G, Gallagher CJ, Dellinger RW, Sun D, Lazarus P. Characterization of UGTs active against SAHA and association between SAHA glucuronidation activity phenotype with UGT genotype. Cancer research. 2009 Apr 1;69(7):2981–2989. doi: 10.1158/0008-5472.CAN-08-4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groenewoud G, Nell A, Potgieter L, Seiler D, Wettmarshausen C. Bioequivalence of exemestane in post-menopausal females. Arzneimittel-Forschung. 2010;60(2):96–100. doi: 10.1055/s-0031-1296255. [DOI] [PubMed] [Google Scholar]
- 18.Jannuzzo MG, Poggesi I, Spinelli R, Rocchetti M, Cicioni P, Buchan P. The effects of degree of hepatic or renal impairment on the pharmacokinetics of exemestane in postmenopausal women. Cancer chemotherapy and pharmacology. 2004 Jun;53(6):475–481. doi: 10.1007/s00280-004-0774-5. [DOI] [PubMed] [Google Scholar]
- 19.Traina TA, Poggesi I, Robson M, et al. Pharmacokinetics and tolerability of exemestane in combination with raloxifene in postmenopausal women with a history of breast cancer. Breast cancer research and treatment. 2008 Sep;111(2):377–388. doi: 10.1007/s10549-007-9787-1. [DOI] [PubMed] [Google Scholar]
- 20.Juul A, Sorensen K, Aksglaede L, et al. A common deletion in the uridine diphosphate glucuronyltransferase (UGT) 2B17 gene is a strong determinant of androgen excretion in healthy pubertal boys. The Journal of clinical endocrinology and metabolism. 2009 Mar;94(3):1005–1011. doi: 10.1210/jc.2008-1984. [DOI] [PubMed] [Google Scholar]
- 21.Yang TL, Chen XD, Guo Y, et al. Genome-wide copy-number-variation study identified a susceptibility gene, UGT2B17, for osteoporosis. American journal of human genetics. 2008 Dec;83(6):663–674. doi: 10.1016/j.ajhg.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deeks ED, Scott LJ. Exemestane: a review of its use in postmenopausal women with breast cancer. Drugs. 2009;69(7):889–918. doi: 10.2165/00003495-200969070-00007. [DOI] [PubMed] [Google Scholar]
- 23.Eisen A, Trudeau M, Shelley W, Messersmith H, Pritchard KI. Aromatase inhibitors in adjuvant therapy for hormone receptor positive breast cancer: a systematic review. Cancer treatment reviews. 2008 Apr;34(2):157–174. doi: 10.1016/j.ctrv.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Untch M, Jackisch C. Exemestane in early breast cancer: a review. Therapeutics and clinical risk management. 2008 Dec;4(6):1295–1304. doi: 10.2147/tcrm.s4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beresford M, Tumur I, Chakrabarti J, Barden J, Rao N, Makris A. A qualitative systematic review of the evidence base for non-cross-resistance between steroidal and non-steroidal aromatase inhibitors in metastatic breast cancer. Clinical oncology (Royal College of Radiologists (Great Britain)) 2011 Apr;23(3):209–215. doi: 10.1016/j.clon.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 26.Goss PE, Ingle JN, Pritchard KI, et al. Exemestane versus anastrozole in postmenopausal women with early breast cancer: NCIC CTG MA.27--a randomized controlled phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013 Apr 10;31(11):1398–1404. doi: 10.1200/JCO.2012.44.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lonning PE. Lack of complete cross-resistance between different aromatase inhibitors; a real finding in search for an explanation? European journal of cancer (Oxford, England : 1990) 2009 Mar;45(4):527–535. doi: 10.1016/j.ejca.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 28.Lonning PE, Bajetta E, Murray R, et al. Activity of exemestane in metastatic breast cancer after failure of nonsteroidal aromatase inhibitors: a phase II trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000 Jun;18(11):2234–2244. doi: 10.1200/JCO.2000.18.11.2234. [DOI] [PubMed] [Google Scholar]
- 29.Cheung AM, Tomlinson G, Goss PE. Bone loss with exemestane: Is the jury still out? Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005 Dec 20;23(36):9433–9434. doi: 10.1200/JCO.2005.04.1376. author reply 9433-9435. [DOI] [PubMed] [Google Scholar]
- 30.Chien AJ, Goss PE. Aromatase inhibitors and bone health in women with breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006 Nov 20;24(33):5305–5312. doi: 10.1200/JCO.2006.07.5382. [DOI] [PubMed] [Google Scholar]
- 31.Goss PE, Hadji P, Subar M, Abreu P, Thomsen T, Banke-Bochita J. Effects of steroidal and nonsteroidal aromatase inhibitors on markers of bone turnover in healthy postmenopausal women. Breast cancer research : BCR. 2007;9(4):R52. doi: 10.1186/bcr1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lonning PE, Geisler J, Krag LE, et al. Effects of exemestane administered for 2 years versus placebo on bone mineral density, bone biomarkers, and plasma lipids in patients with surgically resected early breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005 Aug 1;23(22):5126–5137. doi: 10.1200/JCO.2005.07.097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.