

Abstract
Overexpression of B cell lymphoma 2 (Bcl-2) proteins is associated with therapy resistance in various human cancers. Traditional approaches target the Bcl-2 homology (BH)3 domain of Bcl-2; however, the BH4 domain represents a superior therapeutic target in light of its unique structure and crucial involvement in many cellular functions. In this critical review, we focus on the structural and functional basis of targeting the BH4 domain of Bcl-2, and highlight the recent advances in drug discovery efforts toward small-molecule BH4 domain inhibitors (e.g. BDA-366). The proof-of-concept studies support the hypothesis that targeting the BH4 domain of Bcl-2 holds promise to offer a novel anticancer therapy through the induction of apoptosis and an increased potential to overcome therapeutic resistance.
Keywords: BH4 domain, Bcl-2 proteins, cancer therapy, BH4 antagonists, apoptosis, drug resistance
Introduction
B cell lymphoma 2 (Bcl-2) family proteins play a crucial role in regulating apoptosis, an important biological process that eliminates cells with an increased potential to malignancy such as those with damaged DNA or aberrant cell cycling [1–3]. The Bcl-2 family can be classified into three subfamilies according to the existence of Bcl-2 homology (BH) domains and different functions: antiapoptotic (or prosurvival) multidomain proteins, proapoptotic multidomain proteins and proapoptotic BH3-only proteins. Overexpression of antiapoptotic Bcl-2 family proteins is implicated in the pathology and drug resistance of various hematopoietic malignancies and solid tumors [4,5]. Most antiapoptotic family members (e.g. Bcl-2, Bcl-xL, Bcl-w and Bcl-B) and proapoptotic multidomain proteins (e.g. Bax, Bak and Bok) share BH1–BH4 domains [6] (Figure 1), whereas proapoptotic BH3-only proteins (e.g. Bid, Bim, Bad and Puma) only have the BH3 domain. All Bcl-2 family members contain this death-promoting BH3 domain, which is crucially important to the interactions between proapoptotic and antiapoptotic proteins.
Figure 1.

Sequence and structure of B cell lymphoma 2 (Bcl-2). (a) Bcl-2 contains BH1–4 domains. (b) The sequences of Bcl-2 BH1–4 domains. The colored letters represent different α-helical structures. (c) Left panel: the 3D NMR structure of Bcl-2 with Hα1–8 is displayed in different colors according to the same color coding in (b) (PDB ID codes: 1G5M and 1GJH). Right panel: the surface structure of Bcl-2 is shown with BH4 domain in the form of ribbon. Abbreviations: BH, Bcl-2 homology; TM, transmembrane domain.
Not surprisingly, targeting the BH3 domain to generate Bcl-2 inhibitors for cancer therapy has attracted tremendous attention over the past decades [7,8]. So far, nearly a dozen BH3 mimetics as Bcl-2 inhibitors are under investigation in different phases of human clinical trials such as ABT-737 [9], ABT-263 (navitoclax, orally available derivative of ABT-737) [10], ABT-199 (venetoclax) [11], AT-101 (R-(−)-gossypol), GX15-070 (obatoclax) and TW37. Nevertheless, to date, none of the aforementioned compounds has been approved for use in the clinic for cancer treatment. The limited efficacies of navitoclax, AT-101 and obatoclax reported in clinical trials have excluded them from the potential commercialization either as a single therapeutic agent or in combination with other cancer therapy [12–14]. In addition, the side effects (e.g. dose-limiting thrombocytopenia of navitoclax) and other off-target effects remain an issue [15]. Another challenge of these BH3 mimics is their susceptibility to cancer cell resistance, particularly induced by Mcl-1 and A1 [16,17]. Excitingly, venetoclax has very recently been granted breakthrough therapy designation for the treatment of 17p deletion relapsed-refractory chronic lymphocytic leukemia (CLL) (http://www.roche.com). Thus, identifying novel Bcl-2 inhibitors and relevant new therapeutic targets are urgently needed.
To this end, targeting the BH4 domain of Bcl-2 holds promise to offer a novel and attractive means for anticancer therapy by induction of apoptosis. The BH4 domain is identified as a crucial domain for the antiapoptotic activity of Bcl-2 [18]. Bcl-2 lacking the BH4 domain was found to promote rather than inhibit apoptosis [19]. Different from the BH3 domain, which usually interacts with Bcl-2 relatives, the BH4 domain is involved in many cellular functions that do not belong to the Bcl-2 family [20,21]. In this review, we summarize the current understanding of the structure and physiological functions of the Bcl-2 BH4 domain and focus on recent advances in targeting the Bcl-2 BH4 domain as a new means for anticancer therapy.
Structure of Bcl-2
As the ‘founding member’ of Bcl-2 family proteins, Bcl-2 was originally identified as the proto-oncogene involved in the t(14;18) translocation in human follicular lymphoma in 1985 [22]. The 3D structure of a Bcl-2–Bcl-xL chimeric protein containing a truncated loop derived from Bcl-xL between the Hα1 and Hα2 was first determined by NMR spectroscopy [23]. Bcl-2 exhibits a similar tertiary structure to Bcl-xL (Figure 1) consisting of two predominantly hydrophobic α-helices (Hα5 and Hα6) surrounded by six amphipathic α-helices. From the N terminus to the C terminus, Hα1 to Hα8 are connected one by one with or without a loop. The highly conserved BH4 domain is located on the native N-terminal domain of Bcl-2, comprising a stretch of 20 amino acids (residues 10–30) organized in an α-helical structure. The BH1, BH2 and BH3 domains form a hydrophobic groove where BH3-only proteins can bind, and mutations in this region have been shown to abolish the antiapoptotic activity of Bcl-2 and also block the heterodimerization with other Bcl-2 relatives. By contrast, the deletion of the BH4 domain completely eliminates the antiapoptotic activity of Bcl-2, while having no influence on the ability of Bcl-2 to bind BH3-only proteins [18,24]. Moreover, BH4-domain-deleted Bcl-2 (ΔBH4 Bcl-2) can also function as a dominant killer instead of a survival protein and promote cell death, similar to the protein Bax [19,25]. Moreover, the BH4 domain of Bcl-2 is also involved in angiogenesis and autophagy [25–27].
Functions of Bcl-2 BH4 domain
Bcl-2 regulates apoptosis not only by neutralizing proapoptotic Bcl-2 relatives but also by interacting with other apoptosis regulators that are not members of the Bcl-2 family of proteins [28]. The BH3 domain modulates the interactions with Bcl-2 family members, whereas the BH4 domain can also participate in the interactions with other crucial proteins beyond the Bcl-2 family. The Bcl-2 BH4 domain interacts with Bax, a requisite gateway protein of apoptosis in the Bcl-2 family, and also engages in a growing list of non-Bcl-2 proteins including apoptosis-stimulating of p53 protein 2 (ASPP2), cell death protein 4 (CED-4), human mutS homolog 6 (hMSH6), inositol 1,4,5-trisphosphate receptor (IP3R), c-Myc Ras, human ribonucleotide reductase subunit M2 (hRRM2), ryanodine receptor (RyR) and voltage-dependent anion channel (VDAC) with more details presented below.
Interaction with Bax
In the early 1990s, the NH2-terminal region (residues 1–34), which contains the BH4 domain, was identified as important for Bcl-2 binding to Bax. Substitutions of hydrophobic residues of the BH4 domain (e.g. V15E) or deletion of the BH4 domain can result in the loss of the capability of Bcl-2 to form a heterodimer with Bax and the failure to inhibit staurosporine-induced apoptosis [29]. Very recently, the stapled Bcl-2 BH4 domain helices were revealed to bind directly and inhibit proapoptotic Bax by a noncanonical mechanism [30]. Different from the classical BH3-in-groove interaction, the noncanonical Bcl-2-BH4–Bax interaction blocks the conformational activation of Bax and reinforces its inactive, monomeric state. This novel interaction provides new opportunities in that Bcl-2 BH4 inhibitors interfering with Bcl-2-BH4–Bax interaction could have great potential to combat pathologic cell survival. Furthermore, BH4 mimetics targeting this interaction could serve as novel inhibitors to abolish unwanted or premature cell death.
Interaction with IP3R
Bcl-2 inhibits apoptosis not only by preserving mitochondria integrity but also regulating Ca2+ signals from the endoplasmic reticulum (ER) [31]. The function of regulating Ca2+ signals is mediated through the interaction of the Bcl-2 BH4 domain with the regulatory and coupling domain of IP3R [32,33]. IP3R [34], a membrane glycoprotein complex acting as an intracellular Ca2+-release channel localized predominately in the ER of all cell types, plays a crucial part in the control of cellular and physiological processes including cell division, cell proliferation and apoptosis [35,36]. Bcl-2–IP3R interaction was detected in cell extracts by co-immunoprecipitation and blue native gel electrophoresis in vitro, and also confirmed in live cells using fluorescence resonance energy transfer [37]. Bcl-2–IP3R interaction inhibits IP3R-dependent channel opening diminishing Ca2+ release from the ER and Ca2+-mediated apoptosis. The BH4 domain of Bcl-2 was found to be necessary and sufficient for this interaction by pulldown and co-immunoprecipitation experiments [32]. A peptide, TAT-BH4, was generated by fusing the cell-penetrating peptide of HIV TAT [38] to a synthetic peptide corresponding to the BH4 domain to evaluate the effect of the BH4 domain on Ca2+ release. TAT-BH4 inhibits anti-CD3-induced Ca2+ elevation and anti-CD3-induced apoptosis in the Jurkat human T cell leukemia line, indicating that TAT-BH4 has an antiapoptotic action similar to full-length Bcl-2. Moreover, the interaction of Bcl-xL BH4, which is similar to Bcl-2 BH4 in primary sequence, with IP3R was also explored [39]. Different from Bcl-2 BH4, Bcl-xL BH4 neither binds the modulatory domain of IP3R nor inhibits IP3R-induced Ca2+ release in permeabilized and intact cells. Interestingly, one residue (Lys17 in Bcl-2 BH4 versus Asp11 in Bcl-xL BH4) is crucial for the respective distinct biological properties. Replacement of Lys17 with Asp in Bcl-2 BH4 can completely abolish its IP3R binding and inhibition, whereas changing Asp11 into Lys in Bcl-xL BH4 can acquire the IP3R-binding and -inhibitory properties. Besides, it is reported that the BH4 domain of Bcl-xL, but not that of Bcl-2, inhibits apoptosis by decreasing VDAC1-mediated Ca2+ uptake into the mitochondria via binding and targeting VDAC1 [40]. Thus, it is of great potential to develop inhibitors selectively targeting the Bcl-2 BH4 domain and preserving the essential biological functions of Bcl-xL. This new approach could offer advantages to avoid potential side effects including toxicities caused by the pan-Bcl-2 inhibitory activity of BH3 mimetics.
Interaction with Raf-1
Bcl-2, but not ΔBH4 Bcl-2, was found to target Raf-1 [a serine/threonine protein kinase in the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signal transduction pathway) to mitochondria in cells [41]. The Bcl-2–Raf-1 interaction is dependent on the BH4 domain of Bcl-2 because removal of the BH4 domain from Bcl-2 can abolish its ability to bind Raf-1. This interaction results in phosphorylation of Bad and possibly other protein substrates in the vicinity of Bcl-2. Different from the traditional role of Raf-1 as a promoter of cell proliferation and differentiation within the context of the Ras signaling pathways at the plasma membrane, Raf-1 cooperates with Bcl-2 acting as a blocker of cell death at the mitochondria. Targeting the active Raf-1 to the mitochondria could promote resistance to staurosporine-induced apoptosis through interaction with Bcl-2. Kinase-inactive Raf-1 mutant was found to abolish apoptosis suppression by Bcl-2 [41].
Interaction with calcineurin
The direct binding between Bcl-2 and calcineurin, a calcium- and calmodulin-dependent serine/threonine protein phosphatase, was also revealed [42]. The BH4 domain of Bcl-2 is required for this interaction because calcineurin co-precipitates with transmembrane-domain-deleted (ΔTM Bcl-2) and C-terminus-deleted Bcl-2 (ΔC Bcl-2), but not N-terminus-deleted Bcl-2 (ΔN Bcl-2). Meanwhile, the BH4 domain alone is sufficient for the co-precipitation of calcineurin. The Bcl-2–calcineurin interaction provides an explanation for the Bcl-2 function of suppressing nuclear factor of activated T cells (NF-AT) signaling in T cells of Bcl-2 transgenic mice and blocking cell death resulting from calcineurin overexpression [43].
Interactions with other various proteins
Besides the important proteins highlighted above, Bcl-2 BH4 domain is also involved in interactions with a variety of other proteins. The Bcl-2 BH4 domain binds to Ras and the interaction blocks Ras-mediated apoptotic signaling [44]. The Bcl-2 interaction through its BH4 domain with paxillin plays an important part during nephrogenesis [45]. The Bcl-2 BH4 domain can bind directly to c-Myc, which is also an oncogenic protein that functionally promotes DNA damage, genetic instability and tumorigenesis. This interaction is essential for Bcl-2 to enhance c-Myc transcriptional activity and inhibit DNA repair [46]. Bcl-2 promotes tumor angiogenesis and BH4 deletion abrogates hypoxia-inducible factor (HIF)-1-mediated vascular endothelial growth factor (VEGF) expression in hypoxic tumor cells [27]. Xenografts derived from cells expressing ΔBH4 Bcl-2 showed a reduction of metastatic potential compared with those derived from cells expressing wild-type Bcl-2 [26]. A summary of proteins including those important ones highlighted above associated with the Bcl-2 BH4 domain and their functions is provided in Table 1.
Table 1.
Proteins associated with the Bcl-2 BH4 domain
| Associated protein |
Participating domains |
Functions | Refs |
|---|---|---|---|
| ASPP2 | BH4 of Bcl-2a | Block antiapoptotic function of Bcl-2 and induce apoptosis | [63] |
| Bax | BH1–4 of Bcl-2 | Form a heterodimer with Bax to protect cells from death | [30] |
| Calcineurin | BH4 of Bcl-2 | Suppress NF-AT signaling to block apoptosis | [42] |
| CED-4 | BH4 of Bcl-2 | Sequester CED-4 and prevent apoptosis | [18] |
| HIF-1α | BH4 of Bcl-2 | Increase tumor angiogenesis and progression | [64] |
| hMSH6 | BH4 of Bcl-2 | Enhance DNA damage and suppress DNA repair | [65] |
| hRRM2 | BH4 of Bcl-2 | Display inhibitory effects on RNR activity, dNTP pool level and DNA replication | [66] |
| IP3R | BH4 of Bcl-2 | Inhibit IP3R-dependent channel opening, suppress Ca2+ release from the ER and Ca2+-mediated apoptosis | [32] |
| c-Myc | BH4 of Bcl-2 | Enhance c-Myc transcriptional activity and inhibit DNA repair | [46] |
| Paxillin | BH4 of Bcl-2 | Impair ureteric bud branching and tubulogenesis | [45] |
| Raf-1 | BH4 of Bcl-2 | Block cell death and promote resistance to apoptosis | [41] |
| Ras | BH4 of Bcl-2 | Block the apoptotic signaling mediated by mitochondrial Ras | [44] |
| RyR | BH4 of Bcl-2 | Inhibit RyR-based intracellular Ca2+ release channels | [67] |
| VDAC | BH4 of Bcl-2 | Inhibit VDAC activity, prevent apoptotic mitochondrial changes and prevent apoptotic cell death | [68] |
Whereas all the BH1–4 domains can participate the binding, the binding interaction between BH4 domain of Bcl-2 and ASPP2 is the tightest.
Targeting Bcl-2 BH4 domain as a novel strategy for cancer therapy
Bcl-2 is overexpressed in numerous human cancers and overexpression of Bcl-2 is associated with resistance to various cytotoxic agents and radiotherapy [47]. Despite the identification of Bcl-2 as a drug target over the past decades, targeting the specific BH4 domain of Bcl-2 is a novel strategy for cancer therapy. It is a relatively recent endeavor in comparison with the traditional targeting of the BH3 domain. Importantly, not all of the Bcl-2 family members share the BH4 domain. The structure of the Bcl-2 BH4 domain is unique, and even the highly homologous Bcl-xL BH4 is different in crucial residues and function [39,40]. Thus, developing selective Bcl-2 BH4 inhibitors with low toxicity might be feasible. More importantly, Bcl-2 via its BH4 domain cooperates with numerous proteins that participate in different signaling pathways and could be associated with resistance to apoptosis. For example, the interaction between Bcl-2 BH4 and Raf-1 promotes resistance to staurosporine-induced apoptosis [41]. The acquired generalized resistance to apoptosis in cadmium-transformed cells could be the result of blocking the activation of the c-Jun N-terminal kinase (JNK) signaling pathway by Bcl-2 overexpression. Moreover, the Bcl-2 BH4 domain is required for modulating JNK phosphorylation [48]. Hence, disturbing these interactions by targeting the BH4 domain holds great promise to induce apoptosis of cancer cells with the potential to overcome resistance [49,50].
To date, several BH4 domain inhibitors have been identified as research tools to understand the role of the Bcl-2 BH4 domain and as promising drug candidates as a new class of anticancer agents. Intriguingly, small molecule BDA-366 was recently discovered as a potent and effective BH4 domain antagonist that displays remarkable anticancer activity in vitro and in vivo, providing the proof-of-concept of this approach. Meanwhile, combination of BDA-366 with a mammalian target of rapamycin (mTOR) inhibitor exhibits excellent synergistic effects against lung cancer, demonstrating its superiority in overcoming chemoresistance [50].
Peptides: pep2 (IDP) and TAT-IDPDD/AA
The Bcl-2–IP3R interaction has been investigated for many years to help understand Ca2+ regulation by Bcl-2 at the molecular level [51,52]. The easiest way to generate an inhibitor disturbing a protein–protein interaction is to intercept the amino acid sequence of an interacting epitope. The region of IP3R that interacts with Bcl-2 is the subdomain comprising residues 1347–1426 within the regulatory and coupling domain. Two 20-mer peptides were generated based on this subdomain: peptide 1 (pep1) derived from amino acids 1365–1384; and peptide 2 (pep2) derived from amino acids 1389–1408 [37] (Figure 2a). In co-immunoprecipitation assays employing Jurkat cells and Bcl-2(+) WEHI7.2 cells, pep2 was found to inhibit the Bcl-2–IP3R interaction consistently at the concentration of 400 µM, whereas pep1 and control peptide (ctrlpep; scrambled sequence of pep2) did not inhibit the interaction. It was observed that pep2 disrupts the specific interaction between Bcl-2 and IP3R rather than the BH3-only protein Bim. Furthermore, pep2 reverses the Bcl-2 inhibitory effect on IP3R channel opening in vitro, and reverses Bcl-2 inhibition of anti-CD3-induced Ca2+ elevation in Bcl-2(+) WEHI7.2 cells (Figure 2b), as well as attenuating Bcl-2 inhibition of anti-CD3-induced apoptosis in Jurkat cells [37] (Figure 2c).
Figure 2.
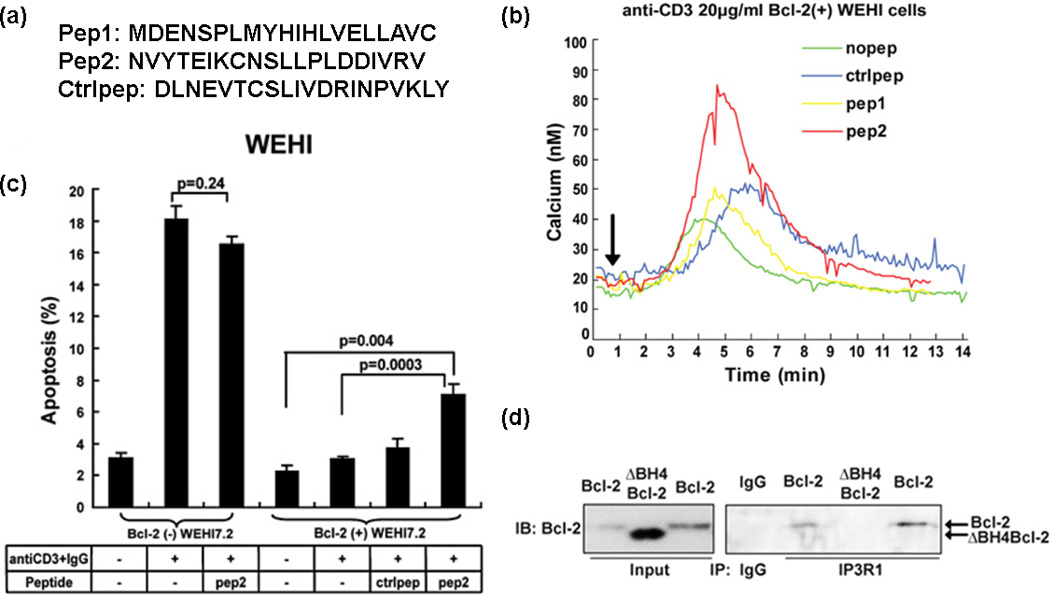
Peptide 2 (pep2) inhibits B cell lymphoma 2 (Bcl-2) BH4–IP3R interaction and IP3R-induced Ca2+ elevation to induce apoptosis. (a) The sequences of pep1, pep2 and ctrlpep are shown. (b) pep2 reverses Bcl-2 inhibition of anti-CD3-induced Ca2+ elevation in Bcl-2(+) WEHI7.2 cells. (c) pep2 enhances anti-CD3-induced apoptosis in Bcl-2(+) WEHI7.2 cells. (d) BH4 domain is required for Bcl-2–IP3R interaction. Reproduced, with permission, from [32,37].
Interestingly, it is the BH4 domain of Bcl-2 that directly interacts with IP3R, because BH4 deletion abrogates the Bcl-2–IP3R interaction (Figure 2d). Together with the fact that fluorescein isothiocyanate (FITC)-labeled TAT-BH4 (fTAT-BH4) can interact with IP3R, the accumulating evidence indicates the BH4 domain is necessary and sufficient to mediate Bcl-2–IP3R interaction. The BH4 domain peptide can inhibit IP3R-mediated Ca2+ release in unidirectional Ca2+-flux assays and pep2 can alleviate this inhibition. In addition, TAT-pep2 prevents the inhibition of anti-CD3-induced apoptosis by TAT-BH4 in the Jurkat human T cell leukemia line. With the assistance of chemical probe pep2, better understanding on the antiapoptotic activity of the BH4 domain has been achieved. These findings illustrate that the BH4 domain is a valuable therapeutic target for cancer therapy offering better potential to overcome Bcl-2-mediated resistance to cancer cell death.
Another synthetic peptide TAT-IDPDD/AA is an analog of TAT-pep2, with Asp–Asp (DD), the aspartyl protease site, replaced with Ala–Ala (AA) [49] (Figure 3a). TAT-IDPDD/AA also functions as a competitive inhibitor of the Bcl-2–IP3R interaction by binding to the BH4 domain of Bcl-2, and is more effective in inducing Ca2+ oscillations than TAT-pep2. This is probably because of its increased cellular uptake and stability as a result of reduced proteolytic cleavage rather than increased affinity of TAT-IDPDD/AA for Bcl-2. More importantly, TAT-IDPDD/AA induces a striking Ca2+ elevation in primary CLL cells, which have great clinical importance, because CLL is always associated with elevated Bcl-2 and Bcl-2 has a crucial role in apoptosis resistance in CLL. Not surprisingly, TAT-IDPDD/AA-evoked Ca2+ elevation induces apoptosis in primary CLL cells as shown by the images of Hoechst-stained nuclei (Figure 3b). This induction of cancer cell death is specific to TAT-IDPDD/AA, because neither TAT-Scr nor TAT-pep2 has similar apoptotic effects against CLL cells. Interestingly, TAT-IDPDD/AA-induced apoptosis is more sensitive toward cancer cells than normal cells. CLL cells are sixfold more sensitive to apoptosis induced by TAT-IDPDD/AA with an IC50 value of 12.7 µM versus normal peripheral blood lymphocytes (IC50 = 77.3 µM) (Figute 3c), indicating that TAT-IDPDD/AA selectively targets leukemia cells that overexpress Bcl-2. It is suggested that the divergent apoptotic sensitivity is caused by different expression levels of IP3R2 [53]. Interestingly, TAT-IDPDD/AA is now also known as BIRD-2 (Bcl-2/IP3 receptor disrupter-2). When this peptide is combined with other Bcl-2 inhibitors (e.g. HA14-1, ABT-263 and ABT-199), the apoptosis induction is enhanced compared with the single agent treatment [54–56].
Figure 3.
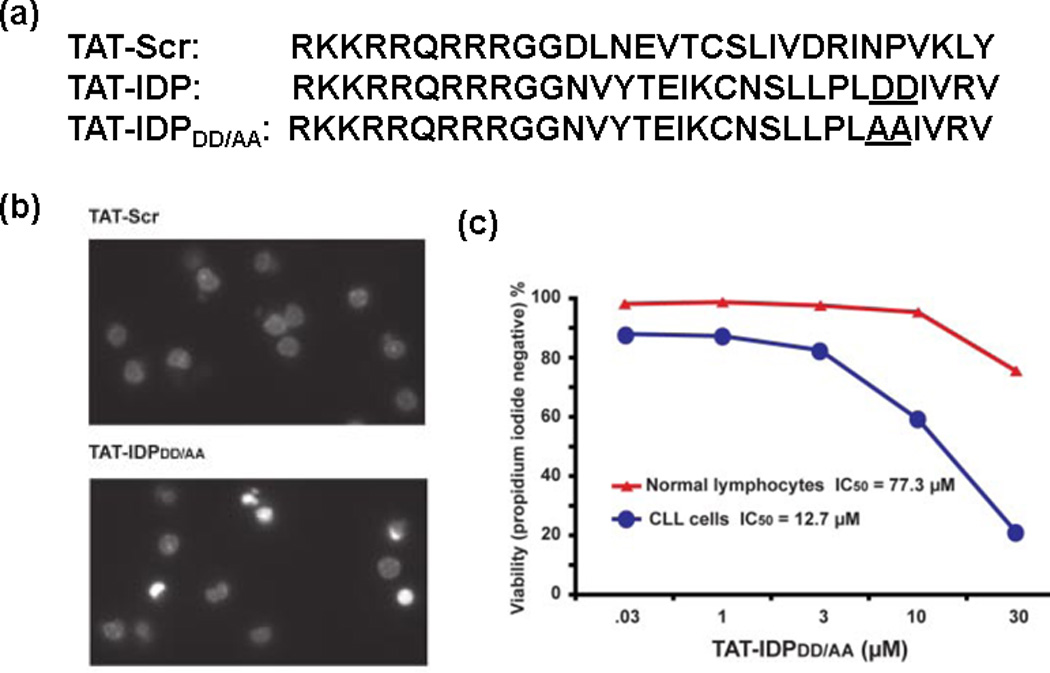
TAT-IDPDD/AA induces Ca2+-driven apoptosis in chronic lymphocytic leukemia (CLL) cells rather than normal lymphocytes. (a) The sequences of TAT-Scr, TAT-IDP and TAT-IDPDD/AA. (b) Images of Hoechst-stained nuclei demonstrate apoptotic morphology 24 h after treatment with 10 µM TAT-IDPDD/AA. (c) CLL cells are sixfold more sensitive to apoptosis induced by TAT-IDPDD/AA than normal lymphocytes. Reproduced, with permission, from [49].
Non-peptide small-molecule inhibitors: BDA-366
Very recently, the non-peptide small-molecule antagonist BDA-366 that targets the BH4 domain of Bcl-2 was discovered by our team by HTS [50]. Approximately 300 000 molecules from the National Cancer Institute (NCI) library were docked into the Bcl-2 BH4 domain (PDB ID codes: 1G5M and 1G5O) using the University of California, San Francisco (UCSF) DOCK 6.1 program suite. The cytotoxicity of the top 200 molecules was further screened against human lung cancer cells using the sulforhodamine B (SRB) assay, and BDA-366 (Figure 4a) was identified as the most potent compound. Among various lung cancer cells, cell lines with relatively higher levels of Bcl-2 (e.g. H157, H358, DMS53 and DMS153) were found more sensitive to BDA-366. Direct binding affinities of BDA-366 with Bcl-2, ΔBH1 Bcl-2, ΔBH2 Bcl-2, ΔBH3 Bcl-2 and ΔBH4 Bcl-2 were measured by a fluorescence polarization assay. BDA-366 binds to Bcl-2 with a Ki value of 3.3 nM, but ΔBH4 Bcl-2 fails to bind BDA-366 even at 0.5 µM. By contrast, deletion of BH1, BH2 or BH3 has little impact on this binding. Moreover, BDA-366 does not engage other Bcl-2 family members including Bcl-xL, Mcl-2 and Bf1/A1 (Figure 4b). It was confirmed by Bcl-2 knockdown cell lines that BDA-366 induces apoptosis in a Bcl-2-dependent fashion. Mutations in the Bcl-2 BH4 domain result in resistance to BDA-366 while maintaining sensitivity to the BH3 mimetic (ABT-199). These findings indicate that BDA-366 selectively binds Bcl-2 at the BH4 domain to induce cancer cell apoptosis. Further investigation suggests that interaction between BDA-366 and Bcl-2 BH4 domain is capable of altering Bcl-2 conformation via exposure of its BH3 domain leading to conversion of Bcl-2 from a survival protein to a killer. BDA-366-treated Bcl-2, rather than Bcl-2 alone, enhances the ability of Bax to bind to 6A7 antibody, whereas BDA-366 itself is not a Bax activator (Figure 4c). This suggests that BDA-366 induces Bcl-2-dependent Bax activation, and Bax is essential for BDA-366 to induce apoptotic cell death, because Bax-deficient MEF cells were significantly resistant to BDA-366. At the same time, BDA-366 reduces Bcl-2–IP3R binding in association with increased Ca2+ release, which could uncover an additional mechanism on how BDA-366 induces apoptosis in a BH4-binding-dependent manner.
Figure 4.
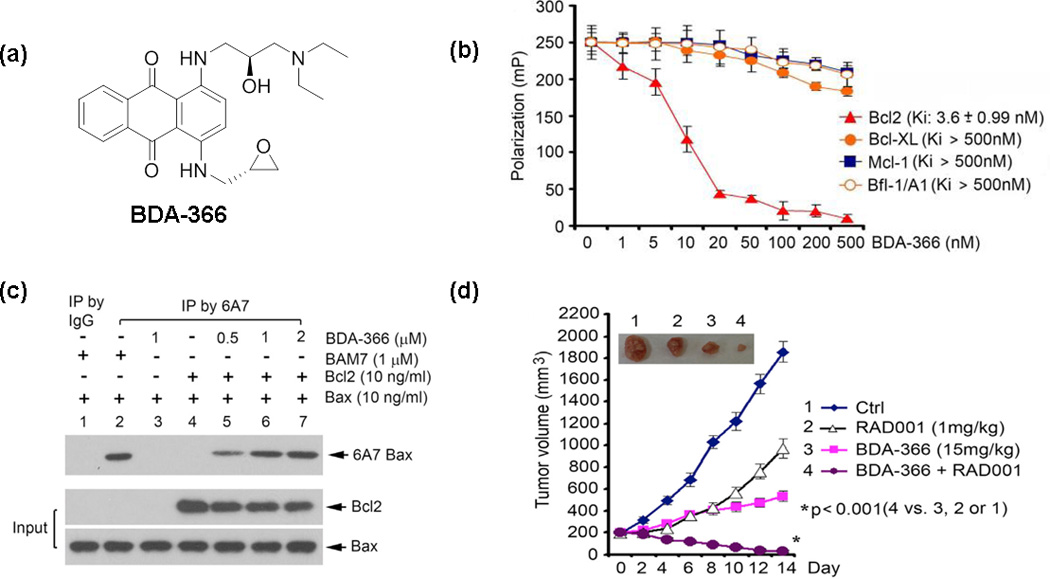
BDA-366 suppresses lung cancer tumor growth by selectively targeting the BH4 domain of Bcl-2 and Bcl-2-dependent Bax activation. (a) The chemical structure of BDA-366 is shown. (b) BDA-366 binds Bcl-2 selectively as illustrated by the competition fluorescence polarization assay. (c) BDA-366 can induce Bcl-2-dependent Bax activation rather than activate Bax directly. (d) BDA-366 strongly synergizes with RAD001 in vivo. Reproduced, with permission, from [50].
The anticancer effect of BDA-366 in vivo was also evaluated. In lung cancer mouse xenografts derived from H460 cells, BDA-366 displayed a dose-dependent regression of lung tumor growth at the doses of 10, 20 and 30 mg/kg via i.p. route for 14 days. Also, BDA-366 efficiently suppressed growth of small-cell lung cancer tumors in a patient-derived xenograft (SCLC PDX) mouse model at the dose of 20 mg/kg for 2 weeks. Moreover, combination of BDA-366 and an mTOR inhibitor RAD001 was explored given that expression of Bcl-2 is associated with resistance of cancer cells to mTOR inhibitors such as RAD001, which displays limited antitumor activity in patients with lung cancer [57,58]. Nu/Nu mice with non-small-cell lung cancer (NSCLC, i.e. H460) xenograft were treated with BDA-366 (15 mg/kg), RAD001 (1 mg/kg) and their combination, respectively (Figure 4d). Their combination exhibits a significantly greater efficacy than BDA-366 or RAD001 alone in suppressing lung tumor growth in vivo with no obvious normal tissue toxicity.
The Bcl-2 BH4 domain antagonist BDA-366 selectively binds the BH4 domain and exposes the BH3 domain of Bcl-2 to induce the conversion of Bcl-2 from a survival to a killer. The exciting pharmacological profile of BDA-366 against human lung cancer in vitro and in vivo demonstrates the great promise to develop Bcl-2 BH4 inhibitors as a new class of anticancer agents for lung cancer and other various types of cancers. Its markedly synergistic effect against lung cancer in combination with RAD001 displays the great potential of BDA-366 to overcome drug resistance. BDA-366 and its optimized analogs designed by our team are currently under preclinical development toward investigational new drug (IND)-enabling studies.
Concluding remarks and future perspectives
In conclusion, targeting the BH4 domain of Bcl-2 represents a novel and attractive strategy for developing a new class of cancer therapy. The BH4 domain is essential for Bcl-2 antiapoptotic function. Furthermore, the BH4 domain of Bcl-2 is involved in direct interactions with Bcl-2 family members and various other non-Bcl-2 proteins associated with different signaling pathways. Whereas peptides such as pep2 (IDP) and TAT-IDPDD/AA provide useful tools for better understanding of the antiapoptotic activity of the BH4 domain, the small molecule BDA-366 as a BH4 domain antagonist offers the first-in-class proof-of-concept toward a drug candidate with the great potential of overcoming drug resistance. We anticipate that more diverse small molecules capable of disturbing the interactions of the Bcl-2 BH4 domain with various proteins will be accessible based on modern drug discovery techniques and strategies, including HTS, to identify hits for hit-to-lead optimization or useful fragments with relatively weaker binding for fragment-based drug design [59–62]. Co-crystal structures of Bcl-2 BH4 and non-peptide small molecular antagonists are needed to aid further rational drug design and optimization. Development of novel small molecules as Bcl-2 BH4 domain inhibitors will not only facilitate the understanding of the crucial role of the BH4 domain but will also generate new anticancer agents that could prove to be a viable therapeutic approach to benefit cancer patients in the future.
Highlights.
No Bcl-2 inhibitors targeting the BH3 domain approved for clinical use
BH4 domain of Bcl-2 is identified as a promising novel target for cancer therapy
Bcl-2 BH4 domain interacts with Bcl-2 family members as well as non-Bcl-2 proteins
Interactions of BH4 domain with various proteins promote resistance to apoptosis
Targeting the BH4 domain of Bcl-2 offers great potential to overcome drug resistance
Acknowledgments
This work was supported by grants P30 DA028821, R01 DA038446 from the National Institutes of Health, Cancer Prevention Research Institute of Texas (CPRIT) award, R.A. Welch Foundation Chemistry and Biology Collaborative Grant from the Gulf Coast Consortia (GCC) and a training fellowship from the Keck Center for Interdisciplinary Bioscience Training of the GCC (NIGMS grant T32 GM089657), John Sealy Memorial Endowment Fund, Institute for Translational Sciences (ITS) and the Center for Addiction Research (CAR) at UTMB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Teaser: Targeting the BH4 domain of Bcl-2 offers a novel and attractive means for anticancer therapy by induction of apoptosis with an improved potential to overcome drug resistance.
Conflicts of interest
The authors declare no competing financial interest.
References
- 1.Czabotar PE, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 2.Xin M, et al. Small-molecule Bax agonists for cancer therapy. Nat. Commun. 2014;5:4935. doi: 10.1038/ncomms5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Z, et al. Direct activation of Bax protein for cancer therapy. Med. Res. Rev. 2015 doi: 10.1002/med.21379. PMID: 26395559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galsky MD, Vogelzang NJ. Docetaxel-based combination therapy for castration-resistant prostate cancer. Annal. Oncol. 2010;21:2135–2144. doi: 10.1093/annonc/mdq050. [DOI] [PubMed] [Google Scholar]
- 5.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kvansakul M, et al. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 2008;15:1564–1571. doi: 10.1038/cdd.2008.83. [DOI] [PubMed] [Google Scholar]
- 7.Besbes S, et al. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget. 2015;6:12862–12871. doi: 10.18632/oncotarget.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas S, et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets. 2013;17:61–75. doi: 10.1517/14728222.2013.733001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 10.Tse C, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 11.Souers AJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 12.Roberts AW, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonpavde G, et al. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Annal. Oncol. 2012;23:1803–1808. doi: 10.1093/annonc/mdr555. [DOI] [PubMed] [Google Scholar]
- 14.Chiappori A, et al. Obatoclax mesylate, a pan-bcl-2 inhibitor, in combination with docetaxel in a phase 1/2 trial in relapsed non-small-cell lung cancer. J. Thorac. Oncol. 2014;9:121–125. doi: 10.1097/JTO.0000000000000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson WH, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yecies D, et al. Acquired resistance to ABT-737 in lymphoma cells that upregulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woo SM, et al. Cafestol overcomes ABT-737 resistance in Mcl-1-overexpressed renal carcinoma Caki cells through downregulation of Mcl-1 expression and upregulation of Bim expression. Cell Death Dis. 2014;5:e1514. doi: 10.1038/cddis.2014.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang DC, et al. The conserved N-terminal BH4 domain of Bcl-2 homologues is essential for inhibition of apoptosis and interaction with CED-4. EMBO J. 1998;17:1029–1039. doi: 10.1093/emboj/17.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng EH, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- 20.Monaco G, et al. The selective BH4-domain biology of Bcl-2-family members: IP3Rs and beyond. Cell Mol. Life Sci. 2013;70:1171–1183. doi: 10.1007/s00018-012-1118-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rong YP, et al. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim. Biophys. Acta. 2009;1793:971–978. doi: 10.1016/j.bbamcr.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakhshi A, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 23.Petros AM, et al. Solution structure of the antiapoptotic protein bcl-2. Proc. Natl. Acad. Sci. U. S. A. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanada M, et al. Structure-function analysis of Bcl-2 protein. Identification of conserved domains important for homodimerization with Bcl-2 and heterodimerization with Bax. J. Biol. Chem. 1995;270:11962–11969. doi: 10.1074/jbc.270.20.11962. [DOI] [PubMed] [Google Scholar]
- 25.Trisciuoglio D, et al. Removal of the BH4 domain from Bcl-2 protein triggers an autophagic process that impairs tumor growth. Neoplasia. 2013;15:315–327. doi: 10.1593/neo.121392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabellini C, et al. BH4 domain of bcl-2 protein is required for its proangiogenic function under hypoxic condition. Carcinogenesis. 2013;34:2558–2567. doi: 10.1093/carcin/bgt242. [DOI] [PubMed] [Google Scholar]
- 27.Trisciuoglio D, et al. Involvement of BH4 domain of bcl-2 in the regulation of HIF-1-mediated VEGF expression in hypoxic tumor cells. Cell Death Differ. 2011;18:1024–1035. doi: 10.1038/cdd.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akl H, et al. A dual role for the anti-apoptotic Bcl-2 protein in cancer: mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta. 2014;1843:2240–2252. doi: 10.1016/j.bbamcr.2014.04.017. [DOI] [PubMed] [Google Scholar]
- 29.Hirotani M, et al. NH2-terminal BH4 domain of Bcl-2 is functional for heterodimerization with Bax and inhibition of apoptosis. J. Biol. Chem. 1999;274:20415–20420. doi: 10.1074/jbc.274.29.20415. [DOI] [PubMed] [Google Scholar]
- 30.Barclay LA, et al. Inhibition of pro-apoptotic BAX by a noncanonical interaction mechanism. Mol. Cell. 2015;57:873–886. doi: 10.1016/j.molcel.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu. Rev. Physiol. 2008;70:73–91. doi: 10.1146/annurev.physiol.70.021507.105852. [DOI] [PubMed] [Google Scholar]
- 32.Rong YP, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14397–14402. doi: 10.1073/pnas.0907555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenberg EF, et al. Bcl-2 regulation of the inositol 1,4,5-trisphosphate receptor and calcium signaling in normal and malignant lymphocytes: potential new target for cancer treatment. Biochim. Biophys. Acta. 2014;1843:2205–2210. doi: 10.1016/j.bbamcr.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bosanac I, et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420:696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 35.Mikoshiba K. Role of IP3 receptor signaling in cell functions and diseases. Adv. Biol. Regul. 2015;57:217–227. doi: 10.1016/j.jbior.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Foskett JK, et al. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rong YP, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol. Cell. 2008;31:255–265. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gump JM, Dowdy SF. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol. Med. 2007;13:443–448. doi: 10.1016/j.molmed.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Monaco G, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012;19:295–309. doi: 10.1038/cdd.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monaco G, et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 2015;290:9150–9161. doi: 10.1074/jbc.M114.622514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang HG, et al. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87:629–638. doi: 10.1016/s0092-8674(00)81383-5. [DOI] [PubMed] [Google Scholar]
- 42.Shibasaki F, et al. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2. Nature. 1997;386:728–731. doi: 10.1038/386728a0. [DOI] [PubMed] [Google Scholar]
- 43.Linette GP, et al. Cross talk between cell death and cell cycle progression: BCL-2 regulates NFAT-mediated activation. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9545–9552. doi: 10.1073/pnas.93.18.9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denis GV, et al. Bcl-2, via its BH4 domain, blocks apoptotic signaling mediated by mitochondrial Ras. J. Biol. Chem. 2003;278:5775–5785. doi: 10.1074/jbc.M210202200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorenson CM. Interaction of bcl-2 with Paxillin through its BH4 domain is important during ureteric bud branching. J. Biol. Chem. 2004;279:11368–11374. doi: 10.1074/jbc.M310079200. [DOI] [PubMed] [Google Scholar]
- 46.Jin Z, et al. Bcl2 suppresses DNA repair by enhancing c-Myc transcriptional activity. J. Biol. Chem. 2006;281:14446–14456. doi: 10.1074/jbc.M511914200. [DOI] [PubMed] [Google Scholar]
- 47.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 48.Qu W, et al. Acquisition of apoptotic resistance in cadmium-transformed human prostate epithelial cells: Bcl-2 overexpression blocks the activation of JNK signal transduction pathway. Environ. Health Perspect. 2007;115:1094–1100. doi: 10.1289/ehp.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong F, et al. Induction of Ca(2)+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood. 2011;117:2924–2934. doi: 10.1182/blood-2010-09-307405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han B, et al. Small-molecule Bcl2 BH4 antagonist for lung cancer therapy. Cancer Cell. 2015;27:852–863. doi: 10.1016/j.ccell.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen R, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanson CJ, et al. Bcl-2 suppresses Ca2+ release through inositol 1,4,5-trisphosphate receptors and inhibits Ca2+ uptake by mitochondria without affecting ER calcium store content. Cell Calcium. 2008;44:324–338. doi: 10.1016/j.ceca.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 53.Akl H, et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013;4:e632. doi: 10.1038/cddis.2013.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lavik AR, et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget. 2015;6:27388–27402. doi: 10.18632/oncotarget.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akl H, et al. HA14-1 potentiates apoptosis in B-cell cancer cells sensitive to a peptide disrupting IP receptor/Bcl-2 complexes. Int. J. Dev. Biol. 2015 doi: 10.1387/ijdb.150213gb. PMID: 26260683. [DOI] [PubMed] [Google Scholar]
- 56.Akl H, et al. HA14-1, but not the BH3 mimetic ABT-737, causes Ca2+ dysregulation in platelets and human cell lines. Haematologica. 2013;98:e49–e51. doi: 10.3324/haematol.2012.080598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Besse B, et al. Phase II study of everolimus-erlotinib in previously treated patients with advanced non-small-cell lung cancer. Annal. Oncol. 2014;25:409–415. doi: 10.1093/annonc/mdt536. [DOI] [PubMed] [Google Scholar]
- 58.Ramalingam SS, et al. Phase II study of docetaxel in combination with everolimus for second- or third-line therapy of advanced non-small-cell lung cancer. J. Thorac. Oncol. 2013;8:369–372. doi: 10.1097/JTO.0b013e318282709c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 60.Chen H, et al. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur. J. Med. Chem. 2013;62:498–507. doi: 10.1016/j.ejmech.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen H, et al. Discovery of potent anticancer agent HJC0416, an orally bioavailable small molecule inhibitor of signal transducer and activator of transcription 3 (STAT3) Eur. J. Med. Chem. 2014;82:195–203. doi: 10.1016/j.ejmech.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen H, et al. Evolutions in fragment-based drug design: the deconstruction-reconstruction approach. Drug Discov. Today. 2015;20:105–113. doi: 10.1016/j.drudis.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katz C, et al. Molecular basis of the interaction between the antiapoptotic Bcl-2 family proteins and the proapoptotic protein ASPP2. Proc. Natl. Acad. Sci. U. S. A. 2008;105:12277–12282. doi: 10.1073/pnas.0711269105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trisciuoglio D, et al. Bcl-2 regulates HIF-1alpha protein stabilization in hypoxic melanoma cells via the molecular chaperone HSP90. PloS One. 2010;5:e11772. doi: 10.1371/journal.pone.0011772. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Hou Y, et al. Bcl2 impedes DNA mismatch repair by directly regulating the hMSH2–hMSH6 heterodimeric complex. J. Biol. Chem. 2007;282:9279–9287. doi: 10.1074/jbc.M608523200. [DOI] [PubMed] [Google Scholar]
- 66.Xie M, et al. Bcl2 induces DNA replication stress by inhibiting ribonucleotide reductase. Cancer Res. 2014;74:212–223. doi: 10.1158/0008-5472.CAN-13-1536-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vervliet T, et al. Bcl-2 binds to and inhibits ryanodine receptors. J. Cell Sci. 2014;127:2782–2792. doi: 10.1242/jcs.150011. [DOI] [PubMed] [Google Scholar]
- 68.Shimizu S, et al. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]